
Abstract
Vitamin D-dependent rickets type IA (VDDR-IA) is caused by biallelic mutations in CYP27B1. Data regarding genotype–phenotype correlation in VDDR-IA are scarce. Here, we aimed to investigate clinical/genotypic features and long-term follow-up of 13 new cases with VDDR-IA and genotype–phenotype correlation of reported cases in the literature. Thirteen patients with VDDR-IA were evaluated. Eight patients had reached their final height at the time of the study and, for whom, long-term outcome data were analyzed. Further, all VDDR-IA patients in the literature (n:183) were analyzed and clinical–genetic features were recorded. The median age of diagnosis was 2.55 ± 1.13 (1.0–12) years. Initial diagnoses before referral to our clinic were nutritional rickets (n:7), hypophosphatemic rickets (n:2), and pseudohypoparathyroidism (n:1). All had biochemical evidence suggestive of VDDR-IA; except one with elevated 1,25(OH)2D3 and another with hyperphosphatemia, in whom pseudohypoparathyroidism was excluded with molecular tests. Combined analyses of our cohort and other series in the literature demonstrated that three most common CYP27B1 mutations are p.F443Pfs*24, c.195 + 2T > G, and p.V88Wfs*71. In Turkish population, p.K192E mutation along with the former two is the most common mutations. Comparison of clinical features demonstrated that c.195 + 2T > G mutation causes the most severe and p.K192E mutation causes the least severe phenotype with respect to age and height at presentation and calcitriol requirement. We found a clear genotype–phenotype correlation in VDDR-IA, notably CYP27B1 intronic c.195 + 2T > G mutation causes a more severe phenotype with lower height SDS at presentation and, higher calcitriol requirement, while less severe phenotype occurs in p.K192E mutation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
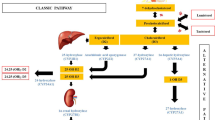
Vitamin D is essential for bone mineral metabolism, maintenance of calcium, and phosphate homeostasis. The 1 α-hydroxyl group of calcitriol (1,25(OH)2D3) is required for the binding of vitamin D to its nuclear receptor [1]. Therefore, two inactive precursors of vitamin D: ergocalciferol (Vitamin D2) and cholecalciferol (Vitamin D3), need sequential 25- and 1α–hydroxylation to form 1,25(OH)2 D3. First, vitamin D is hydroxylated to 25-hydroxy vitamin D (25(OH)D3) by several hepatic enzymes, mainly microsomal CYP2R1 in the liver, then enters mitochondria where it is further hydroxylated to the biologically active metabolite 1,25(OH)2D3 by the 25-OH vitamin D-1α-hydroxylase in kidneys.
The hereditary defects in vitamin D metabolism and its function cause rare forms of rickets, also known as vitamin D-dependent rickets (VDDR) [2]. VDDR type 1 involves VDDR-IA (1α-hydroxylase deficiency) and VDDR-IB (25-hydroxylase deficiency). VDDR type 2 involves vitamin D receptor (VDDR-IIA) and post-receptor (VDDR-IIB) defects [2]. More recently, CYP3A4 mutation as a cause of VDDR type 3 (VDDR-III) by increasing inactivation of vitamin D has been described [3]. VDDR-IA (OMIM; 264700) is the most common type of VDDR and is caused by biallelic inactivating mutations in CYP27B1 gene which results in defective 1α-hydroxylation of 25(OH)D3 to 1,25(OH)2D3 [4]. Despite the paucity of the studies, its prevalence is estimated as around 1-5/10.000 (https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=EN&Expert=289157). Patients with VDDR-IA appear normal at birth and usually present during the first two years of life with hypotonia, muscle weakness, growth failure, delay in walking, and bowing in legs [5]. Laboratory characteristics of the patients with VDDR-IA demonstrate typical calciopenic rickets with inappropriately normal or low levels of 1,25(OH)2D3 and normal or increased concentrations of 25(OH)D3 [6,7,8].
To the best of our knowledge, over 180 patients with 81 mutations in the CYP27B1 gene have been reported from different ethnic groups so far. These mutations span all exons of the gene and mostly include missense and nonsense changes (approximately 70% of CYP27B1 mutations) (http://www.hgmd.cf.ac.uk/ac/index.php) (Fig. 1). However, data regarding genotype–phenotype correlation and long-term follow-up and outcome of patients with VDDR-I-A are scarce.

Schematic presentation of CYP27B1 (NM_000785.4) gene with its exons and introns showing all reported mutations. Arrows show identified variants at respective exons and introns. Mutations identified in Turkish cohorts underlined, and mutations identified in our patients denoted in bold. Frameshift, splice site and missense/nonsense mutations showed in upper, middle, and lower boxes, respectively
In this study, we report 13 new patients with VDDR-IA and analyzed the most common genotypes worldwide with extensive literature review. In addition, genotype–phenotype correlation with special emphasis on clinical presentations, response to treatment, and long-term outcome were investigated.
Subjects and Methods
Patients
Patients presenting with clinical and radiological findings of rickets with elevated serum alkaline phosphatase (ALP) and parathyroid hormone (PTH), normal or increased levels of 25(OH)D3, and inappropriately normal or low levels of 1,25(OH)2D3 if available were included to the study. The diagnosis of VDDR-IA was suggested on clinical ground with unresponsiveness to high-dose cholecalciferol treatment and responsive to standard-dose calcitriol treatment. Eventually, the diagnosis was confirmed by genetic testing in all patients.
A comprehensive search of PubMed, using following keywords in the PubMed search query: “CYP27B1,” “VDDR,” and “Vitamin D-dependent rickets” was conducted to identify all the studies presenting data concerning the genotype–phenotype association and/or long-term follow-up of VDDR-IA patients. We limited our search to human studies. We excluded studies that report cases where the molecular diagnosis of VDDR-IA had not been established. We also checked the bibliographies of included studies for relevant articles. Data were extracted on to a pre-formatted table with details of the study, CYP27B1 variant, and number of patients that carry each individual variant. For the comparison of the clinical features of most common mutations, the clinical and laboratory findings were reviewed and statistical analyses were performed between the mutation groups having more than 20 homozygous patients. Patients with compound heterozygous mutations were excluded from the statistical analyses. The last search was completed on August 24, 2020.
Follow-Up
Serum calcium, phosphate, ALP, PTH concentrations, and urinary calcium excretion were evaluated once per month for the first three months after initiating calcitriol treatment. Thereafter, children were seen every three to four months as long as growth continued. After epiphyseal fusion, follow-up visits occurred every 6 months. Auxological measurements were obtained at each visit. Renal ultrasound was performed yearly to assess the presence of nephrocalcinosis. The patients were transferred to adult endocrinology clinic after the age of 18 years. The patients with closed epiphysis and no height changes for at least one year were accepted at final height.
Biochemical Analysis
Calcium, phosphate, and creatinine in serum and urine samples were analyzed with automated systems (AU 600, Beckman Coulter, USA). Serum ALP levels were measured by using AU 600 systems using methods based on the recommendations of the IFCC (International Federation of Clinical Chemistry) which is a quantitative colorimetric measurement using 2-amino-2-methyl-1-propanol as a buffer. Serum PTH was analyzed by Access Immunoassay Systems (Beckman Coulter, USA).
Plasma 25(OH)D3 measurements were performed by using Thermo Finnigan high-performance liquid chromatography (HPLC) system with ClinRep® HPLC Complete Kit (Recipe Chemicals–Instruments, Germany). Intra-assay and inter-assay coefficients of variability for the levels of 21.5 and 75.5 μg/L were 4.60% and 3.06%, and 3.60% and 4.40%, respectively. The quality evaluation of the method was reported [9].
The measurement of 1,25(OH)2D3 levels was performed in different validated commercial laboratories using ELISA-based kits throughout the years when the patients were presented to our clinic before the initiation of calcitriol who were not on calcitriol. For evaluation of the results, reference ranges provided by the laboratory had been used which were given separately for each patient in Table 2.
Radiological Evaluation
Bone mineral density of lumbar vertebrae was measured by dual energy X-ray absorptiometry (Hologic, Bedford, MA, USA).
CYP27B1 Gene Sequencing
Genomic DNA was extracted using a QIAamp DNA Mini Kit (QIAGEN, Germantown, MD USA) from peripheral blood lymphocytes according to standard protocols after informed consent. All coding exons and exon–intron boundaries of the CYP27B1 gene (NM_000785.4) were sequenced via Sanger sequencing using an ABI 3130XL DNA Sequencer (Thermo Fisher Scientific, Waltham, MA, USA). The primers were designed using Primer3 software (primer3.ut.ee) [10].
Statistical Analysis
Statistical analyses were performed with GraphPad Prism 8.0 (GraphPad Software, Inc., San Diego, CA). The data were presented as mean ± SEM (median; ranges). The median age of presentation, 1,25(OH)2D3 levels, height standard deviation score (SDS), and doses of calcitriol were compared using the nonparametric Kruskal–Wallis test, and Mann–Whitney U test used for pairwise group comparisons. Spearman rank correlation was used to test associations between prior therapy and 1,25(OH)2D3 levels at admission. All tests were two-tailed. A p value ≤ 0.05 was considered statistically significant.
Results
Clinical Characteristics
Thirteen (7 males) patients from 10 unrelated families (6 consanguineous parents, 4 nonconsanguineous parents but who were originated from nearby villages) were evaluated. The mean age of diagnosis was 4.8 ± 1.13 years (Median: 2.55; min: 1,0; max: 12). All patients had a history of receiving routine prophylactic vitamin D replacement (400 IU/day) for the first year of life. Ten patients out of 13 came to medical attention within the first 24 months of life. Initial diagnoses of the patients were nutritional rickets (NR) (n = 7), hypophosphatemic rickets (HR) (n = 2), and pseudohypoparathyroidism (PHP) (n = 1) before the referral to our clinic. Seven patients had a history of high-dose vitamin D intake (300,000–1,500,000 IU); two of them (Pt#1.1, Pt#4.1) had toxic levels of 25(OH)D3 (≥ 150 ng/mL) and three (Pt#3.1, Pt#6.1, Pt#10.1) had hypervitaminosis D (≥ 100 ng/mL). The distribution of complaints and available clinical data prior to referral are presented in Table 1. The mean time from the first medical visit to the diagnosis of VDDR-IA was 29.72 ± 10.14 months (Median: 11.10; min: 0.52; max: 96).
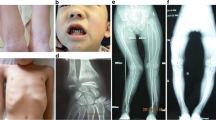
Upon referral to our clinic, all patients were diagnosed with VDDR-IA based on the biochemical and radiological findings of active rickets. Hypophosphatemia, elevated ALP and PTH, and normal/elevated 25(OH)D3 levels were detected in all patients except Pt#8.1. She was presented to the clinic with hypocalcemia (Ca: 1.59 mmol/L) and hyperphosphatemia (PO4: 2 mmol/L) and initially diagnosed as PHP. However, VDDR-IA was subsequently considered upon bone pain and the review of the X-rays on initial presentation which was consistent with rickets (Fig. 2). PHP was excluded also after demonstrating normal GNAS gene sequencing and Gsα levels (104%, N: 85–115%). All measured 1,25(OH)2D3 levels were low or inappropriately normal, except mildly elevated in Pt#3.1.

Radiologic features of the Pt#8.1 at admission showing active rickets with a loss of provisional calcification zone with irregular metaphysis, fraying and widening of growth plate (a1, b1), and after treatment with normal growth plate with mild osteopenia (a2, b2)
The mean height SDS on admission was − 1.71 ± 1.2 (Median: − 1.23; min: − 4.71; max: − 0.25). Six patients had height SDS more than 1 SDS below their mid-parenteral height (MPH). The clinical and biochemical characteristics of the patients are summarized in Table 2.
Treatment and Follow-Up
All patients received calcitriol treatment, which was initiated either at the time of diagnosis (n:6) or after failure of treatment with vitamin D3 (n:7). The initial calcitriol dose was 41.01 ± 7.59 ng/kg/day (median: 40; min: 14.84; max: 86.2) which was determined by severity of the disease and/or hypocalcemia. The mean dose for calcitriol to achieve clinical and biochemical recovery was 59.53 ± 13.68 ng/kg/day (median: 50; min: 12.19; max: 144.23). Clinical, biochemical, and radiological improvement was observed in all patients after initiation of calcitriol therapy. Normocalcemia achieved at 1–3 months of treatment, but normalization of PTH and ALP occurred at a later period (3–12 months). Upon referral to our clinic, one patient with calcitriol and phosphate treatment (Pt#2.1) and one patient who received high-dose vitamin D treatment (Pt#3.1) had microlithiasis on renal ultrasonography, which resolved after initiation of relevant treatment and discontinuation of phosphate therapy. None of the patients developed microlithiasis or nephrocalcinosis during the follow-up.
The onset of walking documented in four children (median age of 14.2 months; range 12–21) who had not started walking before the initiation of treatment. In these children, the median duration until to walk after initiation of calcitriol treatment was 6 months (range: 3–9 months) and the patients could walk at a median age of 20.75 months (range: 16.9–27 months).
The mean duration of calcitriol treatment was 9.45 ± 7.17 years (median: 7.15; min: 1.4; max: 25.16). The mean height SDS change from baseline to last visit was 0.73 ± 0.39 SDS (median:1.04; min: − 1.91; max: 2.82). Ten patients had improved height SDS during follow-up, in whom, the mean increase in height SDS from baseline to last follow-up visit was 1.33 ± 0.26 SDS (median: 1.19; min: 0.25; max: 2.82). In 4 patients with no height SDS improvement during follow-up, two patients (#1.2, #4.1) received treatment in the first two years of life, and they were compliant; however, their parents were short and the final height SDSs were within their MPH SDSs. The other two patients (#7.1 and #7.2) have been diagnosed at older ages (12 and 11.6 years) and noncompliant to treatment.
On the long-term follow-up, eight patients reached adult height. Their mean final height SDS was − 1.38 ± 0.46 (Median: − 1.39; min: − 2.91; max: 0.62), MPH SDS was − 1.12 ± 0.39 (Median: − 1.50; min: −2.46; max: 0.86). Of these, three patients were fully compliant to treatment and had the highest BMD z-scores among patients reaching adult height. Five patients either stopped treatment (n:3) or declared to skip the drug doses (n:2). Lumbar BMD z-scores of the patients were − 0.14 ± 0.42 (Median: 0.05; min: − 1.9; max: 1.7). The clinical characteristics of eight patients who reached final height are summarized in Table 3.
Sequence Analysis of the CYP27B1 Gene
All of the patients had homozygous mutations in CYP27B1 gene. We revealed a homozygous seven-nucleotide duplication (c.1319_1325dupCCCACCC) causing frameshift mutation (p.F443Pfs*24) in 8 patients from 6 unrelated families. A homozygous missense c.574 A > G (p.K192E) mutation was detected in three patients from two unrelated families. Homozygous c.590G > A (p.G197D) and c.1215 + 2T > A (p.L380Afs*57) mutations were detected in Pt#9.1 and Pt#10.1, respectively (Table 2).
Genotype–Phenotype Correlation
Combined analyses of the current cohort (n: 13) and other cases in the literature (n: 183) were performed. Three mutations, namely p.F443Pfs*24, c.195 + 2T > G, and p.V88Wfs* were the most common mutations in the CYP27B1 gene and identified as a homozygous state in 24, 20, and 28 patients, respectively. Comparison of available data regarding clinical features of each mutation group demonstrated that height SDS at presentation was lower and, higher calcitriol treatment doses required during treatment in patients with c.195 + 2T > G mutation, suggesting more severe disease (Table 4). Evaluation of Turkish cohorts separately demonstrated that p.K192E mutation is among the three most common mutation along with p.F443Pfs*24 and c.195 + 2T > G. Comparison of clinical features of p.K192E mutation with the other two mutations demonstrated that patients with p.K192E had older age and better height SDS at presentation and lower calcitriol replacement dose requirement during treatment, suggesting milder disease (Supplementary Table S1).
Discussion
In this report, we presented detailed clinical features and long-term follow-up of 13 new patients with VDDR-IA and delineated genotype–phenotype correlation for the most common mutations thorough the literature review. Based on the mutations registered in the Human Gene Mutation Database (HGMD; January 2020) and literature search, a total of 81 different mutations in CYP27B1 gene have been identified as a cause of VDDR-IA (Fig. 1) [11]. Among them, we identified three most commonly observed pathogenic variants which are a seven-nucleotide duplication in exon 8 (p.F443Pfs*24), a small deletion in exon 2 (p.V88Wfs*71), and a splice donor site mutation in intron 1 (c.195 + 2T > G), all together, observed in 67 [5, 7, 12,13,14,15,16,17,18,19,20,21,22], 63 [5, 12, 13, 20] and 42 [7, 8, 15, 16] alleles, respectively. In Turkish patients cohorts, two most frequently observed pathogenic variants in CYP27B1 are also seven-nucleotide duplication in exon 8 and a splice donor site mutation in intron 1 (c.195 + 2T > G) as in the rest of the world. However, in Turkish patients and in our cohort reported here, a specific mutation p.K192E appears to be one of the most common mutations.
The seven-nucleotide duplication in exon 8 has been reported in different ethnic groups [5, 13, 14, 17,18,19,20,21,22] and has been suggested that it has risen by independent de novo events, probably as the result of a slipped strand mispairing during meiosis [13]. It has also been reported in Turkish cohorts previously [7, 15, 16], which along with our study further confirmed that the seven-nucleotide duplication in exon 8 is a common mutation distributed worldwide.
Splice donor site mutation in intron 1 (c.195 + 2T > G) observed in 42 alleles (20 homozygous, 2 compound heterozygous), which makes it the third most common mutation in CYP27B1 described in the literature and only found in Turkish population [7, 8, 15, 16]. Since this mutation has not been reported in other ethnic groups, it may delineate a founder or common ancestor effect [16]. To date, 63 Turkish patients with VDDR-IA (including our patients) have been studied for CYP27B1 mutations and 11 different mutations have been described. Nine of those described mutations including the common intron 1 (c.195 + 2T > G) are unique to the Turkish population (Supplementary Table S2) [7, 8, 15, 16, 23].
The second most common mutation worldwide is deletion in exon 2 (p.V88Wfs*71) is a known frameshift mutation that results in a severely truncated protein. It is a founder mutation mostly observed in French Canadian patients, originating from Quebec, where the prevalence of VDDR-IA is reported to be unusually high [12, 20]. This mutation has not been described yet in Turkish patients whereas p.K192 E mutation appears to be the third most common mutation in Turkish patients. p.K192E mutation has not been reported in other ethnic groups, as well (Supplementary Table S2).
Previously, some authors suggested that the severity of the CYP27B1 mutation cannot be predicted from the clinical presentation of the disease [17, 24]. However, analyzing the clinical and molecular characteristics of the patients reported to date, we were able to demonstrate a certain genotype–phenotype correlation in VDDR-IA patients. Although we could not find significant differences in biochemical parameters at presentation between the different genotypes, the age and height SDS at presentation and calcitriol doses required to control the disease differed among the genotypes. It appears that intron 1 c.195 + 2T > G mutation causes a more severe phenotype with lower height SDS at presentation and, higher calcitriol requirement, while a less severe phenotype occurs in patients with p.K192E mutation. The previously studied functional assay of the mutations in CYP27B1 showed that almost all mutations cause either complete loss of enzymatic activity [12, 24] or retained just the 5–12% of normal enzymatic activity [18, 25]. Nevertheless, only a few disease causing mutations showed enzymatic activity of more than 20% of normal [25, 26], in which clinically asymptomatic homozygous mutation carriers in older ages had been described with elevated ALP and PTH levels as the biochemical features of rickets [26]. Although we could not perform a functional assay, we predict that p.K192E mutation leads to less severe loss of enzymatic activity, since the patients with this mutation present in older age and with milder biochemical abnormalities compared to other mutations (Supplementary Table S1).
There are limited data about the long-term outcome of the disease [5, 17]. Edouard et al. [5] observed that treatment with calcitriol resulted in the normalization of biochemical parameters within three months while other studies reported a longer period of time (6–12 months) [16, 17]. The difference in the time for normalization of biochemical parameters could be due to the difference in the initial calcitriol dosage. Edouard et al. [5] started calcitriol treatment at a dose of 1.0 μg/day and decreased thereafter, while, similar to us, Chi et al. [17] started calcitriol treatment at a median daily dose of 0.5 μg/day (range 0.25–1.5 μg). In our cohort, normocalcemia was achieved at 1–3 months of treatment, but normalization of PTH and ALP occurred later between 3 and 12 months. Thus, our study further supports the relationship between the initial dose of calcitriol therapy and time for normalization of biochemical parameters. Nephrocalcinosis, a complication of treatment, was not detected in any of the patients during follow-up in our cohort or the patients of Chi et al. [17], while 16% of the cases with higher initial calcitriol treatment doses were found to have nephrocalcinosis [5]. Thus, treatment initiation with lower doses and titrating the dose according to biochemical response could be a more appropriate approach to avoid the treatment-related complications.
It has been reported that patients had normal BMD after three months of treatment, even though they received calcitriol only after puberty, suggested that having low levels of 1,25(OH)2D3 during childhood does not affect the achievement of normal peak bone mass [5]. However, our findings dispute that observation. We have demonstrated in our older patients that bad treatment compliance during childhood before the growth cease leads to short adult stature and low BMD in the long term (Table 3). Thus, we suggest that early initiation of treatment and good compliance are essential in achieving normal height and BMD.
A few clinical points in VDDR-IA deserve further discussion. Clinical and laboratory features of VDRR-IA are similar to nutritional rickets (NR) which may cause initial misdiagnosis and treatment delay as in our patients. Normal/high 25(OH)D3 levels, low or inappropriately normal 1,25(OH)2D3 levels, and unresponsiveness to cholecalciferol treatment are important features of VDDR-IA that differ from NR. Misdiagnosis of HR as in our Pt#3.1, and phosphate treatment may also delay healing of rickets and lead to additional complications such as nephrocalcinosis/nephrolithiasis [27], since the phosphate treatment further increases phosphate excretion due to stimulatory effect on PTH. Discontinuation of phosphate treatment improved microlithiasis in our case. Mild nephrocalcinosis has been detected in 16% of VDRR-IA [5] whereas about 30–70% of patients with HR have been reported to develop nephrocalcinosis [28]. Another patient (Pt#8.1) in our cohort had hyperphosphatemia and for that reason, initially evaluated for PHP. When we evaluated all reported cases in the literature, although they are not discussed in the relevant papers, there are several normo/hyperphosphotemic cases with proven CYP27B1 mutation, similar to our Pt#8.1 [5, 12, 13, 25, 26, 29,30,31]. Falsely elevated serum phosphate concentrations (pseudohyperphosphatemia) have been described in several conditions such as paraproteinemia, hemolysis, and icteric and lipemic serums [32]. However, we could not detect any of these conditions in our patient, and she continued to have upper normal phosphate levels (around 1,6 mmol/L) during follow-up. Although the exact mechanism of normo/hyperphosphatemia in some cases with CYP27B1 mutation is yet unknown, it is important to underline that normo/hyperphosphatemia can be seen rarely in genetically proven VDRRR-1A patients [5, 12, 13, 25, 26, 29,30,31].
Lastly, in VDRR-IA, low 1,25(OH)2D3 levels due to deficient 1α-hydroxylase enzyme activity is the hallmark of the disease. However, several cases reported in the literature having normal 1,25(OH)2D3 levels [6, 14,15,16, 25]. In our study, Pt#3.1 had mildly elevated 1,25(OH)2D3 level as well. A small increase of 1,25(OH)2D3 level has been observed in CYP27B1 knockout mice after given high dietary vitamin D and, normalization of 1,25(OH)2D3 level after vitamin D replacement in a case with VDDR-IA has been also reported [33]. However, we could not detect any correlation between prior cholecalciferol intake (r: 0.487; p: 0.40) or 25(OH)D3 levels (r: − 0.154; p:0.83) and 1,25(OH)2D3 levels at admission in our cohort. Furthermore, it seems that variation in 1,25(OH)2D3 levels could not be explained with genotype, since, in our cohort, the patients with the same mutation (p.F443Pfs*24) having low to high 1,25(OH)2D3 levels independently from serum 25(OH)D3. levels. These findings suggest that 25(OH)D3 could be converted to 1,25(OH)2D3 by a non-CYP27B1 enzyme [34] or other enzymatic pathways involving in vitamin D metabolism or degradation might have a role in 1,25(OH)2D3 levels. Recently, novel CYP11A1-derived hydroxy derivatives of D3 have been described and, 20(OH)D3 is the main metabolite of this reaction, and acts as biased agonists on VDR [35, 36]. It is also possible that 20(OH)D3 might cross reacts with 1,25(OH)2D3 in the assays. It should also be considered that reference ranges for 1,25(OH)2D3 levels are determined in normal subjects with normal PTH and phosphate levels. In situations with elevated PTH and low phosphate levels, due to their stimulatory effect on 1α-hydroxylase, 1,25(OH)2D3 levels should be much higher. Thus, normal or mildly elevated 1,25(OH)2D3 levels in VDDR-IA can be considered as inappropriately normal in that setting. Thus, clinical and biochemical findings of cholecalciferol treatment-resistant calciopenic rickets with normal or increased 25(OH)D3 levels, together with good treatment response to the standard-dose calcitriol treatment, will exclude NR, VDDR-IB,-II, and-III and make the most probable diagnosis of VDRR-IA, even without measurement of 1,25(OH)2D3 levels. We made the clinical diagnosis of VDDR-IA in all of our patients and Pt#3.1 as well with these principles and eventually confirmed with genetic testing. Genetic analysis is required in cases with unusual features to identify the exact type of rickets and/or hypocalcemia as in our case with hyperphosphatemia and elevated 1,25(OH)2D3 level.
In conclusion, early and correct diagnosis of rare genetic forms of rickets is challenging, especially in developing countries where NR is the most common form of the disease. VDDR-IA should be considered when a patient develops signs of rickets while under vitamin D prophylaxis and has a normal 25(OH)D3 level or is unresponsive to cholecalciferol treatment for NR. Genetic analysis is important for making an accurate diagnosis since there are rare cases with VDDR-IA who have normo/hyperphosphatemia or normal 1,25(OH)2 D3 levels. Proper and timely treatment is important for better long-term outcomes as we have demonstrated poor final height and osteopenia in noncompliant patients.
Finally, our cohort together with the reviewed cases in the literature suggests a genotype–phenotype correlation in patients with VDDR-IA notably that intron 1 c.195 + 2T > G mutation causes more severe phenotype with lower height SDS at presentation and, higher calcitriol requirement, while less severe phenotype occurs in patients with p.K192E mutation.
References
Chiellini G, DeLuca F (2011) The importance of stereochemistry on the actions of vitamin D. Curr Top Med Chem 11:840–859
Acar S, Demir K, Shi Y (2017) Genetic causes of rickets. J Clin Res Pediatr Endocrinol 9:88–105. https://doi.org/10.4274/jcrpe.2017.S008
Roizen JD, Li D, O’Lear L et al (2018) CYP3A4 mutation causes vitamin D-dependent rickets type 3. J Clin Invest 128:1913–1918
Fraser D, Kooh SW, Kind HP et al (1973) Pathogenesis of hereditary vitamin-D-dependent rickets: an inborn error of vitamin D metabolism involving defective conversion of 25-hydroxyvitamin D to 1α, 25-dihydroxyvitamin D. N Engl J Med 289:817–822
Edouard T, Alos N, Chabot G et al (2011) Short- and long-term outcome of patients with pseudo-vitamin D deficiency rickets treated with calcitriol. J Clin Endocrinol Metab 96:82–89. https://doi.org/10.1210/jc.2010-1340
Giannakopoulos A, Efthymiadou A, Chrysis D (2017) A case of vitamin-D-dependent rickets type 1A with normal 1,25-dihydroxyvitamin D caused by two novel mutations of the CYP27B1 gene. Horm Res Paediatr 87:58–63. https://doi.org/10.1159/000446774
Tahir S, Demirbilek H, Ozbek MN et al (2016) Genotype and phenotype characteristics in 22 patients with vitamin D-dependent rickets type I. Horm Res Paediatr 85:309–317. https://doi.org/10.1159/000444483
Demir K, Kattan WE, Zou M et al (2015) Novel CYP27B1 gene mutations in patients with vitamin D-dependent rickets type 1A. PLoS ONE 10:1–14. https://doi.org/10.1371/journal.pone.0131376
Baykan O, Yaman A, Arpa M et al (2014) The comparison of serum vitamin D3 measurement with HPLC, HPLC coupled tandem mass spectrometry using atmospheric pressure chemical ionization, and immunoassay methods. Clin Chem Lab Med 52:3. https://doi.org/10.1515/cclm-2014-4042
Untergasser A, Cutcutache I, Koressaar T et al (2012) Primer3—new capabilities and interfaces. Nucleic Acids Res 40:e115–e115
Stenson PD, Ball EV, Mort M et al (2003) Human gene mutation database (HGMD®): 2003 update. Hum Mutat 21:577–581
Wang JT, Lin CJ, Burridge SM et al (1998) Genetics of vitamin D 1α-hydroxylase deficiency in 17 families. Am J Hum Genet 63:1694–1702. https://doi.org/10.1086/302156
Chan JK, Kaplan LE, Perwad F et al (2007) Vitamin D 1α-hydroxylase gene mutations in patients with 1α-hydroxylase deficiency. J Clin Endocrinol Metab 92:3177–3182. https://doi.org/10.1210/jc.2006-2664
Ito N, Peña AS, Perano S et al (2014) First Australian report of vitamin D-dependent rickets type I. Med J Aust 201:420–421. https://doi.org/10.5694/mja13.00220
Durmaz E, Zou M, Al-Rijjal RA et al (2012) Clinical and genetic analysis of patients with vitamin D-dependent rickets type 1A. Clin Endocrinol (Oxf) 77:363–369. https://doi.org/10.1111/j.1365-2265.2012.04394.x
Fatma D, Gamze O, Heves K et al (2019) Genetic and clinical characteristics of patients with vitamin D dependent rickets type 1A. J Clin Res Pediatr Endocrinol 11:34–40. https://doi.org/10.4274/jcrpe.galenos.2018.2018.0121
Chi Y, Sun J, Pang L et al (2018) Mutation update and long-term outcome after treatment with active vitamin D3 in Chinese patients with pseudovitamin D-deficiency rickets (PDDR). Osteoporos Int 30:481–489. https://doi.org/10.1007/s00198-018-4607-5
Cui N, Xia W, Su H et al (2012) Novel mutations of CYP27B1 gene lead to reduced activity of 1α-hydroxylase in Chinese patients. Bone 51:563–569. https://doi.org/10.1016/j.bone.2012.05.006
Smith SJ, Rucka AK, Berry JL et al (1999) Three families with pseudovitamin D-deficiency rickets resulting in loss of functional enzyme activity. J bone Miner Res 14:730–739
Yoshida T, Monkawa T, Tenenhouse HS et al (1998) Two novel 1α-hydroxylase mutations in French-Canadians with vitamin D dependency rickets type I. Kidney Int 54:1437–1443. https://doi.org/10.1046/j.1523-1755.1998.00133.x
Beck-Nielsen SS, Hertel NT, Brock-Jacobsen B (2006) Vitamin D 1 alpha-hydroxylase deficiency as the cause of severe rickets in a 1-year-old-old boy. Ugeskr Laeger 168:700–702
Porcu L, Meloni A, Casula L et al (2002) A novel splicing defect (IVS6+ 1G> T) in a patient with pseudovitamin D deficiency rickets. J Endocrinol Invest 25:557–560
Orbak Z (2017) A novel mutation of CYP27B1 in two siblings with vitamin D-dependent rickets type 1A. In: 8th International Conference on Children
Kitanaka S, Murayama A, MToshioyuki S et al (1999) No enzyme activity of 25-hydroxyvitamin D3 1-a hydroxylase gene product in pseudovitamin D deficiency rickets, including that with mild clinical manifestation. J Clin Endocrinol Metab 84:4111–4117
Wang X, Zhang MYH, Miller WL, Portale AA (2002) Novel gene mutations in patients with 1α-hydroxylase deficiency that confer partial enzyme activity in vitro. J Clin Endocrinol Metab 87:2424–2430. https://doi.org/10.1210/jc.87.6.2424
Alzahrani AS, Zou M, Baitei EY et al (2010) Alzahrani2010 a novel G102E mutation of CYP27B1 in a large family with vitamin D-dependent rickets type 1.pdf. J Clin Endocrinol Metab 95:4176–4183
Füchtbauer L, Brusgaard K, Ledaal P et al (2015) Case report: vitamin D-dependent rickets type 1 caused by a novel CYP27B1 mutation. Clin Case Reports 3:1012–1016. https://doi.org/10.1002/ccr3.406
Haffner D, Emma F, Eastwood DM et al (2019) Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia. Nat Rev Nephrol 15:435–455. https://doi.org/10.1038/s41581-019-0152-5
Hu WW, Ke YH, He JW et al (2014) A novel compound mutation of CYP27B1 in a Chinese family with vitamin D-dependent rickets type 1A. J Pediatr Endocrinol Metab 27:335–341. https://doi.org/10.1515/jpem-2013-0183
Kim CJ (2011) Vitamin D dependent rickets type I. Korean J Pediatr 54:51–54. https://doi.org/10.3345/kjp.2011.54.2.51
Kitanaka S, Takeyama K, Murayama A et al (1998) Inactivating mutations in the 25-hydroxyvitamin D3 1α-hydroxylase gene in patients with pseudovitamin D-deficiency rickets. N Engl J Med 338:653–662
Larner AJ (1995) Pseudohyperphosphatemia. Clin Biochem 28:391–393
Koek WNH, Zillikens MC, van der Eerden BCJ, van Leeuwen JPTM (2016) Novel compound heterozygous mutations in the CYP27B1 gene lead to pseudovitamin D-deficient rickets. Calcif Tissue Int 99:326–331. https://doi.org/10.1007/s00223-016-0165-z
Nishikawa M, Yasuda K, Takamatsu M et al (2019) Generation of 1, 25-dihydroxyvitamin D3 in Cyp27b1 knockout mice by treatment with 25-hydroxyvitamin D3 rescued their rachitic phenotypes. J Steroid Biochem Mol Biol 185:71–79
Slominski AT, Manna PR, Tuckey RC (2015) On the role of skin in the regulation of local and systemic steroidogenic activities. Steroids. https://doi.org/10.1016/j.steroids.2015.04.006.On
Wang J, Slominski A, Tuckey RC, Janjetovic Z (2012) 20-Hydroxyvitamin D3 inhibits proliferation of cancer cells with high efficacy while being non-toxic. Anticancer Res 32:739–746
Acknowledgements
We are deeply grateful to the patients and their families without whom this study could not be performed.
Funding
None.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
Sare Betul Kaygusuz, Ceren Alavanda, Tarik Kirkgoz, Mehmet Eltan, Zehra Yavas Abali, Didem Helvacioglu, Tulay Guran, Pinar Ata, Abdullah Bereket, and Serap Turan declare that they have no conflicts of interest.
Ethical Approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the Marmara University School of Medicine Ethical Committee and with the 1964 Helsinki declaration and its later amendments.
Consent to Participate
Informed consent was obtained from parents of all patients for genetic testing.
Consent for Publication
Informed consent was obtained from parents of all patients for publication of data.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kaygusuz, S.B., Alavanda, C., Kirkgoz, T. et al. Does Genotype–Phenotype Correlation Exist in Vitamin D-Dependent Rickets Type IA: Report of 13 New Cases and Review of the Literature. Calcif Tissue Int 108, 576–586 (2021). https://doi.org/10.1007/s00223-020-00784-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-020-00784-2