
Abstract
A 61-year-old female died in hospital with multiple organ failure 4 weeks following presentation with acute kidney injury, hemolytic anemia and methemoglobinemia. At autopsy, brown to black discoloration of cartilages was observed. Histology revealed brown pigmentation of the hyaline cartilage, with focal full-thickness erosion of the articular hyaline cartilage, characteristic of alkaptonuria (ochronosis). Although alkaptonuria is rarely fatal, this case illustrates a rare acute fatal complication. Accumulation of circulating homgentisic acid secondary to acute derangement of renal function is believed to have overwhelmed the endogenous antioxidant processes, resulting in hemolysis and methemoglobinemia, which were refractory to treatment. Small numbers of cases have previously been reported in the literature in patients known to suffer with the disease, all of which were preceded by acute kidney injury. Whilst the clinical diagnosis of alkaptonuria may be challenging, the autopsy findings of this rare condition are striking and this case illustrates the utility of the autopsy, albeit retrospectively, in arriving at a diagnosis. To our knowledge this is the first reported case where previously undiagnosed alkaptonuria has presented with methemoglobinemia.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alkaptonuria is a rare metabolic disorder defined by a congenital enzyme deficiency which leads to elevated circulating levels of homogentisic acid (HGA) and the gradual deposition of dark melanin-like pigment in connective tissues, known as ochronosis [1]. This process can lead to a variety of long-term sequelae that are rarely fatal. We report the death of a female with acute hemolysis and methemoglobinemia who was found at autopsy to have previously unsuspected alkaptonuria. Undiagnosed alkaptonuria presenting as acute fatal hematologic disturbance has not to our knowledge been previously documented. The features of this case are described below.
Case report
This 61-year-old female died in hospital following a 4 week admission. The deceased was visiting Australia from overseas and presented with a 1 week history of anorexia, nausea and abdominal pain. No other significant medical history was disclosed by the deceased or the only family member who accompanied her on presentation and during her admission to hospital. Investigations revealed hemolysis with methemoglobinemia, deranged liver function and acute kidney injury. She improved following plasmaphoresis and exchange transfusions; however methemoglobinemia was relapsing and its cause remained undiagnosed. She developed multiple organ failure and passed away despite maximal organ support. Her death was referred to the State Coroner because the underlying cause of her methemoglobinemia and multiple organ failure remained undiagnosed.
Prior to autopsy, medical records from her admission to hospital in Australia were reviewed but no previous medical records from her country of origin were available. At autopsy, scars were noted over the hips and knees in keeping with previous arthroplasty. Scleral pigmentation was not seen. Internally, the lungs showed consolidation with pleural effusions. There was generalized pallor of the myocardium and brownish discoloration of the intima of major vessels but there was no significant valvular disease. The kidneys showed sclerotic change, autolysis, and deep brown-to-black discoloration. There were no renal calculi. There was generalized black discoloration of cartilage in keeping with a diagnosis of alkaptonuria (ochronosis). This included the laryngeal cartilages (Fig. 1), tracheal rings and intervertebral discs (Fig. 2), costal cartilages and the articular cartilages including the humeral heads which demonstrated osteoarthritic change (Fig. 3). The post-mortem blood showed the characteristic brown hue of methemoglobin (Fig. 4).

Black pigmentation of the thyroid cartilage

Pigmentation of intervertebral discs

Erosion of the pigmented articular cartilage of the humeral head

Peripheral blood sample taken at autopsy showing the characteristic brown hue of methemoglobin
Histology identified features of alkaptonuria/ochronosis characterized by brown pigmentation of hyaline cartilage and perichondrium. In hyaline cartilage, the pigmentation was diffuse (Fig. 5) whilst in the perichondrium; the pigment was granular (Fig. 6). The pigment was non-refractile and negative for iron using Perls’ stain. Focal erosion of the articular cartilage was noted in the humeral head (Fig. 7) with fibrosis containing fragments of pigmented cartilage in the subchondral bone (Fig. 8). Within the kidneys, there were numerous granular intra-tubular casts which were negative using Perls’ stain. Hemosiderin deposition was noted in the liver, spleen and nodal reticuloendothelial cells secondary to hemolysis. Other findings included diffuse alveolar damage and renal tubular necrosis.

Photomicrograph (H&E) showing diffuse brown pigmentation of the hyaline cartilage (×200)

Photomicrograph (H&E) showing granular brown pigmentation in the perichondrium (×200)

Photomicrograph (H&E) showing erosion of the hyaline cartilage of the humeral head seen macroscopically in Fig. 3 (×100)

Photomicrograph (H&E) showing fragments of pigmented cartilage in the subchondral bone of the humeral head (×400)
DNA was extracted from a post-mortem skin sample. Comparative genomic hybridization revealed a normal female profile with no copy number changes in the HGD gene at 3q13.13. Molecular analysis revealed compound heterozygosity characterized by two mutations (p.Gly270Arg and p.Gly152Arg) supporting the diagnosis of alkaptonuria. Genetic counselling was recommended for the surviving relatives.
The cause of death following autopsy was given as multiple organ failure due to methemoglobinemia and hemolytic anemia due to alkaptonuria.
Discussion
Alkaptonuria is a congenital metabolic disease characterized by a clinical triad of the passage of dark urine, progressive deposition of dark melanin-like pigment in connective tissues and arthropathy [1, 2]. It is autosomal recessive, occurring in an estimated 1 in 250,000 to 1,000,000 in most ethnic groups [3]. Clustering occurs in certain population groups including the Dominican Republic, the Druze of Lebanon, Wales and Slovakia, the latter exhibiting an incidence as high as 1 in 19,000 [4].
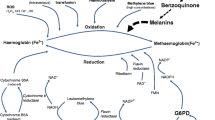
Alkaptonuria results from mutation of the HGD gene on chromosome 3q, which encodes for the enzyme homogentisate 1,2-dioxygenase [5]. This converts homogentisic acid (HGA), a byproduct of phenylalanine and tyrosine metabolism, to maleylacetoacetic acid [6]. Enzyme deficiency results in elevated circulating levels of HGA, which is excreted in the urine and transforms the urine to a characteristic dark color upon oxidation [2]. There is also widespread deposition of HGA as a polymerized melanin-like brown pigment in collagenous connective tissue [1]. The term ochronosis was first coined by Virchow in 1866 after likening the color of the granular pigment seen in post mortem sections to the earth pigment, ochre [1].
HGA has an affinity for fibrillary collagens which are surrounded by abundant mucopolysaccharide ground substance [2]. In the sclera and auricular cartilage this may present clinically as a blue or grey hue [1]. Internally, grey/black discoloration is noted in hyaline, elastic and fibrocartilage including the articular surfaces of the joints, costal cartilages, trachea, epiglottis and intervertebral discs [1, 2, 7]. Discoloration may be observed in blood vessel walls, cardiac valves, endocardium, joint capsules, ligaments and tendons, dura, renal parenchyma, renal calculi and scar tissue. Synovial joints and the aortic valve are sites of predilection [7]. The central nervous system, gastrointestinal tract, hepatobiliary, lymphoreticular and endocrine systems are not typically involved [2, 7]. Bone is not usually pigmented [2, 7].
Although HGA was thought to be responsible for tissue damage in alkaptonuria, in vitro studies indicate benzoquinone-2-acetic acid as the effector molecule, arising through a process of spontaneous oxidation [6]. This conversion produces reactive oxygen species which are thought to be responsible for the melanin-like onchronotic pigment, downstream oxidation and aggregation of proteins and lipid peroxidation promoting inflammation and increasing oxidative stress [6]. In cartilage this induces chondrocyte apoptosis and degradation.
Within cartilage, ochronotic pigment is mainly deposited in the extracellular matrix around chondrocytes in a diffuse distribution [7]. Granular pigment has also been reported within the cytoplasm of chondrocytes [1, 2]. Pigmented shards of fragmented cartilage may be present in the synovium.
Ochronosis results in long term sequelae. In joints, the cartilage becomes brittle and leads to osteoarthritis which may necessitate arthroplasty [1]. In the spine, spondyloarthropathy, ankylosis and disc herniation are common events [1, 2]. Individuals are prone to tendon, ligament and muscle tears. In the cardiovascular system subsequent dystrophic calcification of the aortic valve may manifest as stenosis and/or regurgitation [7, 8]. Disease is less frequent in the other cardiac valves [8]. Renal and prostatic calculi are common, as is ochronotic cast formation within the renal tubules. Pigment deposition in ocular tissue is typically without clinical consequence [2].
The HGD gene comprises 14 exons. Mutations which may result in the disease have been documented in each of the exons, most frequently in the form of missense mutations [4]. Around 138 different reportedly pathogenic mutations have been described in patients from as many as 40 countries [9].
In this case, molecular analysis of the HGD gene identified compound heterozygosity characterized by p.Gly270Arg and p.Gly152Arg mutations. Of the two mutations, the former has been previously described in affected patients from Europe, the Dominican Republic, the USA and Turkey, with the highest number of cases found in Slovakia [4]. To our knowledge, the p.Gly152Arg mutation has not yet been reported in an alkaptonuric patient.
Life expectancy is generally unaffected by the disease [3], however, fatalities from aortic valve disease and end-stage renal disease have been reported [10,11,12]. Rarely, acute fatal hematological derangement has been described. To our knowledge, there exist nine previously published cases of fatal intravascular hemolysis in patients with alkaptonuria accompanied, in all but two cases, by methemoglobinemia [1, 13,14,15,16,17,18,19]. Reports suggest a variety of initiating factors including drug toxicity, exacerbation of chronic renal disease and sepsis. Hemolysis and methemoglobinemia were preceded by acute kidney injury in the previously reported cases.
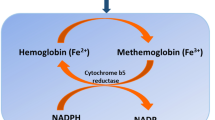
Methemoglobin (MetHb) results from hemoglobin oxidation when iron within the molecule is converted from the ferrous (Fe 2+) to the ferric (Fe3+) state. This results in left-shift of the oxygen-hemoglobin dissociation curve and concomitant cellular hypoxia. In normal individuals, the cellular concentration of MetHb is maintained below 1% by endogenous mechanisms, principally NADH-MetHb reductase (cytochrome-b5 reductase). If exposed to an oxidizing agent, these protective mechanisms may be overwhelmed. Initiating factors include local anesthetic agents such as benzocaine, dapsone, and sulphonamides; substances containing nitrites, nitrates and chlorates; analine dyes and naphthalene (mothballs). Other forms include the methemoglobinopathies where there is abnormal production of hemoglobin more resistant to enzymatic reduction and those where there is reduced or absent function of the endogenous reducing enzymes [20, 21].Although in this case, methemoglobinemia was diagnosed prior to death, if post-mortem samples are utilized for diagnosis artefactual MetHb increases may be observed due to autoxidation during storage, possibly by freezing. Alternatively MetHb may reduce due to the activity of MetHb reductase or microbial activity after death. If post-mortem samples are utilized, delays in sampling and analysis should be reduced as much as possible, collected in EDTA and not frozen, but and stored under refrigeration. Putrefied samples should not be used [22].
In this case, the presenting symptoms suggested a gastrointestinal illness. This may have resulted in dehydration, precipitating acute kidney failure, leading to accumulation of HGA and either directly or through its proposed effector molecule benzoquinone-2-acetic acid, promoted oxidative hemolysis and methemoglobinemia.
It is proposed that elevated HGA levels alone are insufficient to provoke methemoglobinemia and additional factors that disrupt the balance between oxidation and anti-oxidant capacity are necessary [13]. These may include uremia (observed in chronic kidney disease and acute kidney injury), deficiencies of vitamin A, vitamin E and selenium, or genetic deficiencies in enzyme activities. Hemodialysis, transfusion and parenteral iron may contribute to a pro-oxidant state [13]. Therefore, in the deceased, elevated HGA concentrations, acute kidney injury and potentially repeated blood transfusion may have combined to overwhelm endogenous anti-oxidant processes, leading to oxidative hemolysis and relapsing methemoglobinemia.
Methemoglobinemia in the setting of alkaptonuria is resistant to standard therapy, including the administration of the antioxidants methylene blue, ascorbic acid and N-acetyl cysteine and prognosis in the event of this rare metabolic sequela is poor [13, 14]. In the published cases, the ensuing metabolic acidosis and multi-organ failure resulted in 100% mortality.
In this case, no history of alkaptonuria was disclosed by the deceased or her family member. Previous reported cases of alkaptonuria-associated methemoglobinemia were in individuals previously diagnosed with the disease. We are not aware of a case where previously undiagnosed alkaptonuria has presented with methemoglobinemia. Whilst clinical diagnosis may be challenging, the post mortem findings of alkaptonuria are striking and this case illustrates the utility of post mortem examination, albeit retrospectively, in arriving at the diagnosis.
Key points
-
1.
Methemoglobinemia results from oxidation of the hemoglobin molecule from the ferrous (Fe 2+) to the ferric (Fe3+) state and has diverse causes.
-
2.
Elevated levels of circulating homogentisic acid in alkaptonuric patients may overwhelm endogenous anti-oxidant processes resulting in methemoglobinemia.
-
3.
Alkaptonuria may present as otherwise unexplained methemoglobinemia.
-
4.
Alkaptonuria (ochronosis) results in characteristic black pigmentation of hyaline cartilage at autopsy.
-
5.
We report a case of fatal methemoglobinemia where the diagnosis of underlying alkaptonuria was not made until autopsy and where molecular analysis revealed a previously unreported mutation in the HGD gene.
References
Abreo K, Abreo F, Zimmerman SW, Hartman H, Gilbert EF, Katcher ML, et al. A fifty-year-old man with skin pigmentation, arthritis, chronic renal failure and methemoglobinemia. Am J Med Genet. 1983;14:97–114.
Gaines JJ. The pathology of alkaptonuriconchronisis. Hum Pathol. 1989;20:40–6.
Phornphutkul C, Introne WJ, Perry MB, Bernardini I, Murphey MD, Fitzpatrick DL, et al. Natural history of alkaptonuria. N Engl J Med. 2002;347:2111–21.
Zatkova A. An update on molecular genetics of alkaptonuria (AKU). J Inherit Metab Dis. 2011;34:1127–36.
Pollak MR, Chou YH, Cerda JJ, Steinmann B, La Du BN, Seidman JG, et al. Homozygosity mapping of the gene for alkaptonuria to chromosome 3q2. Nat Genet. 1993;5:201–4.
Braconi D, Millucci L, Bernardini G, Santucci A. Oxidative stress and mechanisms of ochronosis in alkaptonuria. Free Radic Biol Med. 2015;88:70–80.
Helliwell TR, Gallagher JA, Ranganath L. Alkaptonuria - a review of surgical and autopsy pathology. Histopathology. 2008;53:503–12.
Pettit SJ, Fisher M, Gallagher JA, Ranganath LR. Cardiovascular manifestations of alkaptonuria. J Inherit Metab Dis. 2011;34:1177–81.
Zatkova A. HGD mutation database. http//:hgddatabase.cvtisr.sk. Accessed 1 Nov 2017.
Gonzales ME. Alkaptonuric aortic stenosis: a case report. AANA J. 1999;67:145–51.
Faria B, Vidinha J, Pego C, Correia H, Sousa T. Impact of chronic kidney disease on the natural history of alkaptonuria. Clin Kidney J. 2012;5:352–5.
Zaraa I, LAbbene I, Trojjet S, Mrabet D, Meddeb N, Chelly, et al. Endogenous ochronosis with a fatal outcome. J Cutan Med Surg. 2012;16:357–60.
Davison AS, Milan AM, Gallagher JA, Ranganath LR. Acute fatal metabolic complications in alkaptonuria. J Inherit Metab Dis. 2016;39:203–10.
Heng AE, Courbebaisse MD, Kemeny JL, Matesan R, Bonniol C, Deteix P, et al. Hemolysis in a patient with alkaptonuria and chronic kidney failure. Am J Kidney Dis. 2010;56:e1–4.
Isa Y, Nihei S, Irifukuhama Y, Ikeda T, Matsumoto H, Nagata K, et al. A rare case of acquired methemoglobinemia associated with alkaptonuria. Intern Med. 2014;53:1797–800.
Bataille S, Moal V, Aquaron RR, Grünfeld JP, Daniel L. Hemolysis: a fatal complication of alkaptonuria in a severe renal failure patient. Clin Nephrol. 2014;81:374–6.
Liu W, Prayson RA. Dura mater involvement in ochronosis (Alkaptonuria). Arch Pathol Lab Med. 2001;125:961–3.
Uchiyama C, Kondoh H, Shintani H. Acute methemoglobinemia associated with ochronotic valvular heart disease: report of a case. Thorac Cardiovasc Surg. 2010;58:113–9.
Mullan A, Cocker D, Taylor G, Millar C, Ranganath L. Fatal oxidative haemolysis and methaemoglobinaemia in a patient with alkaptonuria and acute kidney injury. Clin Kidney J. 2015;8:109–12.
Cortazzo JA, Lichtman AD. Methemoglobinemia: a review and recommendations for management. J Cardiothoracic and Vasc Anaesthesia. 2014;28:1043–7.
Chikezi PC, Ekechukwu CU. Acute patho-toxilogical indicators of methaemoglobinaemia. J Acute Dis. 2016;5:179–84.
Varlet V, Ryser E, Ausberger M, Palmiere C. Stability of post-mortem methemoglobin: Artefactual changes caused by storage conditions. Forensic Sci Int. 2018;283:21–8.
Acknowledgements
We thank Miss Tasma How and the histopathology laboratory at Forensic Science SA for technical support, and Dr. Sui Yu, Mr. Kristian Brion and Mr. Tim Pyragius of SA Pathology for the genetic analysis.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical approval
This article does not contain any studies with human participants performed by any of the authors. Approval to report the case was given by Forensic Science SA, Adelaide, Australia.
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Freeman, A.R., Wills, S.M. Fatal methemoglobinemia complicating alkaptonuria (ochronosis): a rare presentation. Forensic Sci Med Pathol 14, 236–240 (2018). https://doi.org/10.1007/s12024-018-9965-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12024-018-9965-y