
Abstract
Introduction
MicroRNAs (miRNAs), as a family of non-coding RNAs, have opened a new window in cancer biology and transcriptome. It has been revealed that miRNAs post-transcriptionally regulate the gene expression and involve in colorectal cancer (CRC) development and progression. Our aim was to examine the differential expression of miRNAs in a CRC and to correlate their expression levels with mRNA levels of CRC-related genes (K-ras, APC, p53).
Materials and Methods
Seventy-two colorectal tumor tissues from patients with newly diagnosed CRC and 72 matched normal adjacent tissues were analyzed. Relative expression of seven CRC-related miRNAs (miR-21, miR-31, miR-20a, miR-133b, and miR-145, miR-135b and let-7g) and three CRC-related genes (K-ras, APC, p53) was detected using the SYBR Green quantitative real-time PCR technique. The correlation between gene expression levels and clinicopathological features was evaluated.
Results
Our results showed a significant difference between the two groups for the expression level of miR-21, miR-31, miR-145, and miR-20a (P < 0.001). Also, a significant difference between the two groups for the expression level of K-ras was found (P < 0.001). Further analysis revealed an inverse significant correlation between miR-145 and K-ras (R2 = 0.662, P < 0.001), while a positive correlation was observed between miR-21 and K-ras (R2 = 0.732, P < 0.001).
Conclusion
Dysregulation of miRNAs and correlation with molecular signaling pathways designated a biological role for miRNAs in various cellular mechanisms underlying CRC. On the other hand, the pattern of miRNAs expression and its correlation with transcriptional status are helpful to discovery biomarkers and design therapeutics for CRC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Colorectal cancer (CRC), as one of the most common malignancies, is a main cause of cancer-related death worldwide [1]. A number of molecular mechanisms have been documented in CRC development, which might be arising from genetic, epigenetic, and transcriptional changes. These molecular changes include the aberrant gene expression, epigenetic events, and non-coding RNA regulation [1]. Evidently, understanding the cancer development and tumor behavior can be achieved by investigating the dysregulated gene expression, signaling pathways, and the corresponding cellular processes [2, 3]. On the other hand, improving the cancer diagnosis, cancer prognosis, and therapeutic strategies are considerably dependent on depicting the molecular and cellular features of the carcinogenesis [4, 5]. A number of studies have been investigating the gene expression of molecular markers and possible correlations with clinicopathological parameters [5]. These explorations may remarkably improve the accuracy of cancer diagnosis and treatment. Nevertheless, in several tumors, no distinct correlation between the gene expression pattern and histopathological features was reported. Currently, the advanced technologies in molecular sciences have integrated new dysregulated molecular events along with histopathological features of cancers [6, 7].
MicroRNAs (miRNAs) are endogenous non-coding RNAs with about 18–22 nt in length that complimentarily bind to their target mRNA. These small RNA molecules have been well-known to negatively regulate gene expression at the post-transcriptional level [8, 9]. Indeed, the aberrant expression of miRNAs could also involve in gene alterations, including gene mutation, methylation, and amplification. In this background, these molecules have been suggested to involve in cancer signaling pathways as tumor suppressors or oncogenes [10]. Dysregulation of miRNAs has also been confirmed to engage in CRC development and progression. In other words, the altered expression of miRNAs has frequently been correlated with tumor staging, diagnosis, and prognosis [11, 12].
In this context, several investigations, including experimental, functional, and computational studies, have revealed the possible correlation between miRNAs and their mRNA targets [13,14,15]. However, since the miRNAs might be co-transcribed with their target genes, it may positively be correlated together. Therefore, it is obvious that the experimental and functional studies can outstandingly complete and validate the computational data [15]. A range of altered signaling pathways, including APC, K-ras, P53, Wnt, and EGF, was commonly reported as the frequent mechanisms underlying CRC development and progression [16]. Additionally, the regulatory role of miRNAs in these signaling pathways has been demonstrated by various investigators. In order to recognize the potential interactions, current studies incorporated the cancer-related miRNA and gene expression analyses using the large-scale miRNA and mRNA expression profile datasets [17]. Such expression analysis promotes the insights into miRNA–mRNA interaction networks, facilitating the identification of gene regulation at the post-transcriptional level. The experimental data from gene expression and miRNAs analysis might indicate a RNA-mediated regulatory mechanism for gene dysregulation in cancer development [17,18,19]. This study is aimed at examining the expression level of CRC-related miRNAs and key genes, including APC, K-ras, and P53. We performed mRNA and miRNA expression analyses of the colorectal cancer samples using the quantitative real-time PCR technique.
Materials and Methods
Study Population and Sample Collection
After the project was approved by the Research Ethics Committee (Ethical code number: IR.IUMS.REC 94–26699), a prospective study was performed with 74 consecutive colorectal cancer patients and healthy individuals from July 2017 to August 2018. All patients signed a free and informed consent form for enrollment in the study. Exclusion criteria include possible genetic syndromes, inflammatory disease, and any types of gastrointestinal tumor. Clinicopathological features collected from medical records included gender, age, tumor location, TNM staging, and lymphovascular infiltration (LVI) (Table 1). All patients were diagnosed with stages II, III, and IV of CRC, no patient with stage I was available due to late diagnosis. None of the patients have received chemotherapy prior or at the time of tissue sampling. The adjacent non-tumor tissues from CRC patients were collected as control samples.
RNA Extraction
The samples were processed and total RNA was isolated from the frozen tissue samples by TRIzol Reagent (Invitrogen, Life Technologies, Inc., Carlsbad, CA, USA) according to the manufacturer’s protocol. Then, the extracted RNA was treated using RNase-free DNase I and stored at − 80 °C, as described previously [20].
cDNA Synthesis
Reverse transcription (RT) of miRNA templates was performed using a reverse transcription kit. Briefly, a total RNA (1 μg) was transcribed into cDNA using cDNA reverse transcription kit (Amplicon, Denmark). The extracted RNA was mixed with 1 μl of random hexamer primers and 1-μl M-MuLV reverse transcriptase (200 U/μl). Nuclease-free DEPC was added to bring the mixture up to a volume of 15 μl. Then, the mixture was incubated at 65°C for 5 min in a 7500 thermocycler (ABI) and cDNA was synthesized with the program for 5 min at 25 °C, 60 min at 42 °C, and 5 min at 70 °C.
Quantitative Real-Time PCR for Analyzing miRNAs and Gene Expression
The expression of the aforementioned miRNAs was measured by a quantitative poly A reverse transcription real-time polymerase chain reaction (qPCR) using specific primers (Table 2). All PCR reactions were performed in triplicate and the mean Ct data were concluded using cycle threshold settings. cDNA was replicated on a 7500 Fast Real-time PCR System and the comparative quantitation 2−ΔΔCT method was used to compare differences in cycle number thresholds for samples normalized for endogenous controls (details provided in Supplemental Methods).
Data Analysis
The relative expression levels of miRNAs and genes were normalized to those of RNU6B and GAPDH, respectively, as internal controls. The amplification reactions were accomplished using Rotor-Gene Q software. Normalizing was done by subtracting Ct values for genes/miRNAs from Ct values for the target gene/miRNA for each sample. The normalization was completed by the equation:
Bioinformatic Pathway Analysis
Bioinformatic analysis for miRNA–gene interactions and targets was completed by TarBase v7.0 database mirPath v.3 [21]. Furthermore, the predicted targets of miRNAs were determined using the softwares TargetScan, miRanda, and PicTar.
Statistical Analysis
SPSS software version 22.0 was used for statistical analysis. Data were analyzed using the Student’s t test in independent experiments. At first, the normality was checked using the Kolmogorov-Smirnov test. Furthermore, the Pearson correlation coefficient was used to examine the relationship between gene expressions in samples based on miRNA expression level. The level of significance for statistical test was 0.05.
Results
Dysregulation of K-Ras Transcriptional Levels in Colorectal Carcinomas Compared with Healthy Controls
By comparing RNA expression levels in the studied groups based on malignant and non-cancer tissue using Mann–Whitney U test, a statistically significant difference was discovered for K-ras (p = 0.027) (Fig. 1). Additionally, data analysis revealed that invasive CRC with clinical stage III/IV has a higher expression level of K-ras as compared with tumors with a clinical stage II (p = 0.04). No significant difference in p53 and APC mRNA expression levels was observed between the tumor tissues and the adjacent normal tissues (p > 0.05) (Fig. 1).

Gene expression fold-change differences between CRC and healthy groups using RT-qPCR. Relative gene expression was calculated using the 2-ΔΔCt method, using GADPH mRNA expression as the reference gene
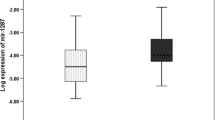
Dysregulation of miRNA Expression Levels in CRC Compared with Normal Tissues
We found the expression of miRNAs (miR-31, miR-21, miR-20a) was upregulated in CRC patients compared with adjacent controls (p < 0.05) while miR-145 showed a significant downregulation in CRC compared with the normal group (p < 0.05). However, the expression level of miR-133b was not significant between CRC and control groups (p = 0.57).
mRNA Expression Levels and Clinicopathological Features of CRC Patients
Data analysis showed that the invasive CRC with a clinical stage III/IV had a higher expression level of K-ras as compared with tumors with a clinical stage II (p = 0.04). A statistical trend towards a higher level of K-ras mRNA expression was found in the higher stages of CRC patients as compared with the lower stages and the normal tissues (p < 0.05).
Diagnostic Value of CRC-Related Genes
ROC curve analysis was performed in order to evaluate the diagnostic value of the genes. Differential expression of K-ras between CRC and healthy individuals showed a cut-off value of 2.5 (mean ± SD = 1.68 ± 0.158) with a sensitivity of 55% and a specificity of 61% and an AUC value of 0.75 (95% CI, 0.86–0.96) (Fig. 2).

ROC curve analysis of the diagnostic value of CRC-related genes to distinguish between CRC patients and healthy individuals
Interrelationship Between mRNA Levels and miRNA Expression Levels
The correlations between the mRNA expression levels and the levels of miRNA expression were analyzed using Spearman’s rank correlation test. A significant positive correlation was found between miR-21 and K-ras mRNA levels (R2 = 0.732, p < 0.001) in the CRC tumor group. The trend towards positive correlation was detected in advanced histological grade III/IV tumors.
Statistically significant negative correlation was observed between miR-145 and K-ras mRNA levels (R2 = − 0.662, p < 0.001) in the CRC tumor group (Fig. 3).

The correlations between the mRNA expression levels and the levels of miRNA expression were analyzed using Spearman’s rank correlation test. A significant positive correlation was found between miR-21 and K-ras mRNA levels (R2 = 0.732, p < 0.001) in the CRC tumor group. Also, a significant negative correlation was observed between miR-145 and K-ras mRNA levels (R2 = − 0.662, p < 0.001) in the CRC tumor group
K-Ras mRNA and miR-21 Relative Expression Levels and Correlation with Clinicopathological Features
To further validate the miRNA/mRNA relationship, we examined the association of the duplexes in colorectal cancer. K-ras (p = 0.034) and miR-21 (p = 0.012) showed a significant association with tumor differentiation. The expression levels of miR-21 and miR-31 progressively increased from well-differentiated to moderately differentiated and poorly differentiated tumors. In the same manner, K-ras mRNA levels increased from well-differentiated to poorly differentiated colorectal tumors.
Bioinformatic Pathway Analysis; miRNA–Gene Signaling Pathway Crosstalk
Bioinformatic analysis revealed a number of experimentally validated miRNA–gene interactions and targets in TarBase v7.0 database mirPath v.3 (Fig. 4.). The predicted targets of the miRNAs in this study also were determined using the softwares TargetScan, miRanda, and PicTar. MiR-21, miR-31, miR-145, miR-20a, and miR-135b were differentially expressed in our dataset. Considering these miRNAs, our miRNA–mRNA correlation studies showed significant correlations for K-ras involved in CRC development, giving miR-21 and miR-145 high priority for assessing their effects on CRC development and progression. The putative binding sites in the 3′ UTRs of miR-21 and miR-145 in mRNA of genes were predicted by the softwares TargetScan, miRanda, and PicTar.

miRNA versus GO (gene ontology) categories heatmap generated from the DIANA-miRPath v3.0 interface. The heatmap depicts the level of enrichment in GO categories of various miRNAs in Homo sapiens. The heatmap enables the identification of miRNA subclasses or GO terms that characterize similar miRNAs, since they are clustered together
Analysis on the miRNA versus mRNA correlation dataset yielded several pathways with known or putative relations to CRC carcinogenesis reaching a p value < 0.05. Although the majority of the significant pathways were unique for each miRNA, the pathways reaching statistical significance were the Wnt/β-catenin and PI3K/AKT signaling pathway for miR-21, and the p53 signaling pathway for miR-145.
Discussion
It is known that miRNAs, as key negative regulators of gene expression, are aberrantly expressed in various malignancies. Numerous studies have indicated a main biological role of miRNAs in most of the signaling pathways in colorectal cancer (CRC) pathogenesis [8, 11, 12]. There are increasing evidences that cancer-related miRNAs act as tumor suppressors and oncogenes in cancer pathogenesis [8]. So far, the function of numerous miRNAs including miR-21, miR-145, and miR-200c has been well-known in CRC development and associated with clinicopathological features of the cancer patients [22,23,24].
The miRNA analysis and correlation with gene expression may be valuable to verify the biology of CRC-related miRNAs and recognition of their target genes, providing valuable insights into understanding CRC development and progression [15, 18]. Additionally, a potential correlation between miRNA expression levels and clinicopathological parameters might substantiate the miRNA/mRNA interaction network data arose from functional and computational investigations. Indeed, the experimental data arose from gene expression and miRNAs analysis also may indicate a miRNA-mediated regulatory mechanism for gene dysregulation in cancer development [10, 15, 17].
We aimed to examine the expression level of CRC-related miRNAs and key genes, including APC, K-ras, and P53. We performed mRNA and miRNA analyses of the colorectal cancer samples using the quantitative real-time PCR technique. The differential expression of a subset of 7 miRNAs in 74 CRC patients was evaluated and correlated the miRNA levels with mRNA levels. It was found that miR-21 had the largest number of significant correlations to mRNA levels of K-RAS.
In the present study, the miRNA expression profile of CRC tissues was analyzed, revealing dysregulation of miRNAs expression compared with adjacent normal tissues. Our results demonstrated a dysregulation of miRNAs in cancer samples compared with non-cancer samples. The expression levels, of miR-21, miR-31, miR-135b, miR-20a, were found to be upregulated in cancer samples compared with non-cancer samples. Also, the expression levels of miR-145 (p = 0.002) and let-7g (p = 0.016) were found to be dowregulated in cancer samples compared with non-cancer samples. We then further validated differential expression of a subset of 5 miRNAs in 74 individuals by qPCR and correlated the miRNA levels with mRNA levels in these same individuals. It was found that miR-21 (R2 = 0.732, p < 0.001) and miR-145 (R2 = 0.662, p < 0.001) had the largest number of significant correlations to mRNA levels.
Our results, in agreement with other studies [25, 26], showed that K-ras expression levels were the highest in tumors and were correlated with the differentiation of tumor (p = 0.034). Further analysis revealed, in agreement with other results, that the expression levels of K-ras are significantly higher in advanced stages (III and IV) tumor compared with stage II. This results are agreeing with the fact that K-ras gene expression is highest in G0 and G1 phase of the cell cycle [26, 27].
According to bioinformatics analysis, the pathways reaching statistical significance were the Wnt/β-catenin and PI3K/AKT signaling pathway for miR-21, and the p53 signaling pathway for miR-145. Commonly, an activated mutation of the Wnt/β-catenin pathway components, especially APC, has been confirmed in colorectal tumors [28,29,30]. Also, it has been confirmed that downregulation of miR-21 results in inhibiting the phosphorylation of extracellular signal-regulated kinases (ERK) and protein kinase B (AKT). Phosphorylation of β-catenin by AKT contributes in promoting the transcription of the β-catenin/TCF axis genes. In other words, upregulation of miR-21 is frequently found and associated with poor prognosis in CRC patients with an APC mutation. In this context, the β-catenin/TCF axis, targeted by miR-21 via the PTEN/AKT pathway, is suggested as a promoter for tumor development and progression in APC-mutated colorectal cancer. These findings might indicate that the miR-21 expression level is valuable for prognosis in colorectal cancer patients harboring APC mutations.
Also, a positive correlation between K-ras mRNA and miR-21 in CRC tissues was detected. Indeed, we found a statistical trend to the positive correlation of K-ras mRNA and miR-21 expression levels in grades III and IV. Controversially, a negative correlation has been reported between K-ras mRNA and miR-21 in grade II colorectal tumors [31]. These discrepancy findings might represent a particular group of miR-21–K-ras translational silencing that needs to be more deeply investigated. These evidences may indicate the relevance of both K-ras and miR-21 in the regulation of the tumor progression in later stages of CRC.
Additionally, we noticed that K-ras mRNA expression levels were positively associated with miR-21 expression levels in the group of advanced tumors. Based on these results, we can assume that miR-21 may have a more significant effect on K-ras mRNA expression levels in advanced tumors than in lower stages of tumors. In the other words, higher K-ras mRNA expression levels are correlated to a worse prognosis for CRC patients.
According to previous investigations, expression levels of the miR-21 molecule were the highest in invasive carcinomas and gradually decreased towards non-transformed tissue [32, 33]. In the present study, K-ras mRNA expression levels were higher in CRC than in normal tissue. It could possibly mean that the miR-21 molecule may be important in promoting invasion by increasing K-ras mRNA translation, as proposed by others, but not in all tumor grades according to invasiveness [34, 35]. An explanation for the differences in various grades is that the compared groups have various signaling pathways, contributing to the heterogeneity in the CRC, and K-ras and miR-21 levels are most likely related to tumor phenotype. Another clarification for these remarkable findings is probably the [36, 37].
The positive correlation between miR-21 and K-ras mRNA levels, partly, might be due to multifunctional and complex role of K-ras in epithelial cells. Another reason might be related to complex interactions between miRNA and mRNA, considering the fact that one miRNA can target several mRNAs, and the translation of a single mRNA can be regulated by multiple miRNA molecules and the heterogeneity of miRNA behavior in the biology of cancer types/subtypes [38]. Moreover, it has been demonstrated that the level of K-ras is different in tumors of different phenotypes and might be silenced by promoter methylation [39]. Our findings also denote the complexity of various epigenetic events, so expression levels of K-ras seem to be the result of the summary of the combinations of RNA interference mechanisms and promoter methylation. These rationales may partly explain different and some unexpected results in various tumor groups.
MiR-135b has been revealed to dysregulate in a number of malignancies including lung cancer and colorectal cancer, indicating an oncogenic role for this miRNA. MiR-135b could promote cancer cell proliferation, invasion, and metastasis in tumors by targeting several key tumor suppressors. It has been reported that transcriptional dysregulation of miR-135b may be caused by the crosstalking between various CRC-associated signaling pathways such as K-RAS, p53, and APC [40, 41]. On the other hand, the regulation of miR-135b has been confirmed to depend on the alteration in the key genes frequently reported in CRC development and progression. Another study reported a significant upregulation of miR-135b in colorectal cancer that was reversely correlated with the expression levels of APC. It has also been confirmed that it promotes downstream Wnt signaling pathway. The putative regulatory mechanism seems to be completed regardless of the mutational status of APC. These findings suggested a miR-135b-mediated mechanism in CRC carcinogenesis by the regulation of APC expression and Wnt pathway activity [42].
MiR-145 is well-known as a downregulated miRNA with a biological role and a tumor suppressor in CRC. Experimental and functional studies have revealed that miR-145 inhibits cell proliferation, invasion, and metastasis by targeting multiple oncogenes. In disagreement with this, it has been reported that miR-145 may function as an oncogene in CRC by targeting E-cadherin. Indeed, miR-145 may act as either a tumor suppressor or an oncogene depends on the cancer stage and various cellular contexts [43, 44]. Our results showed that miR-145 is negatively correlated with K-ras indicating a suppressing effect on the K-ras and corresponding downstream pathways. This finding needs further validation by functional studies at the protein levels. Recently, miR-145 has been demonstrated to interact with some p53 target genes and regulate the p53 signaling pathway in CRC. This gene regulatory miR-145/p53 axis could mediate cell cycle, apoptosis, migration and invasion. P53 mutually may improve miR-145 maturation via modulation of Drosha-mediated miRNA processing. Corresponding to a regulatory role for p53, miR-145 expression levels are significantly lower in colorectal tumors that harbor p53 mutations [45].
Furthermore, miRNA–gene interactions and validation of miRNA targets were completed using TarBase v6.0 database [46]. The predicted targets of the miRNAs in this study were verified using the softwares TargetScan, miRanda, and PicTar. miR-21, miR-135b, and miR-200c, miR-20a, and miR-145 were differentially expressed in our dataset and for these miRNAs, our miRNA–mRNA correlation studies show a number of statistically significant correlations for genes involved in CRC, giving these four miRNAs high priority for assessing their effects on CRC development and progression. Some studies have suggested that the miRNAs–mRNAs regulation pattern includes both the coherent and incoherent [47].
The identified miRNA/mRNA combinations not only will help in the understanding of the molecular pathology of colorectal cancer but also may have a potential therapeutic capacity for the disease. Among these, some targets show upregulated expression compared with miRNAs, while other targets show downregulated expression compared with miRNAs. Indeed, miRNAs may have a certain specific target mRNA and contribute to the regulation of miRNA [48]. However, target genes may be regulated by several upregulated and downregulated miRNAs. Altogether, the mRNAs–miRNAs regulation pattern has been found to be a potential key regulator in the development of CRC. Even though this approach improves our understanding of miRNA–gene relationships, mRNA expression profiles alone may not be sufficient to represent protein translation processes, involving several regulatory steps [15, 38, 48]. Therefore, the determination of relationships between genes and miRNAs using only mRNA expression data is limited. On the other hand, protein pattern datasets and the relationships with miRNA expression profile are suggested to extensively be explored.
We supposed, based on the previous experimental and bioinformatics data, that the modulation of key markers, including K-ras and miR-21, will improve the development of new therapeutic possibilities, making the treatment of CRC more effective. In spite of the paradigm that miRNAs repress the putative targets, our miRNA versus mRNA correlation data showed a significant positive correlation between miR-21 and K-ras, which is attractive. The positive correlations in our data could be due to downstream effects, perhaps due to inhibited repression or perturbations of feedback loops [49]. Correlations between miRNA and miRNA and gene regulatory networks often include feedback motifs, and miRNAs are interwoven into such complex regulatory networks, which coordinate transcriptional regulation in signaling networks [50, 51]. Our integrated analysis of global mRNA–miRNA correlation and bioinformatics analysis needs to be incorporated in functional studies to identify novel target genes for miRNAs with the potential to involve in CRC.
Conclusion
The integrated data suggested a miR-K-ras pathway significantly enriched in related target genes from the miRNA–mRNA network in CRC development. Our results might indicate that signaling pathways involved in the formation and progression of CRC might arbitrate to miRNA-associated epigenetic mechanisms, determining K-ras expression levels. Since the relationship between cancer-related miRNA and mRNA is dynamic and multifaceted, further studies on the miRNA–mRNA network are required to deeply analyze the dominance of CRC.
References
Agah S, Akbari A, Talebi A, Masoudi M, Sarveazad A, Mirzaei A, et al. Quantification of plasma cell-free circulating DNA at different stages of colorectal cancer. Cancer Investig. 2017;35(10):625–32.
Akbari A, Farahnejad Z, Akhtari J, Abastabar M, Mobini GR, Mehbod ASA. Staphylococcus aureus enterotoxin B downregulates the expression of transforming growth factor-beta (TGF-β) signaling transducers in human glioblastoma Jundishapur Journal of Microbiology 2016;5;9(5):e27297. https://doi.org/10.5812/jjm.27297.
Giancotti FG. Deregulation of cell signaling in cancer. FEBS Lett. 2014;588(16):2558–70.
Wang X, et al. The molecular landscape of synchronous colorectal cancer reveals genetic heterogeneity. Carcinogenesis. 2018;39(5):708–18.
Kheirelseid EA, et al. Clinical applications of gene expression in colorectal cancer. J Gastrointest Oncol. 2013;4(2):144.
Inamura K. Colorectal cancers: an update on their molecular pathology. Cancers. 2018;10(1):26.
Mobini GR, Ghahremani MH, Amanpour S, Dehpour AR, Akbari A, Hoseiniharouni SM, et al. Transforming growth factor beta-induced factor 2-linked X (TGIF2LX) regulates two morphogenesis genes, Nir1 and Nir2 in human colorectal. Acta Med Iran. 2016;54(5):302–7.
Catalanotto C, Cogoni C, Zardo G. MicroRNA in control of gene expression: an overview of nuclear functions. Int J Mol Sci. 2016;17(10):1712.
Eslamizadeh S, Heidari M, Sh A, Faghihloo E, Ghazi H, Mirzaei A, et al. The role of microRNA signature as diagnostic biomarkers in different clinical stages of colorectal cancer. Cell J. 2018;20:220–30.
Zhou K, Liu M, Cao Y. New insight into microRNA functions in cancer: oncogene–microRNA–tumor suppressor gene network. Frontiers in molecular biosciences, vol. 4; 2017. p. 46.
Dong Y, Yu J, Ng SS. MicroRNA dysregulation as a prognostic biomarker in colorectal cancer. Cancer Manag Res. 2014;6:405.
Corsini LR, Bronte G, Terrasi M, Amodeo V, Fanale D, Fiorentino E, et al. The role of microRNAs in cancer: diagnostic and prognostic biomarkers and targets of therapies. Expert Opin Ther Targets. 2012;16(sup2):S103–9.
Oh M, Rhee S, Moon JH, Chae H, Lee S, Kang J, et al. Literature-based condition-specific miRNA-mRNA target prediction. PLoS One. 2017;12(3):e0174999.
Hashimoto Y, Akiyama Y, Yuasa Y. Multiple-to-multiple relationships between microRNAs and target genes in gastric cancer. PLoS One. 2013;8(5):e62589.
Kandhavelu J, et al. Computational analysis of miRNA and their gene targets significantly involved in colorectal cancer progression. MicroRNA (Shariqah, United Arab Emirates). 2018.
Tariq K, Ghias K. Colorectal cancer carcinogenesis: a review of mechanisms. Cancer Biol Med. 2016;13(1):120–35.
Seo J, Jin D, Choi CH, Lee H. Integration of MicroRNA, mRNA, and protein expression data for the identification of cancer-related MicroRNAs. PLoS One. 2017;12(1):e0168412.
Wu X-m, Shao XQ, Meng XX, Zhang XN, Zhu L, Liu SX, et al. Genome-wide analysis of microRNA and mRNA expression signatures in hydroxycamptothecin-resistant gastric cancer cells. Acta Pharmacol Sin. 2011;32(2):259–69.
Li M-h, Fu S-b, Xiao H-s. Genome-wide analysis of microRNA and mRNA expression signatures in cancer. Acta Pharmacol Sin. 2015;36(10):1200–11.
Akbari A, Ghahremani MH, Mobini GR, Abastabar M, Akhtari J, Bolhassani M, et al. Down-regulation of miR-135b in colon adenocarcinoma induced by a TGF-β receptor I kinase inhibitor (SD-208). Iran J Basic Med Sci. 2015;18(9):856–61.
Vlachos IS, Zagganas K, Paraskevopoulou MD, Georgakilas G, Karagkouni D, Vergoulis T, et al. DIANA-miRPath v3. 0: deciphering microRNA function with experimental support. Nucleic Acids Res. 2015;43(W1):W460–6.
Cekaite L, et al. MicroRNAs as growth regulators, their function and biomarker status in colorectal cancer. Oncotarget. 2016;7(6):6476.
Chen J, Wang W, Zhang Y, Hu T, Chen Y. The roles of miR-200c in colon cancer and associated molecular mechanisms. Tumor Biol. 2014;35(7):6475–83.
Yu Y, Nangia-Makker P, Farhana L, G. Rajendra S, Levi E, Majumdar APN. miR-21 and miR-145 cooperation in regulation of colon cancer stem cells. Mol Cancer. 2015;14(1):98.
Gao XH, et al. Differences of protein expression profiles, KRAS and BRAF mutation, and prognosis in right-sided colon, left-sided colon and rectal cancer. Sci Rep. 2017;7(1):7882.
Boutin AT, Liao WT, Wang M, Hwang SS, Karpinets TV, Cheung H, et al. Oncogenic Kras drives invasion and maintains metastases in colorectal cancer. Genes Dev. 2017;31:370–82.
Nussinov R, Tsai C-J, Jang H. Independent and core pathways in oncogenic KRAS signaling. In: Independent and core pathways in oncogenic KRAS signaling: Taylor & Francis; 2016.
Chen Y, Gruidl M, Remily-Wood E, Liu RZ, Eschrich S, Lloyd M, et al. Quantification of β-catenin signaling components in colon cancer cell lines, tissue sections, and microdissected tumor cells using reaction monitoring mass spectrometry. J Proteome Res. 2010;9(8):4215–27.
Najdi R, Holcombe RF, Waterman ML. Wnt signaling and colon carcinogenesis: beyond APC. J Carcinog. 2011;10:5.
Novellasdemunt L, Antas P, Li VS. Targeting Wnt signaling in colorectal cancer. A review in the theme: cell signaling: proteins, pathways and mechanisms. Am J Phys Cell Phys. 2015;309(8):C511–21.
Ye J-J, Cao J. MicroRNAs in colorectal cancer as markers and targets: recent advances. World J Gastroenterol: WJG. 2014;20(15):4288.
Ferraro A, Kontos CK, Boni T, Bantounas I, Siakouli D, Kosmidou V, et al. Epigenetic regulation of miR-21 in colorectal cancer: ITGB4 as a novel miR-21 target and a three-gene network (miR-21-ITGΒ4-PDCD4) as predictor of metastatic tumor potential. Epigenetics. 2014;9(1):129–41.
Wu Y, Song Y, Xiong Y, Wang X, Xu K, Han B, et al. MicroRNA-21 (Mir-21) promotes cell growth and invasion by repressing tumor suppressor PTEN in colorectal cancer. Cell Physiol Biochem. 2017;43(3):945–58.
Li T, et al. MicroRNA-21 as a potential colon and rectal cancer biomarker. World J Gastroenterol: WJG. 2013;19(34):5615.
Oliveto S, Mancino M, Manfrini N, Biffo S. Role of microRNAs in translation regulation and cancer. World J Biol Chem. 2017;8(1):45–56.
Aizer A, Shav-Tal Y. Intracellular trafficking and dynamics of P bodies. Prion. 2008;2(4):131–4.
Stalder L, Mühlemann O. Processing bodies are not required for mammalian nonsense-mediated mRNA decay. Rna. 2009;15:1265–73.
Cantini L, et al. A review of computational approaches detecting microRNAs involved in cancer. Frontiers in Bioscience (Landmark edition). 2017;22:1774–91.
Serra RW, Fang M, Park SM, Hutchinson L, Green MR. A KRAS-directed transcriptional silencing pathway that mediates the CpG island methylator phenotype. Elife. 2014;3:e02313.
Valeri N, et al. MicroRNA-135b promotes cancer progression by acting as a downstream effector of oncogenic pathways in colon cancer. Cancer Cell. 2014;25(4):469–83.
Aslam MI, Hussein S, West K, Singh B, Jameson JS, Pringle JH. MicroRNAs associated with initiation and progression of colonic polyp: a feasibility study. Int J Surg. 2015;13:272–9.
Nagel R, le Sage C, Diosdado B, van der Waal M, Oude Vrielink JAF, Bolijn A, et al. Regulation of the adenomatous polyposis coli gene by the miR-135 family in colorectal cancer. Cancer Res. 2008;68(14):5795–802.
Chivukula RR, Shi G, Acharya A, Mills EW, Zeitels LR, Anandam JL, et al. An essential mesenchymal function for miR-143/145 in intestinal epithelial regeneration. Cell. 2014;157(5):1104–16.
Li S, Wu X, Xu Y, Wu S, Li Z, Chen R, et al. miR-145 suppresses colorectal cancer cell migration and invasion by targeting an ETS-related gene. Oncol Rep. 2016;36(4):1917–26.
Rokavec M, et al. The p53/microRNA connection in gastrointestinal cancer. Clin Exp Gastroenterol. 2014;7:395.
Vergoulis T, et al. TarBase 6.0: capturing the exponential growth of miRNA targets with experimental support. Nucleic Acids Res. 2011;40(D1):D222–9.
Zhang G, Yin S, Mao J, Liang F, Zhao C, Li P, et al. Integrated analysis of mRNA-seq and miRNA-seq in the liver of Pelteobagrus vachelli in response to hypoxia. Sci Rep. 2016;6:22907.
Guo L, et al. Integrative analysis of miRNA-mRNA and miRNA-miRNA interactions. Biomed Res Int. 2014;2014.
Martinez-Pastor M, Lancaster WA, Tonner PD, Adams MWW, Schmid AK. A transcription network of interlocking positive feedback loops maintains intracellular iron balance in archaea. Nucleic Acids Res. 2017;45(17):9990–10001.
Herranz H, Cohen SM. MicroRNAs and gene regulatory networks: managing the impact of noise in biological systems. Genes Dev. 2010;24(13):1339–44.
Lai X, Wolkenhauer O, Vera J. Understanding microRNA-mediated gene regulatory networks through mathematical modelling. Nucleic Acids Res. 2016;44(13):6019–35.
Acknowledgments
We would like to thank the patients who participated in the study.
Funding
This work was financially supported by Deputy of Research, Iran University of Medical Sciences (Grant No. 26699).
Author information
Authors and Affiliations
Contributions
PG and AA contributed to the study design and conception. FM and SM performed experiments. AA and AT assisted with the analysis of the data. AA prepared the manuscript which PG and AT significantly revised. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
The project was approved by the Research Ethics Committee (Ethical code number: IR.IUMS.REC 94–26699.) All patients signed a free and informed consent form for enrollment in the study.
Conflict of Interest
All authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Moghadamnia, F., Ghoraeian, P., Minaeian, S. et al. MicroRNA Expression and Correlation with mRNA Levels of Colorectal Cancer-Related Genes. J Gastrointest Canc 51, 271–279 (2020). https://doi.org/10.1007/s12029-019-00249-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12029-019-00249-2