
Abstract
Background
Aneurysmal subarachnoid hemorrhage (aSAH) is associated with high morbidity and mortality. EG-1962 is a sustained-release microparticle formulation of nimodipine that has shown preclinical efficacy when administered intraventricularly or intracisternally to dogs with SAH, without evidence of toxicity at doses in the anticipated therapeutic range. Thus, we propose to administer EG-1962 to humans in order to assess safety and tolerability and determine a dose to investigate efficacy in subsequent clinical studies.
Methods
We describe a Phase 1/2a multicenter, controlled, randomized, open-label, dose escalation study to determine the maximum tolerated dose (MTD) and assess the safety and tolerability of EG-1962 in patients with aSAH. The study will comprise two parts: a dose escalation period (Part 1) to determine the MTD of EG-1962 and a treatment period (Part 2) to assess the safety and tolerability of the selected dose of EG-1962. Patients with a ruptured saccular aneurysm treated by neurosurgical clipping or endovascular coiling will be considered for enrollment. Patients will be randomized to receive either EG-1962 (study drug: nimodipine microparticles) or oral nimodipine in the approved dose regimen (active control) within 60 h of aSAH.
Results
Primary objectives are to determine the MTD and the safety and tolerability of the selected dose of intraventricular EG-1962 as compared to enteral nimodipine. The secondary objective is to determine release and distribution by measuring plasma and CSF concentrations of nimodipine. Exploratory objectives are to determine the incidence of delayed cerebral infarction on computed tomography, clinical features of delayed cerebral ischemia, angiographic vasospasm, and incidence of rescue therapy and clinical outcome. Clinical outcome will be determined at 90 days after aSAH using the extended Glasgow outcome scale, modified Rankin scale, Montreal cognitive assessment, telephone interview of cognitive status, and Barthel index.
Conclusion
Here, we describe a Phase 1/2a multicenter, controlled, randomized, open-label, dose escalation study to determine the MTD and assess the safety and tolerability of EG-1962 in patients with aSAH.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
This report endeavors to comply with standard protocol items: recommendations for interventional trials (SPIRIT) [1]. Nimodipine remains the only drug in North America and Europe to successfully improve outcome after aneurysmal subarachnoid hemorrhage (aSAH). Meta-analysis of eight studies of nimodipine reported that it reduced the odds of poor outcome [2]. Serious adverse reactions to nimodipine were not described in all studies but in two studies that did report them, hypotension occurred in 2.1 % of nimodipine and 1.4 % of placebo groups [3]. On the other hand, nimodipine was associated with dose-limiting hypotension in up to 50 % of patients when administered intravenously and 5–8 % after oral intake [4, 5]. Plasma concentrations can exceed those associated with hypotension, yet cerebrospinal fluid (CSF) concentrations remain well below therapeutic concentrations [6]. Hypotension is deleterious to patients with aSAH because it may lower cerebral perfusion pressure and worsen DCI [7, 8].
Local delivery of sustained-release formulations of dihydropyridines, such as nicardipine, into the subarachnoid space next to cerebral arteries markedly reduces the incidence of DCI and improves outcome [9, 10]. This provides evidence supporting intracranial delivery of sustained-release formulations of dihydropyridines to prevent DCI and improve outcome of patients with aSAH. Some sustained-release nicardipine formulations, however, were prepared with the neurotoxin, dichloromethane, were of unknown stability and also not easily injectable for use in patients not undergoing craniotomy. A formulation of the dihydropyridine nimodipine in a biodegradable polymer is available from Edge Therapeutics, Incorporated®. This formulation allows for sustained release of nimodipine for at least 21 days. This paper reviews data supporting use of EG-1962 in humans and the design of the first human clinical trial.
Rationale
Aneurysmal SAH causes poor outcome by producing immediate brain injury due to hemorrhage into the subarachnoid space, brain, and ventricles, as well as by causing global ischemia. Additionally, up to 30 % of patients deteriorate neurologically days after the SAH secondary to DCI [11]. DCI is the most common, potentially reversible complication of aSAH; however, to date, there is no highly effective treatment to prevent DCI. How SAH causes DCI is not entirely understood, but multiple processes are hypothesized to contribute, including angiographic vasospasm, cortical spreading ischemia, microthromboembolism, loss of autoregulation, and capillary transit time heterogeneity, which all share a common mechanism: vasoconstriction [11–15].
Nimodipine, a dihydropyridine L-type calcium channel antagonist, reduced the risk of DCI and improved outcome in patients with aSAH [3]. When nimodipine is administered orally or intravenously, the CSF concentration is at least 2 orders of magnitude lower than the plasma concentration [6]. The CSF concentrations are about 2–7 nmol/L (0.84–3 ng/mL) when plasma concentrations are 70–96 nmol/L (30–40 ng/mL). Hypotension begins to occur at plasma concentrations of 70 nmol/L (30 ng/mL), when CSF concentrations are still below the concentration that will dilate cerebral arteries [16]. Experimental and clinical evidence suggests that nimodipine can reduce angiographic vasospasm, cortical spreading ischemia, and microthromboemboli [17–19]. Higher doses of nimodipine may be more efficacious, but that they cannot be administered systemically due to hypotension [4, 20].
Intraoperative local delivery of sustained-release pellets of dihydropyridines, such as nicardipine, into the subarachnoid space next to cerebral arteries reduced the incidence of DCI and improved outcome [9, 21–23]. One issue with pellet-based therapeutics is that they are difficult to administer intraventricularly, which limits their use in patients who undergo endovascular coiling to repair the ruptured aneurysm [24]. Only limited amounts could be injected through a cannula in the ventricles. Second, they would not be expected to circulate into the basal cisterns and release drug there. The nicardipine would be released in the ventricles and thus be much diluted by the time it reached the basal cisterns.
Additional support for use of high doses of dihydropyridines comes from reports of use of intrathecal and intraventricular injections of nimodipine or nicardipine to reverse angiographic vasospasm [25–29]. Furthermore, intra-arterial infusion of nimodipine or other dihydropyridine calcium channel antagonists reversed established angiographic vasospasm and improved clinical condition in multiple retrospective reviews of uncontrolled patient series [27, 30–34]. The limitation of local injections of native dihydropyridines is the need for repeated or continuous injection, which is technically difficult and invasive, as well as the risk of hypotension [27, 35].
EG-1962 was developed with a specific target profile to overcome these limitations. Nimodipine was selected as the active ingredient since it is already approved for use in patients with aSAH. A biocompatible, bioabsorbable polymer that could release nimodipine for up to 21 days was required, and poly(dl-lactide-co-glycolide) (PLGA) was selected. It is safe and nontoxic in humans, even when used intracranially [36]. A formulation had to be developed that had no solvents that were unacceptable for intracranial use. The size of the microparticles needed to be between 20 and 125 μm. Smaller microparticles (<10 μm) can be taken up by macrophages and cleared rapidly, and larger microparticles are difficult to inject [37, 38]. Since the ultimate clinical dose was unknown, drug loading was maximized in order to minimize the injection volume. The release of nimodipine from the microparticles in the first hours after implantation (burst release) was kept to a minimum, so that there would not be potential dose dumping into plasma causing side effects. Achieving this product profile required several years of experimentation, resulting in EG-1962, a sustained-release delivery system of nimodipine in PLGA microparticles that are reconstituted in hyaluronic acid. EG-1962 can be injected into the ventricles through an external ventricular catheter (EVD) in patients undergoing endovascular coiling or neurosurgical clipping of ruptured cerebral aneurysms. It can be injected into the basal cisterns after craniotomy and clipping of ruptured intracranial aneurysms. Once injected, the bioabsorbable microparticle formulation circulates into the basal cisternal subarachnoid space and hydrolyzes, releasing nimodipine locally. Nimodipine concentrations are highest in the CSF and lower in the plasma, so that systemic complications, such as hypotension, are less likely to occur.
Preclinical Development
EG-1962 was studied in a double hemorrhage model of SAH in mongrel dogs. Forty dogs underwent baseline angiography and creation of SAH (day 1) by injection of blood into the cisterna magna. EG-1962 (40 or 100 mg) was injected into the cisterna magna or into the lateral ventricle (100 mg, n = 8 per group). Two other groups received placebo PLGA microparticles with or without oral nimodipine. EG-1962 reduced angiographic vasospasm in all groups compared to placebo and oral nimodipine and was not associated with adverse effects, systemic hypotension, or pathologic effects in the brain. Analysis of plasma and CSF samples demonstrated sustained plasma concentrations of nimodipine for 21 days. Reduction in angiographic vasospasm was observed with 40 mg EG-1962 and was associated with plasma maximal nimodipine concentration (C max) of 8.3 ng/mL and a steady-state concentration (C ss) of 4.6 ng/mL.
Safety and tolerability studies of intraventricular or intracisternal injection of EG-1962 were conducted in beagles. Doses of 17, 51, and 103 mg nimodipine were administered with 51 mg equivalent to an estimated human dose of 400 mg and scaled between species based on relative CSF volumes [39]. Microscopically, there was a granulomatous foreign body-type reaction to EG-1962 in the ventricular system that was dose related, more marked at 15 days after injection, and decreasing 29 days after injection. Inflammation, fibrosis, and cardiac myofiber degeneration and necrosis of the left ventricle and interventricular septum of the heart were observed in several beagles administered 103 mg EG-1962 intraventricularly, and in one animal given this dose intracisternally. There was partial resolution of the microscopic findings in the heart by 29 days. Administration of 103 mg EG-1962 also was occasionally associated with increased size of the lateral ventricles. This study established a no observable adverse effect level (NOAEL) of 51 mg of EG-1962 administered intraventricularly in beagles. Scaling on a CSF volume/volume basis, this would be 612 mg in humans and was associated with nimodipine plasma C max of 23 ng/mL and C ss of 14 ng/mL.
A toxicity study of intraventricular EG-1962 also was conducted in rats. EG-1962 was administered at doses of 0.33, 1, or 2 mg of nimodipine. The only potential EG-1962-related microscopic finding consisted of increased incidence/severity of hemorrhage in the brain 15 days after administration of 2 mg EG-1962. This was partly reversed by 29 days. The NOAEL from a single intraventricular injection of EG-1962 in rats was 2 mg. Scaling on a CSF volume/volume basis, this would be 1,200 mg in humans. Mean plasma C max at the NOAEL was 41 ng/mL and C ss was 14 ng/mL.
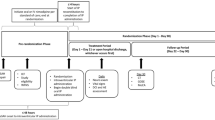
Study Design
Administration
The protocol was developed by Daniel Hänggi, M.D., R. Loch Macdonald, M.D., Ph.D., Stephan A. Mayer, M.D., Francois Aldrich, M.D., Michael N. Diringer, M.D., Brian L. Hoh, M.D., J. Mocco, M.D., Poul Strange, M.D., Ph.D., Herbert J. Faleck, D.O., and Michael Miller, Ph.D. The study is funded by Edge Therapeutics, Inc. and data collection is by a contract research organization (Medpace, Inc., Cincinnati, OH, USA). A steering committee (Daniel Hänggi, M.D., Stephan A. Mayer, M.D., Francois Aldrich, M.D., Michael N. Diringer, M.D., Brian L. Hoh, M.D., J. Mocco, M.D., Poul Strange, M.D., Ph.D.) will have access to the final trial data set and will write a report of the study for publication, which has to be approved by Edge Therapeutics, Inc. The authors are the authors of this paper. No professional writers will be used. The disclosures of these individuals are published with this paper. For patients who suffer complications of aSAH and possibly related to the study, they seek medical attention as required and, according to institutional ethics boards, undergo treatment as necessary which is paid for by their health care plans. In some cases, consent forms for the study include verbage indicating that Edge Therapeutics, Inc. will pay for study-related complications. Patient confidentiality follows the guidelines of the United States Health Insurance Portability and Accountability Act and the personal information protection and electronic documents act (of Canada). Public and scientific inquiries can be directed to the world-wide principle investigator, Professor Dr. med. Daniel Hänggi, or to Edge Therapeutics, Inc. The data collection is monitored independently by Medpace with input from Edge Therapeutics, Inc., who review the data entered into online case report forms. Details of monitoring operating procedures, range checks, and such are available from Edge Therapeutics, Inc. and Medpace, Inc. Protocol modifications will be discussed with the study steering committee and the Data Safety Monitoring Committee (DSMC) and consensus reached prior to implementation.
Objectives
The primary objective of this study is to determine the maximum tolerated dose (MTD) of intraventricular EG-1962 and then to determine the safety and tolerability of the selected dose of intraventricular EG-1962 as compared to enteral nimodipine in patients with aSAH. The secondary objective is to measure plasma and CSF concentrations of nimodipine. Information also will be collected on DCI, cerebral infarction due to DCI, use of rescue therapy, angiographic vasospasm, and clinical outcome measured at 90 days after aSAH.
Synopsis
This study will be conducted according to the principles of the “Declaration of Helsinki” (Somerset-West) and with the laws and regulations of their country. The Investigator will follow the International Conference on Harmonization (ICH) Good Clinical Practices (GCP) Guidelines. The study protocol must be approved by the local Institutional Review Board or Independent Ethics Committee, as appropriate, before any study-related procedures are performed.
This is a phase 1/2a multicenter, controlled, randomized, open-label, dose escalation study to determine the MTD and assess the safety and tolerability of EG-1962 in patients with aSAH. The study will comprise two parts: a dose escalation period (Part 1) to determine the MTD of EG-1962 and a treatment period (Part 2) to assess the safety and tolerability of the selected dose of EG-1962. Patients will be randomized to receive either EG-1962 or nimodipine (60 mg every 4 h orally for 21 days) within 60 h of aSAH. Randomization is performed by an independent company and uses a computer-generated randomization stratified by world federation of neurological surgeons (WFNS) grade implemented by central telephone and the study sites interacting with the contract research organization (Phase 1 to 4, LLC, Princeton Junction, NJ, USA). EG-1962 will be administered once intraventricularly. Intraventricular administration was chosen for this study so that patients undergoing endovascular repair of the ruptured aneurysm could be enrolled and based on efficacy against angiographic vasospasm after intraventricular injection in dogs. Prior to randomization, all patients receive oral or intravenous nimodipine according to the standard-of-care at each study site. EG-1962 is to be administered within 60 h after the onset of aSAH. After enrollment in the study, patients randomized to EG-1962 do not receive oral or intravenous nimodipine. All patients can receive oral or intravenous nimodipine as rescue therapy.
Up to six dose-level cohorts of EG-1962 will be assessed during the dose escalation period. Each dose-level cohort will enroll up to 12 patients in a 3:1 (study drug: active control) randomization scheme: 9 patients on EG-1962 and three patients on nimodipine. Randomization in each cohort is stratified by WFNS grade. There are too few patients to stratify by other variables. The starting dose of EG-1962 is 100 mg and the maximum dose will be 1,200 mg. Dose escalation will be guided by safety and the occurrence of pre-defined dose-limiting toxicities (DLTs). The study has a DSMC. A maximum of 96 patients are approved to be enrolled.
Inclusion and Exclusion Criteria (Table 1)
An upper age limit of 75 was chosen due to the decreasing likelihood of good outcome, as defined by an extended Glasgow outcome score (eGOS) of 6–8, in those over 75 [40]. Patients can undergo neurosurgical clipping or endovascular coiling of the aneurysm since there is no evidence that nimodipine efficacy differs depending on method of aneurysm repair. A time limit of 60 h from aSAH within which to administer EG-1962 was set based on evidence that DCI is rare before 3 days from aSAH and it was found that plasma nimodipine concentrations reached the C ss within 24 h of administration of EG-1962 in preclinical studies. The WFNS score was selected to grade patients because it has lower inter- and intraobserver variability than the Hunt and Hess scale [41]. Patients must have an EVD in place as part of standard-of-care and this, along with sample size estimates of outcomes on the eGOS, guided the decision to include WFNS grade 2–4 patients (Table 2). Analysis of data from the SAH international trialist repository found that 21 % of WFNS grade 1 patients had EVDs in place and fewer had high volumes of SAH that would put them at risk for DCI [42]. The early mortality of WFNS grade 5 patients is high which makes assessment of safety and pharmacokinetics (PK) difficult. Other key inclusion criteria are SAH on baseline computed tomography (CT) scan that is diffuse (clot present in both hemispheres) thick (>4 mm) or thin, or local thick. Clot thickness will be classified by the modified Fisher score [43]. Exclusion criteria were selected to remove those who may not tolerate intraventricular injections (if they have increased intracranial pressure [ICP] >30 mm Hg in sedated patients lasting >4 h anytime since admission), those at low risk of DCI, patients with complications of aneurysm repair that would lead to infarction and poor outcome or death unrelated to EG-1962, and cardiac and hemodynamic criteria that would increase the risk of hypotension and confound interpretation of safety.
There are 17 active sites in the United States and Canada, and seven more sites in various stages of obtaining approval to participate in NEWTON (Supplementary data). The sites include academic and community hospitals. The first patient was enrolled in October 29, 2013.
Dose-Limiting Toxicity and Monitoring
The MTD will be considered reached when three or more patients who are receiving EG-1962 experience a DLT (Table 3). These DLTs were selected since they reflect known (hypotension) or possible side effects (renal, liver toxicity) of nimodipine or other side effects that could occur after intraventricular injection. Additional safety and tolerability data that will be collected are death within 90 days of aSAH and its cause, adverse events of specific interest (i.e., hydrocephalus, meningitis, ventriculitis, hypotension, elevated liver enzymes, renal injury, and average daily change from baseline in systolic and diastolic blood pressure and heart rate).
The study is monitored by an independent DSMC. The DSMC receives reports of all serious adverse events (SAEs) and reviews DLTs and unexpected SAEs related to EG-1962 in an ongoing manner and makes recommendations as necessary. The DSMC convenes to review all safety, tolerability, and PK data obtained for up to 14 days after the administration of EG-1962 to the last patient in the previous cohort. If DLTs are reported in three patients in a given cohort, the DSMC could be convened before enrollment for that cohort is completed to review the safety data and make a recommendation on the dose. The DSMC may recommend dose modification based on the ongoing analysis of the study data. The dose and sample size of Part 2 (treatment period) of the study will be based on the interim analysis of the data from Part 1 (dose escalation period).
Management of DLTs and adverse events is outlined in the protocol and follows guidelines for management of aSAH (Supplementary data) [44]. The main issue with EG-1962 is that once administered there is no mechanism to remove it should an unexpected adverse event occur that is related to it. According to the package insert, the main side effect of nimodipine is hypotension. Allergic reactions are nonexistent or exceedingly rare. The diluent for EG-1962, hyaluronic acid, has been approved for intraarticular injection in Europe since 1987. In post-marketing experience, cases of anaphylactic or anaphylactic-like reaction are reported and all have resolved. Hyaluronic acid injections are often given repeatedly, and there is no evidence that adverse reactions increase in frequency with use. Furthermore, hyaluronic acid approved for intraarticular injection also cannot be removed once injected. The PLGA also is already used in neurosurgery as a dural substitute.
Safety monitoring is described in the Supplementary data and follow those recommended by the International Conference on Harmonization and current GCP.
Definitions, Radiology, and Protocol Details
The definitions for angiographic vasospasm and DCI were based on recommendations of a multidisciplinary committee [45]. Angiographic vasospasm will be assessed by comparison of catheter or CT angiography (CTA) performed at baseline within 60 h of aSAH and a similar study 7–11 days after aSAH. Rescue therapy is defined as induced hypertension (intravenous vasopressors such as dopamine, dobutamine, phenylephrine, epinephrine, and norepinephrine), superselective intra-arterial infusion of vasodilator drugs (nimodipine, nicardipine, verapamil), or balloon angioplasty performed for DCI. The use of rescue therapy in the absence of documented DCI is discouraged, in keeping with current guidelines for management of aSAH and with the limited evidence that it is efficacious [46].
In addition to angiograms, CT scans obtained at baseline, after the aneurysm repair procedure and 30 days after aSAH, will be collected. If there is neurological worsening and rescue therapy is used or DCI is diagnosed, imaging to support this will be submitted. Imaging will be reviewed by a radiology review committee. The CT scans will be reviewed to determine the SAH category at baseline as well as the etiology of any hypodensities [47]. Angiographic vasospasm also will be assessed qualitatively as none/mild (can be more than two major cerebral arteries [segments] with mild (less than 1/3 arterial narrowing) angiographic vasospasm and/or 1 segment with moderate angiographic vasospasm (1/3 to 2/3 arterial narrowing), moderate (at least 2 segments with moderate angiographic vasospasm and/or 1 or 2 segments with severe angiographic vasospasm [more than 2/3 arterial narrowing]), or severe (at least 3 segments with severe angiographic vasospasm). A segment corresponds to a major cerebral artery. This will be done for several reasons. One endpoint of this study is DCI-related infarction. We reported that in other studies, there was a discrepancy between the etiology of infarction according to investigators and as judged by independent review [48, 49]. Rescue therapy was sometimes initiated in patients diagnosed with angiographic vasospasm, but central review did not diagnose angiographic vasospasm. These are important data to have in order to ascertain the contribution of angiographic vasospasm to DCI and to help elucidate the potential mechanism of action of nimodipine. Since DCI-related infarction is potentially modifiable by EG-1962, it is useful to have an independent opinion about the etiology of cerebral infarctions observed on the CT scan obtained after 30 days.
The protocol contains recommendations about administration of concomitant medications that are derived verbatim from the package insert on nimodipine and that are based on guidelines for management of aSAH [44, 46]. Antiepileptic drugs are not recommended unless the patient has a seizure disorder or was thought to have had seizures associated with the aSAH.
The protocol recommends avoiding treatments that are not based on high-level evidence, such as prophylactic balloon angioplasty, selective arterial infusions of vasodilator drugs such as verapamil, nicardipine, or milrinone in the absence of documented change in neurological condition, therapeutic magnesium infusion, and other intrathecal or intraventricular vasodilators or thrombolytics.
Administration of EG-1962
EG-1962 is injected into the ventricles through an EVD. Once flushed into the ventricles, it is recommended that the ventricular drain remain closed and the ICP is monitored. If the ICP increases or the cerebral perfusion pressure decreases to values set by the investigator, then the drain can be opened and CSF is drained. The rationale for leaving the drain closed and only draining CSF if needed is that this will promote circulation of EG-1962 into the basal cisterns. There also is evidence that this method of management of the EVD is associated with fewer complications [50, 51].
If DCI is diagnosed during the study, the investigator can initiate treatment for DCI, including hemodynamic therapy, intra-arterial vasodilators, and/or endovascular balloon dilation.
Follow-Up
Follow-up visits will occur at day 30 (±7 days) and day 90 (±10 days). Outcomes assessed will be a CT scan at 30 days and clinical assessment including Barthel index, eGOS, Montreal cognitive assessment (MoCA), modified Rankin scale (mRS), National Institutes of Health Stroke Scale (NIHSS), and telephone interview of cognitive status (TICS) at 90 days [40, 52–56]. These outcome scales and their assessment at 90 days were selected based on discussions with the Food and Drug Administration and their use in other trials. The Barthel index and NIHSS focus on focal neurologic deficits that are uncommon after aSAH. The eGOS and mRS are commonly used in aSAH trials. Cognitive assessment is increasingly used in aSAH studies and has been advocated because patients who are classified as having favorable outcomes (mRS < 3) have been shown to have deficits in verbal memory, executive functioning, and other areas [57, 58].
Analyses, Statistical Methods, and Sample Size Calculation
The analysis sets will include all randomized (all randomized patients, whether or not they receive EG-1962 or oral nimodipine after randomization), safety (all patients who were randomized and received EG-1962 or oral nimodipine and were assessed for safety at least once), treated (all patients who were randomized and received EG-1962 or at least one dose of oral nimodipine after randomization), and per protocol sets (all patients in the treated set who did not have protocol deviations that would affect assessment of the MTD or safety or tolerability of EG-1962 or of secondary outcomes).
Primary Safety Outcomes
Adverse events will be summarized by crude incidence rates and compared to those reported in the package insert for nimodipine, and those reported in patients on active control in this study. Definitions of adverse events, severity, and seriousness follow current recommendations. Comparisons will be made by appropriated statistical tests although the small number of patients precludes rigorous analysis. Nevertheless, this will allow some estimation of whether there is any difference in serious unexpected adverse events.
Pharmacokinetic and Pharmacodynamic Analyses
The PK analysis will use mixed-effects population PK modeling using commercially available software. Pharmacokinetic evaluations may include, but will not be limited to the following parameters: C max, time to C max, apparent total body clearance of drug from plasma (CL), apparent terminal half-life (t ½), and mean residence time (MRT) in plasma and CSF, where appropriate areas under the curve. Analysis will be done on patients in each dose group and may be repeated for combined dose groups.
Secondary Objective Variables and Exploratory Analysis
The first exploratory endpoint will be the eGOS, analyzed as a dichotomous outcome (favorable outcome 6–8, unfavorable outcome 0–5). This cut point was determined based on analysis of data from the SAH international trialists repository where a cut point that could differentiate favorable and unfavorable outcomes was selected (Table 2) [42]. Analysis will be on an intention-to-treat basis.
When the dose escalation period is complete, this exploratory analysis will help to evaluate the appropriate dose for a future trial by estimating the sample size per group for the second trial. This analysis will use an assumed linear model for the natural log of the odds for success versus the dose (logistic response vs dose). The objective is to forecast, for each of the tested doses, the sample sizes required to achieve an 80 % average power for the favorable outcome rate comparison of the selected dose of EG-1962 to control.
The forecasting method will be based on a Bayesian model using the binomial distribution for the observed favorable response frequencies and a logit link to a bivariate normal model for the slope and intercept of the dose response of the population log-odds for favorable response. The Bayesian method targets this slope and intercept as random variables whose joint distribution is contingent on the observed sample estimates of these parameters from the observed Phase 1 frequencies. Once this joint distribution is determined (termed the posterior distribution) for the slope and intercept, then the posterior probability distributions for each favorable response probability are obtained from the logit model. A second hypothetical study is simulated by first sampling success probabilities for a given dose group and control from the posterior distributions, then the power obtained from a 2-tailed p = 0.05 comparison of the dose and control groups is computed for a given sample size. This is repeated many 1,000 times (Monte-Carlo) to obtain the average power for that dose and sample size. For each dose, the average power versus sample size power curve can then be used to evaluate promising candidates for a dose or doses to test in the next study. Doses requiring a prohibitive sample to achieve at least 80 % would likely be eliminated. The specific details of the decision criteria and associated operating characteristics will be agreed upon and recorded in the statistical analysis plan.
References
Chan AW, Tetzlaff JM, Gotzsche PC, et al. SPIRIT 2013 explanation and elaboration: guidance for protocols of clinical trials. BMJ. 2013;346:e7586.
Liu X, Rinkel GJ. Aneurysmal and clinical characteristics as risk factors for intracerebral haematoma from aneurysmal rupture. J Neurol. 2011;258(5):862–5.
Dorhout Mees SM, Rinkel GJ, Feigin VL, et al. Calcium antagonists for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev. 2007;3:CD000277.
Porchet F, Chiolero R, de Tribolet N. Hypotensive effect of nimodipine during treatment for aneurysmal subarachnoid haemorrhage. Acta Neurochir. 1995;137(1–2):62–9.
Petruk KC, West M, Mohr G, et al. Nimodipine treatment in poor-grade aneurysm patients. Results of a multicenter double-blind placebo-controlled trial. J Neurosurg. 1988;68(4):505–17.
Allen GS, Ahn HS, Preziosi TJ, et al. Cerebral arterial spasm–a controlled trial of nimodipine in patients with subarachnoid hemorrhage. N Engl J Med. 1983;308(11):619–24.
Dankbaar JW, Slooter AJ, Rinkel GJ, van der Schaaf IC. Effect of different components of triple-H therapy on cerebral perfusion in patients with aneurysmal subarachnoid haemorrhage: a systematic review. Crit Care. 2010;14(1):R23.
Darby JM, Yonas H, Marks EC, Durham S, Snyder RW, Nemoto EM. Acute cerebral blood flow response to dopamine-induced hypertension after subarachnoid hemorrhage. J Neurosurg. 1994;80(5):857–64.
Barth M, Capelle HH, Weidauer S, et al. Effect of nicardipine prolonged-release implants on cerebral vasospasm and clinical outcome after severe aneurysmal subarachnoid hemorrhage: a prospective, randomized, double-blind phase IIa study. Stroke. 2007;38(2):330–6.
Kasuya H, Onda H, Sasahara A, Takeshita M, Hori T. Application of nicardipine prolonged-release implants: analysis of 97 consecutive patients with acute subarachnoid hemorrhage. Neurosurgery. 2005;56(5):895–902.
Macdonald RL. Delayed neurological deterioration after subarachnoid haemorrhage. Nat Rev Neurol. 2014;10(1):44–58.
Dreier JP, Major S, Manning A, et al. Cortical spreading ischaemia is a novel process involved in ischaemic damage in patients with aneurysmal subarachnoid haemorrhage. Brain. 2009;132(Pt 7):1866–81.
Pisapia JM, Xu X, Kelly J, et al. Microthrombosis after experimental subarachnoid hemorrhage: time course and effect of red blood cell-bound thrombin-activated pro-urokinase and clazosentan. Exp Neurol. 2012;233(1):357–63.
Budohoski KP, Czosnyka M, Smielewski P, et al. Impairment of cerebral autoregulation predicts delayed cerebral ischemia after subarachnoid hemorrhage: a prospective observational study. Stroke. 2012;43(12):3230–7.
Ostergaard L, Aamand R, Karabegovic S, et al. The role of the microcirculation in delayed cerebral ischemia and chronic degenerative changes after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2013;33(12):1825–37.
Vinge E, Andersson KE, Brandt L, Ljunggren B, Nilsson LG, Rosendal-Helgesen S. Pharmacokinetics of nimodipine in patients with aneurysmal subarachnoid haemorrhage. Eur J Clin Pharmacol. 1986;30(4):421–5.
Cook DJ, Kan S, Ai J, Kasuya H, Macdonald RL. Cisternal sustained release dihydropyridines for subarachnoid hemorrhage. Curr Neurovasc Res. 2012;9(2):139–48.
Dreier JP, Windmuller O, Petzold G, Lindauer U, Einhaupl KM, Dirnagl U. Ischemia triggered by red blood cell products in the subarachnoid space is inhibited by nimodipine administration or moderate volume expansion/hemodilution in rats. Neurosurgery. 2002;51(6):1457–65.
Vergouwen MD, Vermeulen M, de Haan RJ, Levi M, Roos YB. Dihydropyridine calcium antagonists increase fibrinolytic activity: a systematic review. J Cereb Blood Flow Metab. 2007;27(7):1293–308.
Radhakrishnan D, Menon DK. Haemodynamic effects of intravenous nimodipine following aneurysmal subarachnoid haemorrhage: implications for monitoring. Anaesthesia. 1997;52(5):489–91.
Kasuya H, Onda H, Takeshita M, Okada Y, Hori T. Efficacy and safety of nicardipine prolonged-release implants for preventing vasospasm in humans. Stroke. 2002;33(4):1011–5.
Kasuya H. Clinical trial of nicardipine prolonged-release implants for preventing cerebral vasospasm: multicenter cooperative study in Tokyo. Acta Neurochir Suppl. 2011;110(2):165–7.
Krischek B, Kasuya H, Onda H, Hori T. Nicardipine prolonged-release implants for preventing cerebral vasospasm after subarachnoid hemorrhage: effect and outcome in the first 100 patients. Neurol Med Chir. 2007;47(9):389–94.
Barth M, Pena P, Seiz M, et al. Feasibility of intraventricular nicardipine prolonged release implants in patients following aneurysmal subarachnoid haemorrhage. Br J Neurosurg. 2011;25(6):677–83.
Ehtisham A, Taylor S, Bayless L, Samuels OB, Klein MW, Janzen JM. Use of intrathecal nicardipine for aneurysmal subarachnoid hemorrhage-induced cerebral vasospasm. South Med J. 2009;102(2):150–3.
Goodson K, Lapointe M, Monroe T, Chalela JA. Intraventricular nicardipine for refractory cerebral vasospasm after subarachnoid hemorrhage. Neurocrit Care. 2008;8(2):247–52.
Hanggi D, Beseoglu K, Turowski B, Steiger HJ. Feasibility and safety of intrathecal nimodipine on posthaemorrhagic cerebral vasospasm refractory to medical and endovascular therapy. Clin Neurol Neurosurg. 2008;110(8):784–90.
Lu N, Jackson D, Luke S, Festic E, Hanel RA, Freeman WD. Intraventricular nicardipine for aneurysmal subarachnoid hemorrhage related vasospasm: assessment of 90 days outcome. Neurocrit Care. 2012;16(3):368–75.
Shibuya M, Suzuki Y, Enomoto H, Okada T, Ogura K, Sugita K. Effects of prophylactic intrathecal administrations of nicardipine on vasospasm in patients with severe aneurysmal subarachnoid haemorrhage. Acta Neurochir. 1994;131(1–2):19–25.
Biondi A, Ricciardi GK, Puybasset L, et al. Intra-arterial nimodipine for the treatment of symptomatic cerebral vasospasm after aneurysmal subarachnoid hemorrhage: preliminary results. Am J Neuroradiol. 2004;25(6):1067–76.
Cho WS, Kang HS, Kim JE, et al. Intra-arterial nimodipine infusion for cerebral vasospasm in patients with aneurysmal subarachnoid hemorrhage. Interv Neuroradiol. 2011;17(2):169–78.
Kim SS, Park DH, Lim DJ, Kang SH, Cho TH, Chung YG. Angiographic features and clinical outcomes of intra-arterial nimodipine injection in patients with subarachnoid hemorrhage-induced vasospasm. J Korean Neurosurg Soc. 2012;52(3):172–8.
Musahl C, Henkes H, Vajda Z, Coburger J, Hopf N. Continuous local intra-arterial nimodipine administration in severe symptomatic vasospasm after subarachnoid hemorrhage. Neurosurgery. 2011;68(6):1541–7.
Wolf S, Martin H, Landscheidt JF, Rodiek SO, Schurer L, Lumenta CB. Continuous selective intraarterial infusion of nimodipine for therapy of refractory cerebral vasospasm. Neurocrit Care. 2010;12(3):346–51.
Rosenberg N, Lazzaro MA, Lopes DK, Prabhakaran S. High-dose intra-arterial nicardipine results in hypotension following vasospasm treatment in subarachnoid hemorrhage. Neurocrit Care. 2011;15(3):400–4.
Menei P, Montero-Menei C, Venier MC, Benoit JP. Drug delivery into the brain using poly(lactide-co-glycolide) microspheres. Expert Opin Drug Deliv. 2005;2(2):363–76.
Lemperle G, Morhenn VB, Pestonjamasp V, Gallo RL. Migration studies and histology of injectable microspheres of different sizes in mice. Plast Reconstr Surg. 2004;113(5):1380–90.
Nicholas AP, McInnis C, Gupta KB, et al. The fate of biodegradable microspheres injected into rat brain. Neurosci Lett. 2002;323(2):85–8.
Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J. 2008;22(3):659–61.
Wilson JT, Pettigrew LE, Teasdale GM. Structured interviews for the Glasgow Outcome Scale and the extended Glasgow Outcome Scale: guidelines for their use. J Neurotrauma. 1998;15(8):573–85.
Degen LA, Dorhout Mees SM, Algra A, Rinkel GJ. Interobserver variability of grading scales for aneurysmal subarachnoid hemorrhage. Stroke. 2011;42(6):1546–9.
Jaja BN, Attalla D, Macdonald RL, et al. The subarachnoid hemorrhage international trialists (SAHIT) repository: advancing clinical research in subarachnoid hemorrhage. Neurocrit Care. 2014;21(3):551–9.
Frontera JA, Claassen J, Schmidt JM, et al. Prediction of symptomatic vasospasm after subarachnoid hemorrhage: the modified Fisher scale. Neurosurgery. 2006;59(1):21–7.
Diringer MN, Bleck TP, Claude HJ III, et al. Critical care management of patients following aneurysmal subarachnoid hemorrhage: recommendations from the Neurocritical Care Society’s Multidisciplinary Consensus Conference. Neurocrit Care. 2011;15(2):211–40.
Vergouwen MD, Vermeulen M, van GJ, et al. Definition of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage as an outcome event in clinical trials and observational studies: proposal of a multidisciplinary research group. Stroke. 2010;41(10):2391–5.
Connolly ES Jr, Rabinstein AA, Carhuapoma JR, et al. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2012;43(6):1711–37.
Macdonald RL, Higashida RT, Keller E, et al. Preventing vasospasm improves outcome after aneurysmal subarachnoid hemorrhage: rationale and design of CONSCIOUS-2 and CONSCIOUS-3 trials. Neurocrit Care. 2010;13(3):416–24.
Ibrahim GM, Weidauer S, Vatter H, Raabe A, Macdonald RL. Attributing hypodensities on CT to angiographic vasospasm is not sensitive and unreliable. Stroke. 2012;43(1):109–12.
Vergouwen MD, Ilodigwe D, Macdonald RL. Cerebral infarction after subarachnoid hemorrhage contributes to poor outcome by vasospasm-dependent and -independent effects. Stroke. 2011;42(4):924–9.
Klopfenstein JD, Spetzler RF, Kim LJ, et al. Comparison of routine and selective use of intraoperative angiography during aneurysm surgery: a prospective assessment. J Neurosurg. 2004;100(2):230–5.
Olson DM, Zomorodi M, Britz GW, Zomorodi AR, Amato A, Graffagnino C. Continuous cerebral spinal fluid drainage associated with complications in patients admitted with subarachnoid hemorrhage. J Neurosurg. 2013;119(4):974–80.
Brandt J, Spencer M, Folstein M. The telephone interval for cognitive status. Neuropsychiatry Neuropsychol Behav Neurol. 1988;1:111–7.
Mahoney FI, Barthel DW. Functional evaluation: the Barthel index. Md State Med J. 1965;14:61–5.
Nasreddine ZS, Phillips NA, Bedirian V, et al. The montreal cognitive assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc. 2005;53(4):695–9.
Brott T, Adams HP Jr, Olinger CP, et al. Measurements of acute cerebral infarction: a clinical examination scale. Stroke. 1989;20:864–70.
van Swieten JC, Koudstaal PJ, Visser MC, Schouten HJ, Van GJ. Interobserver agreement for the assessment of handicap in stroke patients. Stroke. 1988;19(5):604–7.
Scott RB, Eccles F, Molyneux AJ, Kerr RS, Rothwell PM, Carpenter K. Improved cognitive outcomes with endovascular coiling of ruptured intracranial aneurysms: neuropsychological outcomes from the international subarachnoid aneurysm trial (ISAT). Stroke. 2010;41(8):1743–7.
Al-Khindi T, Macdonald RL, Schweizer TA. Cognitive and functional outcome after aneurysmal subarachnoid hemorrhage. Stroke. 2010;41(8):e519–36.
Suarez JI, Martin RH. Treatment of subarachnoid hemorrhage with human albumin: ALISAH study. Rationale and design. Neurocrit Care. 2010;13(2):263–77.
Hunt SA, Baker DW, Chin MH, et al. ACC/AHA guidelines for the evaluation and management of chronic heart failure in the adult: executive summary a report of the American College of Cardiology/American Heart Association task force on practice guidelines (committee to revise the 1995 guidelines for the evaluation and management of heart failure): developed in collaboration with the international society for heart and lung transplantation; endorsed by the heart failure society of America. Circulation. 2001;104(24):2996–3007.
DeWitt CR, Waksman JC. Pharmacology, pathophysiology and management of calcium channel blocker and beta-blocker toxicity. Toxicol Rev. 2004;23(4):223–38.
Van GJ, Hijdra A, Wijdicks EF, Vermeulen M, Van CH. Acute hydrocephalus after aneurysmal subarachnoid hemorrhage. J Neurosurg. 1985;63(3):355–62.
Horan TC, Gaynes R. Surveillance of nosocomial infections. In: Mayhall CG, editor. Hospital epidemiology and infection control. Philadelphia: Lippincott Williams & Wilkins; 2004. p. 1659–702.
Conflict of interest
Dr. Macdonald receives grant support from the Physicians Services Incorporated Foundation, Brain Aneurysm Foundation, Canadian Institutes for Health Research, and the Heart and Stroke Foundation of Canada; and is Chief Scientific Officer of Edge Therapeutics, Inc. Drs. Hänggi, Etminan, Aldrich, Mayer, Diringer, Hoh, and Mocco receive consulting fees from Edge Therapeutics, Inc. for serving on the steering committee for this trial and for advising Edge Therapeutics, Inc. Dr. Faleck is an employee of Edge Therapeutics, Inc. Drs. Strange and Miller are paid consultants for Edge Therapeutics, Inc.
Author information
Authors and Affiliations
Corresponding author
Additional information
For the NEWTON Investigators.
Trial registration: www.clinicaltrials.gov Identifier: NCT01893190
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Hänggi, D., Etminan, N., Macdonald, R.L. et al. NEWTON: Nimodipine Microparticles to Enhance Recovery While Reducing Toxicity After Subarachnoid Hemorrhage. Neurocrit Care 23, 274–284 (2015). https://doi.org/10.1007/s12028-015-0112-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12028-015-0112-2