
Abstract
Alzheimer’s disease (AD) is a neurodegenerative disease characterized by impaired memory function and oxidative damage. NO is a major signaling molecule produced in the central nervous system to modulate neurological activity through modulating nitric oxide synthase. Recently, PPAR-γ agonists have shown neuroprotective effects in neurodegenerative disorders. However, there have been only a few studies identifying mechanisms through which cognitive benefits may be exerted. The present study was designed to investigate the possible nitric oxide mechanism in the protective effect of pioglitazone against streptozotocin (STZ)-induced memory dysfunction. Wistar rats were intracerebroventricularly (ICV) injected with STZ. Then rats were treated with pioglitazone, NO modulators [l-arginine and nitro-l-arginine methyl ester (L-NAME)] for 21 days. Behavioral alterations were assessed in between the study period. Animals were sacrificed immediately after behavioral session, and mito-oxidative parameters, TNF-α, IL-6, and caspase-3 activity were measured. STZ-treated rats showed a memory deficit and significantly increased in mito-oxidative damage and inflammatory mediators and apoptosis in the hippocampus. Chronic treatment of pioglitazone significantly improved memory retention and attenuated mito-oxidative damage parameters, inflammatory markers, and apoptosis in STZ-treated rats. However, l-arginine pretreatment with lower dose of pioglitazone has not produced any protective effect as compared to per se. Furthermore, pretreatment of L-NAME significantly potentiated its protective effect, which indicates the involvement of nitric oxide for activation of PPAR-γ action. These results demonstrate that pioglitazone offers protection against STZ-induced memory dysfunction possibly due to its antioxidant, anti-inflammatory, and anti-apoptotic action mediating nitric oxide pathways and, therefore, could have a therapeutic potential in AD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Memory impairment is a prominent feature of old age and of several neurodegenerative disorders like AD, Huntington’s chorea, and Down’s syndrome (Siemers 2011; Pike et al. 2011; Duff et al. 2010). Several pathomechanisms of memory impairment have been documented, out of which nitrosative oxidative stress plays a critical role in etiology of the above diseases. Nitric oxide (NO) is a highly reactive and diffusible signaling molecule that plays key roles in modulating both physiologic and pathologic processes in promoting cell survival and inducing cell death (Choi et al. 2002). It is well established that NO formed in the hippocampus has been suggested to have a significant role in learning and memory processes (Steinert et al. 2010). Furthermore, NO-cGMP pathway has been implicated in the induction of hippocampal long-term potentiation (LTP), which is known to be the predominant mechanisms of learning and memory processes (Feil and Kleppisch 2008; Steinert et al. 2010). Several behavioral studies have demonstrated the involvement of NO in certain forms of memory formation and blockade of learning by NO synthase (NOS) inhibitors (Meyer et al. 1998; Yamada et al. 1995).
Streptozotocin (STZ), a glucosamine-nitrosourea compound, generates nitrosative stress by reacting with NO-associated free radicals and destroys DNA of β cells in pancreatic islet (Van Dyke et al. 2010). It has been demonstrated that administration of STZ through intracerebroventricular route in rats has provided a relevant animal model for sporadic dementia of the Alzheimer’s type (SDAT), which is characterized by progressive deterioration of cognition, cerebral glucose and energy metabolism, along with oxidative stress (Hoyer et al. 1994). Even more interesting, brains intracerebroventricularly (ICV) injected with STZ (ICV-STZ) display many pathological features of SDAT including amyloid-β (Aβ) and tau pathologies (Grunblatt et al. 2007; Salkovic-Petrisic and Hoyer 2007). ICV-STZ causes a cholinergic deficiency, supported by reduced choline acetyltransferase (ChAT) activity in the hippocampus (Prickaerts et al. 1999).
Peroxisome proliferator-activated receptor gamma (PPAR-γ) was recognized as a therapeutic target for AD about a decade ago because of not only its effects on insulin sensitization and energy metabolism but also its multiple actions on the brain (Kaundal and Sharma 2010; Nicolakakis and Hamel 2010). Several in vitro and in vivo studies have demonstrated that PPAR-γ is widely expressed in several regions of the brain including piriform cortex, basal ganglia, and dentate gyrus, both in neuronal and non-neuronal cells (Moreno et al. 2004). Moreover, it has been demonstrated that PPAR-γ agonists reduce neuronal cell loss in in vitro models of neurotoxicity (Fuenzalida et al. 2007) and in in vivo models of cerebral ischemia-reperfusion injury (Culman et al. 2007), Parkinson’s disease (Dehmer et al. 2004), and amyotrophic lateral sclerosis (Kiaei et al. 2005). Pioglitazone has been shown to reduce neuroinflammation, attenuate mitochondrial dysfunction-associated oxidative damage, and reduce cell death following CNS injury (Prakash and Kumar 2013; Kapadia et al. 2008). Pioglitazone’s ability to target multiple cellular mechanisms may provide an advantage over other therapeutics for cognitive dysfunction which targets a single secondary mechanism (Sauerbeck et al. 2011). Recently, we have reported the neuroprotective potential of pioglitazone in different neurological states like brain aging and chronic fatigue syndrome (Prakash and Kumar 2013; Kumar et al. 2010). However, the exact protective mechanism of pioglitazone is still unclear. Recently, PPAR-γ ligands have been reported to increase endothelial NO, but the signaling mechanisms are still unclear so far (Allami et al. 2011). Although the role of NO has been demonstrated in pioglitazone-induced peripheral effects (Matsumoto et al. 2007), its role in cognitive dysfunction is still far from our understanding.
Therefore, the present study has been designed to investigate the probable role of nitric oxide mechanism in the neuroprotective effect of pioglitazone against streptozotocin-induced memory dysfunction.
Materials and Methods
Animals
Male young Wistar rats (180–200 g) of age 3.5 months bred in the Central Animal House, Panjab University, Chandigarh, India were used in the study. Animals were acclimatized to laboratory conditions at room temperature prior to experimentation. Following surgery, animals were kept under standard conditions of a 12-h light/dark cycle with food and water ad libitum in groups of two in plastic cages with soft bedding. All the experiments were carried out between 09.00 and 16.00 h. The experimental protocol was approved by the Institutional Animal Ethics Committee (IAEC) of Panjab University and carried out in accordance with the guidelines of Committee for Control and Supervision of Experimentation on Animals (CPCSEA), Government of India on animal experimentation.
Surgery and Intracerebroventricular Administration of Streptozotocin
Surgery was performed as per the previously described protocol (Prakash and Kumar 2009). All animals were anesthetized with thiopental sodium (45 mg/kg, i.p.) and positioned in a stereotaxic apparatus. The head was positioned in a frame and a midline sagittal incision was made in the scalp. Two holes were drilled in the skull for the placement of the injection cannula on both sides over the lateral cerebral ventricle using the following coordinates as described by Paxinos and Watson: 0.8 mm posterior to bregma, 1.5 mm lateral to sagittal suture, and 3.6 mm beneath the cortical surface of brain. The scalp was then closed with a suture and dental cement. After surgery, all animals received gentamicin (5 mg/kg, i.p.) to prevent sepsis after 1 week of surgery. Animals were given bilateral ICV injection of STZ (3 mg/kg) with two divided doses (1 day and 3 days) with the help of a Hamilton microsyringe through the cannula. Streptozotocin was prepared in artificial cerebrospinal fluid and a solution of 25 mg/ml was made. This solution was made freshly and just before injection. In the sham group, artificial cerebrospinal fluid (ACSF; in mmol/l—147 NaCl, 2.9 KCl, 1.6 MgCl2, 1.7 CaCl2, and 2.2 dextrose) was injected on the same days as in the streptozotocin group. To promote diffusion, the microsyringe was left in place for a period of 1 min following injection. Special care of the animals was taken during the postoperative period.
Drugs and Treatment Schedule
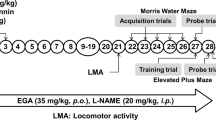
Streptozotocin (STZ), nitro-l-arginine methyl ester (L-NAME) and l-arginine (LA) were purchased from Sigma Chemicals Co., St. Louis, MO, USA. Pioglitazone (PIO) was gifted from Pancea Biotech, Mohali. STZ was prepared in ACSF and delivered in a 10-μl injection volume for ICV administration. Pioglitazone was suspended in 0.5 % w/v sodium-carboxy-methyl-cellulose (Na-CMC) and administered orally in the dose of 5 ml/kg body weight for 21 days. LA and L-NAME were administered 30 min before pioglitazone treatment. Animals were selected randomly based on their body weights into ten groups of six animals each. The study was performed in multiple phases as shown in Scheme 1. Three animals died during the experimental period due to infection.

Protocol design
Treatment group
-
1.
Control (vehicle)
-
2.
Sham-operated group (ACSF, ICV)
-
3.
STZ-treated group (3 mg/kg, ICV)
-
4.
PIO (30 mg/kg p.o.) per se
-
5.
LA (50 mg/kg i.p.) + STZ
-
6.
L-NAME (5 mg/kg i.p.) + STZ
-
7.
PIO (15 mg/kg p.o.) + STZ
-
8.
PIO (30 mg/kg p.o.) + STZ
-
9.
LA (50 mg/kg i.p.) + PIO (15 mg/kg p.o.) + STZ
-
10.
L-NAME (5 mg/kg i.p.) + PIO (15 mg/kg p.o.) + STZ
Behavioral Assessment
Assessment of Cognitive Performance (Morris Water Maze Task)
The acquisition and retention of memory was evaluated by using the Morris water maze (Prakash and Kumar 2009). Morris water maze consisted of a large circular pool (150 cm in diameter, 45 cm in height, filled to a depth of 30 cm with water at 28 ± 1 °C). The pool was divided into four equal quadrants with the help of two threads, fixed at right angle to each other. The pool was placed in an illuminated light room among the several colored clues. These external clues remained unchanged throughout the experimental period and used as reference memory. A circular platform (4.5 cm diameter) was placed in one quadrant of the pool, 1 cm above the water level, during the acquisition phase. The same platform was placed 1 cm below the water level for retention phase. The position of the platform was not changed in any quadrant during assessment of both the phases. Each animal was subjected to four consecutive trials with a gap of 5 min. The animal was gently placed in the water of the pool between quadrants, facing the wall of pool with drop location, changed for each trial, and allowed 120 s to locate the platform. Then, the animal was allowed to stay on the platform for 20 s. If the animal failed to reach the platform within 120 s, the same was guided to reach the platform and remained there for the next 20 s.
-
(a)
Maze acquisition phase (training)
Animals received two consecutive daily training sessions in the following days 12 and 13. During the acquisition phase, each rat was put into the water in any one of four starting positions, the sequence of which was selected randomly. The time latency to reach the visual platform (acquisition latency) was measured.
-
(b)
Maze retention phase (testing for retention of the learned task)
After completion of the acquisition phase, the animal was released randomly from one of the edges facing the wall of the pool to assess for memory retention. Time latency to find the hidden platform on days 14 and 21 following the start of STZ administration was recorded, termed as first retention latency (1st RL) (14th day) and second retention latency (2nd RL) (21st day), respectively.
Biochemical Tests
Biochemical tests were conducted 24 h after the last behavioral test. The animals were sacrificed by decapitation. The brains were removed and rinsed with ice-cold isotonic saline. Brains were then homogenized with ice-cold 0.1 mmol/l phosphate buffer (pH 7.4). The homogenate (10 % w/v) was then centrifuged at 10,000×g for 15 min and the supernatant so formed was used for the biochemical estimations.
Measurement of Lipid Peroxidation
The extent of lipid peroxidation in the brain was determined quantitatively by performing the method as described by Wills (1966). The amount of malondialdehyde (MDA), a measure of lipid peroxidation, was measured by reaction with thiobarbituric acid at 532 nm using a Perkin Elmer Lambda 20 spectrophotometer. The values were calculated using the molar extinction coefficient of chromophore [1.56 × 105 (mol/l)−1 cm−1].
Estimation of Nitrite
The accumulation of nitrite in the supernatant, an indicator of the production of nitric oxide, was determined by a colorimetric assay with Greiss reagent [0.1 % N-(1-napththyl) ethylene diamine dihydrochloride, 1 % sulfanilamide, and 5 % phosphoric acid] according to Green et al. (1982). Equal volumes of the supernatant and the Greiss reagent were mixed and the mixture was incubated for 10 min at room temperature in the dark. The absorbance was measured at 540 nm using a Perkin Elmer Lambda 20 spectrophotometer. The concentration of nitrite in the supernatant was determined from sodium nitrite standard curve.
Superoxide Dismutase Activity
Superoxide dismutase (SOD) activity was assayed by the method of Kono (1978). The assay system consisted of EDTA 0.1 mM, sodium carbonate 50 mM, and 96 mM of nitro blue tetrazolium (NBT). In the cuvette, 2 ml of the above mixture, 0.05 ml of hydroxylamine, and 0.05 ml of the supernatant were added and the auto-oxidation of hydroxylamine was measured for 2 min at 30-s interval by measuring the absorbance at 560 nm using a Perkin Elmer Lambda 20 spectrophotometer.
Estimation of Glutathione Levels
Reduced glutathione (GSH) in hippocampus and cortex was estimated according to the method described by Ellman (1959). Results were calculated using molar extinction coefficient of chromophore (1.36 × 104 M−1 cm−1) and expressed as percentage of control.
Estimation of Acetyl Cholinesterase (AChE) Activity
AChE is a marker of extensive loss of cholinergic neurons in the forebrain. The AChE activity was assessed by the Ellman method (Ellman et al. 1961). The assay mixture contained 0.05 ml of supernatant, 3 ml of sodium phosphate buffer (pH 8), 0.1 ml of acetylthiocholine iodide, and 0.1 ml of DTNB (Ellman reagent). The change in absorbance was measured for 2 min at 30-s interval at 412 nm using a Perkin Elmer Lambda 20 spectrophotometer. Results were expressed as micromoles of acetylthiocholine iodide hydrolyzed per minute per milligram of protein.
Mitochondrial Complex Estimation
Isolation of Rat Brain Mitochondria
Rat brain mitochondria were isolated by the method of Berman and Hastings (1999). The hippocampus was homogenized in isolation buffer with EGTA (215 mM mannitol, 75 mM sucrose, 0.1 % BSA, 20 mM HEPES, 1 mM EGTA, pH 7.2). Homogenates were centrifuged at 13,000×g for 5 min at 4 °C. The pellet was resuspended in isolation buffer with EGTA and spun again at 13,000×g for 5 min. The resulting supernatants were transferred to new tubes and topped off with isolation buffer with EGTA and again spun at 13,000×g for 10 min. Pellets containing pure mitochondria were resuspended in isolation buffer without EGTA.
Estimation of NADH Dehydrogenase Activity
NADH dehydrogenase activity was measured spectrophotometrically (UV-Pharmaspec 1700; Shimadzu, Japan) by the method of King and Howard (1967). The method involves catalytic oxidation of NADH to NAD+ with subsequent reduction of cytochrome c.
Estimation of Succinate Dehydrogenase (SDH) Activity
Succinate dehydrogenase (SDH) activity was measured spectrophotometrically (UV-Pharmaspec 1700; Shimadzu, Japan) according to King (1967). The method involves oxidation of succinate by an artificial electron acceptor, potassium ferricyanide.
Estimation of MTT (Mitochondrial Redox Activity) Assay
The method employed in the present study is based on the in vitro studies to evaluate mitochondrial redox activity through the conversion of MTT tetrazolium salt to formazan crystals by mitochondrial respiratory chain reactions in isolated mitochondria by the method of Liu (Liu et al. 1997). The absorbance of the resulting medium was measured by an ELISA reader at 580 nm wavelength.
Estimation of Cytochrome Oxidase Activity
Cytochrome oxidase activity was assayed in brain mitochondria according to the method of Sottocasa (Sottocasa et al. 1967). The assay mixture contained 0.3 mM reduced cytochrome c in 75 mM phosphate buffer. The reaction was started by the addition of solubilized mitochondrial sample and absorbance change was recorded at 550 nm for 2 min.
Estimations of TNF-α and IL-6
The quantification of TNF-α and IL-6 was done by the help and instructions provided by R&D Systems Quantikine rat TNF-α and IL-6 immunoassay kits. The Quantikine rat TNF-α and IL-6 immunoassays are 4.5 h solid-phase ELISA designed to measure rat TNF-α and IL-6 levels. The assays employ the sandwich enzyme immunoassay technique. A monoclonal antibody specific for rat TNF-α and IL-6 have been pre-coated in the microplate. Standard control and samples are pipetted into the wells and any rat TNF-α and IL-6 present are bound by the immobilized antibody. After washing away any unbound substance, an enzyme-linked polyclonal antibody specific for rat TNF-α and IL-6 is added to the wells. Following a wash to remove any unbound antibody-enzyme reagent, a substrate solution is added to the wells. The enzyme reaction yields a blue product that turns yellow when the stop solution is added. The intensity of the color measured is in proportion to the amount of rat TNF-α and IL-6 bound in the initial steps. The sample values are then read off the standard curve.
Estimation of Caspase-3 Colorimetric Assay
Caspase-3, also known as CPP-32, Yama, or Apopain, is an intracellular cysteine protease that exists as a pro-enzyme, becoming activated during the cascade of events associated with apoptosis. The tissue lysates/homogenates can then be tested for protease activity by the addition of a caspase-specific peptide that is conjugated to the color reporter molecule p-nitroanaline (pNA). The cleavage of the peptide by the caspase releases the chromophore pNA, which can be quantitated spectrophotometrically at a wavelength of 405 nm. The level of caspase enzymatic activity in the cell lysate/homogenate is directly proportional to the color reaction. The enzymatic reaction for caspase activity was carried out using the Imgenex (San Diego, USA) caspase-3 colorimetric kit.
Data Analysis
Values are expressed as mean ± SEM. The behavioral assessment data were analyzed by a repeated measures two-way analysis of variance (ANOVA) with drug-treated groups as between-subjects factors and sessions as the within-subjects factors. The biochemical estimations were separately analyzed by one-way ANOVA. Post hoc comparisons between groups were made using Tukey’s test. P <0.05 was considered significant.
Results
Effect of Pioglitazone and its Interaction with Nitric Oxide Modulators on Spatial Navigation Task in Streptozotocin-Injected Rats
There was a significant difference in the mean IAL of streptozotocin-injected group as compared to sham treatment on day 13 indicating streptozotocin induced impaired acquisition of spatial navigation task. In contrast, chronic pioglitazone (15 and 30 mg/kg) treatment significantly decreased IAL to reach the platform in the pre-trained rats as compared to STZ rats on day 13 following streptozotocin injection [a two-way ANOVA revealed on IAL on group (F 14,8 = 15.36, P < 0.001), session (F 25,14 = 76.45, P < 0.001), and interaction drug treatment × session group (F 14,8 = 132.5, P < 0.001)] (Table 1).
Following training, the mean retention latencies (1st and 2nd RL) to escape onto the hidden platform was significantly decreased in sham-operated and ACSF-injected rats on days 14 and 21, respectively, as compared to IAL on day 13 following streptozotocin injection. On the contrary, the performance in the STZ-injected rats was significantly altered (increased mean retention latencies) after initial training in the water maze on days 14 and 21, as compared to IAL on day 13. The results suggest that streptozotocin caused significant cognitive impairment. However, chronic pioglitazone (15 and 30 mg/kg, p.o.) treatment showed a significant decline and improved the retention performance of the spatial navigation task in the 1st and 2nd RL on days 14 and 21, respectively, as compared to STZ-injected rats following streptozotocin injection (Table 1). Further, LA (50 mg/kg) pretreatment with sub-effective dose of pioglitazone (15 mg/kg) has not produced a significant effect as compared to pioglitazone (low dose) + STZ group. However, L-NAME (5 mg/kg) pretreatment with sub-effective dose of pioglitazone (15 mg/kg) significantly potentiated its protective effect which was significant as compared to pioglitazone (low dose) + STZ group. PIO (30 mg/kg), LA (50 mg/kg), and L-NAME (5 mg/kg) per se did not produce any significant effect in STZ-treated groups as compared to sham treatment.
Effect of Pioglitazone and its Interaction with Nitric Oxide Modulators on Oxidative Parameters in Streptozotocin-Injected Rats
Central streptozotocin administration caused a significant rise in brain MDA level (F 9,40 = 92.28, P < 0.0001) and nitrite concentration (F 9,40 = 18.39, P < 0.0001) and depletion of reduced GSH (F 9,40 = 34.94, P < 0.0001), SOD (F 9,40 = 33.04, P < 0.0001), and catalase (F 9,40 = 45.77, P < 0.0001) activities as compared to sham rats. However, chronic pioglitazone (15 and 30 mg/kg) treatment significantly attenuated oxidative stress (reduced the rise in malondialdehyde, nitrite and restored SOD, catalase and reduced glutathione levels), as compared to streptozotocin-treated group. Further, LA (50 mg/kg) pretreatment with sub-effective dose of pioglitazone (15 mg/kg, p.o.) has not produced a significant effect as compared to pioglitazone (low dose) + STZ group. However, L-NAME (5 mg/kg) pretreatment with sub-effective dose of pioglitazone (15 mg/kg) significantly potentiated its protective effect which was significant as compared to pioglitazone (low dose) + STZ group. Besides, PIO (30 mg/kg), LA (50 mg/kg), and L-NAME (5 mg/kg) per se treatment did not produce any significant effect on oxidative damage as compared to sham treatment (Fig. 1a, b).

a, b Effect of pioglitazone and their interaction with nitric oxide modulators on oxidative parameter in streptozotocin injected rats. Values are mean ± SEM. a P <0.05 as compared to sham group; b P <0.05 as compared to streptozotocin-treated group; c P <0.05 as compared to PIO (15) + STZ group; d P <0.05 as compared to L-NAME (5 mg/kg) + STZ group; e P <0.05 as compared to PIO per se. Repeated measures one-way ANOVA followed by Tukey’s test for multiple comparisons
Effect of Pioglitazone and its Interaction with Nitric Oxide Modulators on Mitochondria Respiratory Enzymes Activities in Streptozotocin-Treated Rats
ICV streptozotocin administration significantly impaired mitochondrial complexes enzyme [NADH dehydrogenase (F 9,40 = 16.12, P < 0.0001), succinate dehydrogenase (F 9,40 = 25.41, P < 0.0001), and cytochrome oxidase (F 9,40 = 22.75, P < 0.0001)] and cell viabilities (F 9,40 = 24.9, P < 0.0001) in hippocampus of STZ rats as compared to sham-operated groups. However, pioglitazone (15 and 30 mg/kg) administration significantly restored the levels of mitochondrial enzymes and increased the cell viabilities as compared to streptozotocin-treated rats. Further, LA (50 mg/kg) pretreatment with sub-effective dose of pioglitazone (15 mg/kg) has not produced a significant effect as compared to pioglitazone (low dose) + STZ group. However, L-NAME (5 mg/kg) pretreatment with sub-effective dose of pioglitazone (15 mg/kg) significantly potentiated its protective effect which was significant as compared to pioglitazone (low dose) + STZ group. Besides, PIO (30 mg/kg), LA (50 mg/kg), and L-NAME (5 mg/kg) per se did not produce any significant effect on mitochondrial enzymes dysfunction as compared to sham treatment (Fig. 2).

Effect of pioglitazone and their interaction with nitric oxide modulators on a complex I, b complex II, c MTT ability, and d complex IV in streptozotocin-treated rats. Values are mean ± SEM. a P <0.05 as compared to sham group; b P <0.05 as compared to streptozotocin-treated group; c P <0.05 as compared to PIO (15) + STZ group; d P <0.05 as compared to L-NAME (5) + STZ group. Repeated measures one-way ANOVA followed by Tukey’s test for multiple comparisons
Effect of Pioglitazone and its Interaction with Nitric Oxide Modulators on Inflammatory Markers (TNF-α and IL-6) in Streptozotocin-Injected Rats
There was a significant increase in the level TNF-α and IL-6 in streptozotocin-treated rats as compared to the control group. However, chronic pioglitazone (15 and 30 mg/kg) administration significantly attenuated the elevated levels of TNF-α (F 9,40 = 15.64, P < 0.0001) and IL-6 (F 9,40 = 34.94, P < 0.001) in streptozotocin-treated rats. Further, LA (50 mg/kg i.p.) pretreatment with sub-effective dose of PIO (15 mg/kg) has not produced a significant effect as compared to pioglitazone (low dose) + STZ group. However, L-NAME (5 mg/kg) pretreatment with sub-effective dose of PIO (15 mg/kg) significantly potentiated the protective effect of pioglitazone which was significant as compared to pioglitazone (low dose) + STZ group (Fig. 3).

Effect of pioglitazone and their interaction with nitric oxide modulators on inflammatory markers (TNF-α and IL-6) in streptozotocin-treated rats. Values are mean ± SEM. a P <0.05 as compared to sham group; b P <0.05 as compared to streptozotocin-treated group; c P <0.05 as compared to PIO (15) + STZ group; d P <0.05 as compared to L-NAME (5) + STZ group. Repeated measures one-way ANOVA followed by Tukey’s test for multiple comparisons
Effect of Pioglitazone and its Interaction with Nitric Oxide Modulators on Caspase-3 Activity in Streptozotocin-Injected Rats
Central administration of streptozotocin showed significant increase of caspase-3 activity in streptozotocin-treated rats as compared to the control group. However, chronic pioglitazone (15 and 30 mg/kg) administration significantly attenuated the increased activity of caspase-3 (F 9,40 = 20.38, P < 0.001) in streptozotocin-treated rats (P < 0.01). Further, LA (50 mg/kg i.p.) pretreatment with sub-effective dose of PIO (15 mg/kg) has not produced a significant effect as compared to pioglitazone (low dose) + STZ group. However, L-NAME (5 mg/kg) pretreatment with sub-effective dose of PIO (15 mg/kg) significantly potentiated the protective effect of pioglitazone which was significant as compared to pioglitazone (low dose) + STZ group (Fig. 4).

Effect of pioglitazone and their interaction with nitric oxide modulators on caspase-3 activity in streptozotocin-treated rats. Values are mean ± SEM. a P <0.05 as compared to sham group; b P <0.05 as compared to streptozotocin-treated group; c P <0.05 as compared to PIO (15) + STZ group; d P <0.05 as compared to L-NAME (5) + STZ group. Repeated measures one-way ANOVA followed by Tukey’s test for multiple comparisons
Effect of Pioglitazone and its Interaction with Nitric Oxide Modulators on Acetylcholinesterase Activity in Streptozotocin-Injected Rats
Intracerebroventricular administration of ACSF did not produce any significant effect on acetylcholinesterase activity as compared to vehicle control group. Central streptozotocin administration significantly increased acetylcholinesterase activity leading to memory impairment as compared to sham-operated rats. However, pioglitazone (15 and 30 mg/kg) administration significantly attenuated acetylcholinesterase activity (F 9,40 = 68.89, P < 0.0001) as compared to streptozotocin-treated group. Further, LA (50 mg/kg) pretreatment with sub-effective dose of PIO (15 mg/kg) has not produced a significant effect as compared pioglitazone (low dose) + STZ group. However, L-NAME (5 mg/kg) pretreatment with sub-effective dose of PIO (15 mg/kg) significantly potentiated the protective effect of pioglitazone which was significant as compared to pioglitazone (low dose) + STZ group. Besides, PIO (30 mg/kg), LA (50 mg/kg), and L-NAME (5 mg/kg) per se did not produce any significant effect on acetylcholinesterase enzymes as compared to sham treatment (Figs. 5 and 6).

Effect of pioglitazone and their interaction with nitric oxide modulators on acetylcholinesterase activity in streptozotocin-treated rats. Values are mean ± SEM. a P <0.05 as compared to sham group; b P <0.05 as compared to streptozotocin-treated group; c P <0.05 as compared to PIO (15) + STZ group; d P <0.05 as compared to L-NAME (5) + STZ group; e P <0.05 as compared to PIO per se. Repeated measures one-way ANOVA followed by Tukey’s test for multiple comparisons

Possible mechanism of action of pioglitazone-mediated nitric oxide pathways against ICV-STZ-induced memory dysfunction
Discussion
Evidence suggested that there is an intimate link between an excessive generation of reactive oxygen, nitrogen species (ROS/RNS) and mitochondrial enzyme alteration (Penna et al. 2012). ROS/RNS can damage various cellular components, such as proteins, lipids, and DNAs, and modulate various pathways, particularly those involving MAPKs (e.g., JNK, ERK, and p38 MAPK) and PKCs (Penna et al. 2012). Mitochondria are particularly vulnerable to oxidative stress because of excessive ROS generation that constantly occurs during oxidative phosphorylation. It has been suggested that ROS-mediated signals (mainly by H2O2) arising within mitochondria can also generate a retrograde response that is conveyed to the nucleus, causing the upregulation of nuclear genes encoding mitochondrial proteins and leading to the induction of mitochondrial biogenesis (Butow and Avadhani 2004). The results of the present study indicate that central administration of streptozotocin impaired mitochondrial enzyme complex activities as indicated by a decrease in the NADH dehydrogenase and succinate dehydrogenase activity and MTT ability and cytochrome c oxidase activities. A recent cell culture study has also suggested that streptozotocin has produced mitochondrial dysfunction in human hepatoma HepG2 cells by increasing ROS/RNS production (Raza and John 2012). It can suggest that NO can directly cause the release of cytochrome c that results in the loss of the mitochondrial transmembrane potential. NO binds with CytCoxidase (complex IV) in the mitochondrial chain of electron transfer (Riobo et al. 2001). Inflammation plays a pivotal role in the pathogenesis of several neurological disorders (Amor et al. 2010). The ROS/RNS are important contributors in activation of microglia, which are capable of generating vast amounts of oxidizing radicals such as superoxide, hydrogen peroxide, and nitric oxide as well as proinflammatory cytokines such as TNF-α and IL-6 (Liu et al. 2003; Merrill and Benveniste 1996). Activated microglia oxidative stress is associated with both neuroinflammation and programmed cell death (apoptosis) (Kubera et al. 2011). We found evidences of each of these processes in the hippocampus following streptozotocin administration. Thus, increases in proinflammatory cytokines TNF-α and IL-6 were noted after ICV administration of STZ, demonstrating neuroinflammation. This was associated with a persistent increase in caspase-3 activity, a characteristic response to apoptosis. Furthermore, it has been depicted that caspase-3 activity, a key enzyme in apoptotic cell death, increased in the hippocampus of streptozotocin-induced diabetic rats (Piotrowski et al. 2001). These data showed that streptozotocin results in oxidative stress and probably induced apoptosis in the hippocampus. The increase in levels of malondialdehyde (MDA), the end product of lipid peroxidation, and decrease in levels of glutathione, an antioxidant, were taken as markers of oxidative stress. In our study, there was a significant increase in MDA level along with marked reduction in reduced glutathione and enzymatic activity of superoxide dismutase and catalase that has been observed in the brain of ICV streptozotocin-treated rats. Nitric oxide combines with superoxide to form peroxynitrite, which rapidly causes protein nitration or nitrosylation, lipid peroxidation, DNA damage, and cell death and has direct toxic effects on the nerve tissue leading to neurotoxicity (Law et al. 2001). In the present study, significantly increased nitrite levels in hippocampus of streptozotocin rats were observed, indicating the role of nitric oxide in the pathomechanism of AD. Multiple mechanisms of streptozotocin are known to be involved in the etiology of AD. Therefore, streptozotocin is thought to be responsible for the pathogenesis of the neurodegenerative changes as observed in this in vivo model of Alzheimer’s disease, even in the absence of amyloid β (Aβ) aggregation (Grunblatt et al. 2007). The present study clearly demonstrated that ICV-STZ produced memory impairment as indicated by the delay in escape latency and retention latency to find out the hidden platform in the Morris water maze task. We have also found that ICV-STZ showed a significant increase in acetylcholinesterase (AChE) activity suggesting cholinergic deficiency in the brain (Prakash and Kumar 2009). Furthermore, it has been reported that administration of STZ altered mRNA expression of AChE and α7 nicotinic acetylcholine receptor (α7 nAChR) in mice brain (Tota et al. 2011).
Pioglitazone is a well-known PPAR-γ agonist expressed widely in the hippocampus and entorhinal cortex (Berger and Moller 2002). The role of PPAR-γ has been proposed in modulation of learning and memory, aging, and neurodegeneration (Moreno et al. 2004). Chronic treatment of pioglitazone significantly improved retention latency suggesting its beneficial effect in cognitive dysfunction. Our data indicated that pioglitazone attenuated oxidative damage (decreased LPO and nitrite and increased antioxidant levels) in hippocampus of streptozotocin-treated rats. NO has been previously suggested as a probable mechanism involved in some effects of PPAR-γ agonists. Recent reports have suggested that pioglitazone produces a neuroprotective effect by downregulation of iNOS in AD (Landreth and Heneka 2001; Feinstein 2003). In the present study, chronic administration of pioglitazone significantly attenuated the increased concentration of nitric in streptozotocin-induced rats. These findings provide evidence in agreement with previous reports indicating the probable NO-dependant mechanism in protective role of pioglitazone in streptozotocin-induced neurotoxicity. It has been demonstrated that nitric oxide, by damaging DNA, could also lead to poly ADP-ribose polymerase overactivation, causing neuronal ATP depletion and death (Ha and Snyder 1999). Cell culture study also suggested that pioglitazone upregulated mitochondrial electron transport proteins in SH-SY5Y human neuroblastoma cells (Miglio et al. 2009). The present study indicated that chronic administration of pioglitazone restored the decreased mitochondrial enzyme activity in streptozotocin-treated rats. An earlier report from our groups has shown that pioglitazone ameliorated the oxidative damage as well as restored mitochondrial dysfunction in animal models of Huntington’s disease. It seems that the beneficial effect of pioglitazone on cognitive performance might be due to its protective effect on mitochondria respiratory enzyme activity. Mitochondrial oxidative damage is based on the fact that electron leakage from electron transport chain (ETC) occurs, mainly through complex I and complex III, and the amount of O2·− increases dramatically if these complexes are inhibited (Turrens 1997). Thus, it is reasonable to suppose that mitochondrial enzyme protection could be the mechanism involved in the neuroprotective effect of pioglitazone. However, the exact mechanism(s) through which pioglitazone restored mitochondrial enzyme complexes activities remains to be explored. Induction of apoptosis often converges on the mitochondria to induce MPT and activation of caspase-3 proteins into the cytoplasm, resulting in a biochemical and morphological alteration of apoptosis (Li et al. 2010). Interestingly, chronic administration of pioglitazone significantly attenuated the caspase-3 activation in streptozotocin-treated rats.
Another mechanism for the protective effects of pioglitazone is the downregulation of inflammatory responses (Schnegg and Robbins 2011). PPAR-γ is widely expressed in microglia and astrocytes and regulates inflammation in the central nervous system (Storer et al. 2005). As shown in the present study, PPAR-γ agonists like pioglitazone significantly inhibited the streptozotocin-induced stimulation of IL-6 and TNF-α expression in STZ-treated rats, which indicate the involvement of anti-inflammatory activity. We have also observed that pioglitazone attenuated AChE activity in streptozotocin-treated rats, which is a marker of cholinergic functions. It has been demonstrated that in streptozotocin-treated rats, there was a decrease in the number of new neurons in the subgranular zone in the dentate gyrus, a reduction of migration of neural progenitor cells, and a decline in spatial learning and memory (Guo et al. 2010). This could be one of the possible pathways of pioglitazone’s protective effect on cognitive performance. It seems that multiple mechanisms might have contributed to the neuroprotective effect of pioglitazone. Recently, it has been also reported that l-arginine itself exacerbates Aβ-induced toxicity and oxidative stress in models of AD (Luo et al. 2015; Liu et al. 2014). The rationale behind the pre-treatment of l-arginine is to know the direct and/or indirect involvement of nitrergic pathways in the modulation of PPAR-γ receptors. Considering the fact that NO transmission can alter the therapeutic effect of pioglitazone, we studied the interaction of pioglitazone with L-NAME (NOS inhibitor) and LA (NOS inducer). NO is involved in synaptic plasticity contributing to learning and memory in several brain areas including the cerebellum and hippocampus (Susswein et al. 2004). It has also been reported that there is a facilitating role for hippocampal NO in an inhibitory avoidance learning task in chicks and rats which is triggered via the calcium-/calmodulin-dependent protein kinase II activation in the hippocampus (Tan 2007). Nitric oxide is a very important signaling molecule which is involved in various physiological functions. However, when generated in excess in the presence of free radicals, it can be neurotoxic. Nitric oxide exerts its neurotoxic effects via several mechanisms. It has been reported that excess NO is in part responsible for glutamate neurotoxicity in primary neuronal cell culture (Valina and Ted 1996). Nitric oxide is a free radical and can combine with superoxide anions to form peroxynitrite, a highly destructive radical moiety (Eliasson et al. 1999). The resultant reactive oxygen/nitrogen species can induce significant oxidative/nitrergic stress that causes lipid peroxidation and produces functional alterations in proteins and DNA, eventually leading to neuronal death (Beckman et al. 1994). l-Arginine pretreatment with pioglitazone significantly reversed its protective effect, providing an evidence for excess involvement of nitric oxide. Further, pre-treatment of selective NOS inhibitor, L-NAME, with pioglitazone significantly potentiated the protective effect of pioglitazone suggesting nitric oxide mechanism. Evidences suggest that thiazolidinediones activate both PPAR-γ-dependent and -independent pathways in the brain (Feinstein et al. 2005). The PPAR-γ-dependent action is mediated through activation of nuclear transcription factors which occurs within days; instead, the receptor-independent effect of thiazolidinediones occurs rapidly (within minutes to hours) (Collino et al. 2006; Feinstein et al. 2005). Our results, although preliminary, might be relevant to understand the neuroprotective effects of pioglitazone (PPAR-γ agonist). This could be one of the possible proposed mechanisms in the neuroprotective effect of pioglitazone, which have been shown in Fig. 5.
The present study highlights the nitric oxide modulatory mechanisms in the neuroprotective effect of pioglitazone against ICV-STZ-induced cognitive dysfunction. Further study provides hope that pioglitazone could be used successfully in the treatment and management of cognitive dysfunction.
References
Allami N et al (2011) Suppression of nitric oxide synthesis by L-NAME reverses the beneficial effects of pioglitazone on scopolamine-induced memory impairment in mice. Eur J Pharmacol 650:240–248
Amor S, Puentes F, Baker D, van der Valk P (2010) Inflammation in neurodegenerative diseases. Immunology 129:154–169
Beckman JS, Chen J, Crow JP, Ye YZ (1994) Reactions of nitric-oxide, superoxide and peroxynitrite with superoxide-dismutase in neurodegeneration. Neural Regen 103:371–380
Berger J, Moller DE (2002) The mechanisms of action of PPARs. Annu Rev Med 53:409–435
Berman SB, Hastings TG (1999) Dopamine oxidation alters mitochondrial respiration and induces permeability transition in brain mitochondria: implications for Parkinson’s disease. J Neurochem 73:1127–1137
Butow RA, Avadhani NG (2004) Mitochondrial signaling: the retrograde response. Mol Cell 14:1–15
Choi BM, Pae HO, Jang SI, Kim YM, Chung HT (2002) Nitric oxide as a pro-apoptotic as well as anti-apoptotic modulator. J Biochem Mol Biol 35:116–126
Collino M et al (2006) Modulation of the oxidative stress and inflammatory response by PPAR-gamma agonists in the hippocampus of rats exposed to cerebral ischemia/reperfusion. Eur J Pharmacol 530:70–80
Culman J, Zhao Y, Gohlke P, Herdegen T (2007) PPAR-gamma: therapeutic target for ischemic stroke. Trends Pharmacol Sci 28:244–249
Dehmer T, Heneka MT, Sastre M, Dichgans J, Schulz JB (2004) Protection by pioglitazone in the MPTP model of Parkinson’s disease correlates with I kappa B alpha induction and block of NF kappa B and iNOS activation. J Neurochem 88:494–501
Duff K et al (2010) Mild cognitive impairment in prediagnosed Huntington disease. Neurology 75:500–507
Eliasson MJL et al (1999) Neuronal nitric oxide synthase activation and peroxynitrite formation in ischemic stroke linked to neural damage. J Neurosci 19:5910–5918
Ellman GL (1959) Tissue Sulfhydryl Groups. Arch Biochem Biophys 82:70–77
Ellman Gl, Courtney KD, Andres V Jr, Feather-Stone RM (1961) A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 7:88–95
Feil R, Kleppisch T (2008) NO/cGMP-dependent modulation of synaptic transmission. Handb Exp Pharmacol. 184:529–560
Feinstein DL (2003) Therapeutic potential of peroxisome proliferator-activated receptor agonists for neurological disease. Diabetes Technol Ther 5:67–73
Feinstein DL et al (2005) Receptor-independent actions of PPAR thiazolidinedione agonists: is mitochondrial function the key? Biochem Pharmacol 70:177–188
Fuenzalida K et al (2007) Peroxisome proliferator-activated receptor gamma up-regulates the Bcl-2 anti-apoptotic protein in neurons and induces mitochondrial stabilization and protection against oxidative stress and apoptosis. J Biol Chem 282:37006–37015
Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR (1982) Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal Biochem 126:131–138
Grunblatt E, Salkovic-Petrisic M, Osmanovic J, Riederer P, Hoyer S (2007) Brain insulin system dysfunction in streptozotocin intracerebroventricularly treated rats generates hyperphosphorylated tau protein. J Neurochem 101:757–770
Guo J et al (2010) Impaired neural stem/progenitor cell proliferation in streptozotocin-induced and spontaneous diabetic mice. Neurosci Res 68:329–336
Ha HC, Snyder SH (1999) Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc Natl Acad Sci U S A 96:13978–13982
Hoyer S, Muller D, Plaschke K (1994) Desensitization of brain insulin receptor. Effect on glucose/energy and related metabolism. J Neural Transm Suppl 44:259–268
Kapadia R, Yi JH, Vemuganti R (2008) Mechanisms of anti-inflammatory and neuroprotective actions of PPAR-gamma agonists. Front Biosci 13:1813–1826
Kaundal RK, Sharma SS (2010) Peroxisome proliferator-activated receptor gamma agonists as neuroprotective agents. Drug News Perspect 23:241–256
Kiaei M, Kipiani K, Chen J, Calingasan NY, Beal MF (2005) Peroxisome proliferator-activated receptor-gamma agonist extends survival in transgenic mouse model of amyotrophic lateral sclerosis. Exp Neurol 191:331–336
King TE (1967) Preparation of succinate dehydrogenase and reconstitution of succinate oxidase. Methods Enzymol 10:322-331
King TE, Howard RL (1967) Preparations and properties of soluble NADH dehydrogenases from cardiac muscle. Methods Enzymol 10:275-294
Kono Y (1978) Generation of superoxide radical during autoxidation of hydroxylamine and an assay for superoxide dismutase. Arch Biochem Biophys 186:189–195
Kubera M, Obuchowicz E, Goehler L, Brzeszcz J, Maes M (2011) In animal models, psychosocial stress-induced (neuro)inflammation, apoptosis and reduced neurogenesis are associated to the onset of depression. Prog Neuropsychopharmacol Biol Psychiatry 35:744–759
Kumar A, Vashist A, Kumar P (2010) Potential role of pioglitazone, caffeic acid and their combination against fatigue syndrome-induced behavioural, biochemical and mitochondrial alterations in mice. Inflammopharmacology 18:241–251
Landreth GE, Heneka MT (2001) Anti-inflammatory actions of peroxisome proliferator-activated receptor gamma agonists in Alzheimer’s disease. Neurobiol Aging 22:937–944
Law A, Gauthier S, Quirion R (2001) Say NO to Alzheimer’s disease: the putative links between nitric oxide and dementia of the Alzheimer’s type. Brain Res Rev 35:73–96
Li Z et al (2010) Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell 141:859–871
Liu Y, Peterson DA, Kimura H, Schubert D (1997) Mechanism of cellular 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction. J Neurochem 69:581–593
Liu RL, Liu W, Doctrow SR, Baudry M (2003) Iron toxicity in organotypic cultures of hippocampal slices: role of reactive oxygen species. J Neurochem 85:492–502
Liu P, Fleete MS, Jing Y, Collie ND, Curtis MA, Waldvogel HJ, Faull RL, Abraham WC, Zhang H (2014) Altered arginine metabolism in Alzheimer’s disease brains. Neurobiol Aging 35:1992–2003
Luo Y, Yue W, Quan X, Wang Y, Zhao B, Lu Z (2015) Asymmetric dimethylarginine exacerbates Aβ-induced toxicity and oxidative stress in human cell and Caenorhabditis elegans models of Alzheimer disease. Free Radic Biol Med 79:117–126
Matsumoto T, Noguchi E, Kobayashi T, Kamata K (2007) Mechanisms underlying the chronic pioglitazone treatment-induced improvement in the impaired endothelium-dependent relaxation seen in aortas from diabetic rats. Free Radic Biol Med 42:993–1007
Merrill JE, Benveniste EN (1996) Cytokines in inflammatory brain lesions: helpful and harmful. Trends Neurosci 19:331–338
Meyer RC, Spangler EL, Patel N, London ED, Ingram DK (1998) Impaired learning in rats in a 14-unit T-maze by 7-nitroindazole, a neuronal nitric oxide synthase inhibitor, is attenuated by the nitric oxide donor, molsidomine. Eur J Pharmacol 341:17–22
Miglio G, Rosa AC, Rattazzi L, Collino M, Lombardi G, Fantozzi R (2009) PPAR gamma stimulation promotes mitochondrial biogenesis and prevents glucose deprivation-induced neuronal cell loss. Neurochem Int 55:496–504
Moreno S, Farioli-Vecchioli S, Ceru MP (2004) Immunolocalization of peroxisome proliferator-activated receptors and retinoid X receptors in the adult rat CNS. Neuroscience 123:131–145
Nicolakakis N, Hamel E (2010) The nuclear receptor PPARgamma as a therapeutic target for cerebrovascular and brain dysfunction in Alzheimer’s disease. Front Aging Neurosci. doi:10.3389/fnagi.2010.00021
Penna C, Perrelli MG, Pagliaro P (2012) Mitochondrial pathways, permeability transition pore and redox signaling in cardioprotection: therapeutic implications. Antioxid Redox Signal 18(5):556–599
Pike KE et al (2011) Cognition and beta-amyloid in preclinical Alzheimer’s disease: data from the AIBL study. Neuropsychologia 49:2384–2390
Piotrowski P, Wierzbicka K, Smialek M (2001) Neuronal death in the rat hippocampus in experimental diabetes and cerebral ischaemia treated with antioxidants. Folia Neuropathol 39:147–154
Prakash AK, Kumar A (2009) Effect of chronic treatment of carvedilol on oxidative stress in an intracerebroventricular streptozotocin induced model of dementia in rats. J Pharm Pharmacol 61:1665–1672
Prakash A, Kumar A (2013) Pioglitazone alleviates the mitochondrial apoptotic pathway and mito-oxidative damage in the d-galactose-induced mouse model. Clin Exp Pharmacol Physiol 40:644–651
Prickaerts J, Fahrig T, Blokland A (1999) Cognitive performance and biochemical markers in septum, hippocampus and striatum of rats after an i.c.v. injection of streptozotocin: a correlation analysis. Behav Brain Res 102:73–88
Raza H, John A (2012) Streptozotocin-induced cytotoxicity, oxidative stress and mitochondrial dysfunction in human hepatoma HepG2 cells. Int J Mol Sci 13:5751–5767
Riobo NA et al (2001) Nitric oxide inhibits mitochondrial NADH:ubiquinone reductase activity through peroxynitrite formation. Biochem J 359:139–145
Salkovic-Petrisic M, Hoyer S (2007) Central insulin resistance as a trigger for sporadic Alzheimer-like pathology: an experimental approach. J Neural Transm Suppl 72:217–233
Sauerbeck A et al (2011) Pioglitazone attenuates mitochondrial dysfunction, cognitive impairment, cortical tissue loss, and inflammation following traumatic brain injury. Exp Neurol 227:128–135
Schnegg CI, Robbins ME (2011) Neuroprotective mechanisms of PPARdelta: modulation of oxidative stress and inflammatory processes. PPAR Res 2011:373560
Siemers E (2011) Designing clinical trials for early (pre-dementia) Alzheimer’s disease: determining the appropriate population for treatment. J Nutr Health Aging 15:22–24
Sottocasa GL, Kuylenstierna B, Ernster L, Bergstrand A (1967) An electron-transport system associated with the outer membrane of liver mitochondria. A biochemical and morphological study. J Cell Biol 32:415–438
Steinert JR, Chernova T, Forsythe ID (2010) Nitric oxide signaling in brain function, dysfunction, and dementia. Neuroscientist 16:435–452
Storer PD, Xu J, Chavis J, Drew PD (2005) Peroxisome proliferator-activated receptor-gamma agonists inhibit the activation of microglia and astrocytes: implications for multiple sclerosis. J Neuroimmunol 161:113–122
Susswein AJ, Katzoff A, Miller N, Hurwitz I (2004) Nitric oxide and memory. Neuroscientist 10:153–162
Tan SE (2007) Roles of hippocampal nitric oxide and calcium/calmodulin-dependent protein kinase II in inhibitory avoidance learning in rats. Behav Pharmacol 18:29–38
Tota S, Kamat PK, Shukla R, Nath C (2011) Improvement of brain energy metabolism and cholinergic functions contributes to the beneficial effects of silibinin against streptozotocin induced memory impairment. Behav Brain Res 221:207–215
Turrens JF (1997) Superoxide production by the mitochondrial respiratory chain. Biosci Rep 17:3–8
Valina LD, Ted MD (1996) Nitric oxide neurotoxicity. J Chem Neuroanat 10:179–190
Van Dyke K, Jabbour N, Hoeldtke R, Van Dyke C, Van Dyke M (2010) Oxidative/nitrosative stresses trigger type I diabetes: preventable in streptozotocin rats and detectable in human disease. Ann N Y Acad Sci 1203:138–145
Wills ED (1966) Mechanisms of lipid peroxide formation in animal tissues. Biochem J 99:667–676
Yamada K et al (1995) Role of nitric oxide in learning and memory and in monoamine metabolism in the rat brain. Br J Pharmacol 115:852–858
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Prakash, A., Kumar, A., Ming, L.C. et al. Modulation of the Nitrergic Pathway via Activation of PPAR-γ Contributes to the Neuroprotective Effect of Pioglitazone Against Streptozotocin-Induced Memory Dysfunction. J Mol Neurosci 56, 739–750 (2015). https://doi.org/10.1007/s12031-015-0508-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-015-0508-7