
Abstract
Purpose
Bone may regulate glucose homeostasis via uncarboxylated bioactive osteocalcin (ucOCN). This study explored whether changes in ucOCN and bone remodeling are associated with change in glucose homeostasis after biliopancreatic diversion (BPD).
Methods
In this secondary exploratory analysis of a 1-year prospective observational study, 16 participants (11 men/5 women; 69% with type 2 diabetes; mean BMI 49.4 kg/m2) were assessed before, 3 days, 3 months and 12 months after BPD. Changes in plasma ucOCN and bone markers (C-terminal telopeptide (CTX), total osteocalcin (OCN)) were correlated with changes in insulin resistance or sensitivity indices (HOMA-IR; adipose tissue insulin resistance index (ADIPO-IR) and insulin sensitivity index (SI) from the hyperinsulinemic-euglycemic clamp), insulin secretion rate (ISR) from the hyperglycemic clamp, and disposition index (DI: SI × ISR) using Spearman correlations before and after adjustment for weight loss.
Results
ucOCN was unchanged at 3 days but increased dramatically at 3 months (+257%) and 12 months (+498%). Change in ucOCN correlated significantly with change in CTX at 3 months (r = 0.62, p = 0.015) and 12 months (r = 0.64, p = 0.025) before adjustment for weight loss. It also correlated significantly with change in fasting insulin (r = −0.53, p = 0.035), HOMA-IR (r = −0.54, p = 0.033) and SI (r = 0.52, p = 0.041) at 3 days, and ADIPO-IR (r = −0.69, p = 0.003) and HbA1c (r = −0.69, p = 0.005) at 3 months. Change in OCN did not correlate with any glucose homeostasis indices. Results were similar after adjustment for weight loss.
Conclusion
The increase in ucOCN may be associated with the improvement in insulin resistance after BPD, independently of weight loss. These findings need to be confirmed in larger, less heterogeneous populations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Biliopancreatic diversion with duodenal switch (BPD) is the most effective bariatric procedure to induce substantial long-term weight loss and type 2 diabetes remission [1,2,3]. It is reported that between 96 and 100% of individuals undergoing BPD will be free of diabetes at 12 months [3]. Interestingly, reversion of diabetes has been observed as early as 1 week after BPD [4]. This could be explained by enhanced hepatic and adipose tissue insulin sensitivity as well as improved insulin secretion rate (ISR) and perhaps β-cell glucose sensitivity and function in the first few days after BPD [4,5,6,7]. Increased muscle insulin sensitivity is observed later, at 3 months postoperatively [6]. Thus, in the absence of clear mechanisms, several hypotheses have been put forward to explain the early improvement in glucose homeostasis after BPD that is observed before significant weight loss occurs [7]. These include profound caloric restriction [8], increased incretin response, change in bile acid composition and flow, alteration of gut microbiota and intestinal glucose metabolism as well as increased brown adipose tissue metabolic activity [9].
Another possible mechanism that has not been yet explored is the contribution of the bone-derived hormone uncarboxylated osteocalcin (ucOCN). Animal studies have revealed that bone is an endocrine organ able to regulate insulin secretion and insulin sensitivity via ucOCN, the bioactive form of osteocalcin (OCN) [10,11,12]. Indeed, OCN knockout mice display reduced β-cell mass and insulin secretion, and decreased insulin sensitivity [12, 13]. OCN is secreted in its intact carboxylated form by the bone-forming cells (osteoblasts). The process of decarboxylation of OCN into ucOCN requires the acidity of the bone milieu, which is provided by the activity of the bone-resorbing cells (osteoclasts) [11, 14]. In humans, an inverse association between gamma-carboxylated OCN/total OCN ratio (a reflection of the metabolically inactive form of OCN) and several indices of insulin resistance has been corroborated in a cross-sectional study [15]. There is however very limited evidence associating ucOCN and glucose homeostasis in humans due to the unavailability of robust measurement methods for ucOCN [15,16,17,18,19].
In light of preclinical studies and cross-sectional human studies suggesting that bone may alter glucose homeostasis via ucOCN [10,11,12, 15], and of our previous findings demonstrating that bone turnover (mainly bone resorption) increases rapidly and dramatically after BPD even prior to significant weight loss [20], coupled with the availability of a new robust assay to measure directly ucOCN in humans [21], we aimed to explore whether the increase in bone resorption and ucOCN contributes to the improvement in glucose homeostasis observed after BPD. We hypothesized that (1) ucOCN increases after BPD; (2) the increase in ucOCN correlates with the increase in the bone resorption marker C-terminal telopeptide (CTX); (3) the increase in ucOCN and CTX correlate with improved indices of insulin sensitivity, insulin secretion and β-cell function after BPD.
Materials and methods
Study design and population
This is an ancillary study from a 1-year prospective observational study that aimed at evaluating fatty acid metabolism during type 2 diabetes remission after BPD [6]. The study population included 16 men and women aged 18–65 years with severe obesity (BMI ≥ 35 kg/m2), and with or without type 2 diabetes (according to the American Diabetes Association’s criteria) [22]. All participants underwent BPD at the Institut universitaire de cardiologie et de pneumologie de Québec (IUCPQ) between 2012 and 2014. Details on the BPD procedure have been published previously [23]. The 1-year frozen sample of one participant was missing. Moreover, two individuals did not complete the 1-year follow-up due to diagnosis of low-grade breast cancer after the 3-month visit (n = 1) or pregnancy during follow-up (n = 1), leaving 13 participants who completed the study.
Main exclusion criteria were: pharmacological treatment with fibrate, insulin, thiazolidinedione, beta-blocker, corticosteroid, oral contraceptive pill, hormone replacement therapy, treatment for osteoporosis, or any other medication known to affect bone or lipid metabolism, uncontrolled thyroid disease, decompensated or severe renal or hepatic disease other than non-alcoholic fatty liver disease, or other serious medical condition or surgical complication following BPD. None of the participants received warfarin. Before surgery, participants were prescribed vitamin D3 to ensure normal preoperative 25-hydroxyvitamin D (25OHD) levels. After BPD, a standard prescription was given to all participants, including daily multivitamin (Centrum Forte®), calcium carbonate 1000 mg, vitamin D2 50,000 IU, vitamin A and iron. Doses were adjusted based on blood results at 3 months and 12 months. As reported previously, 25OHD remained unchanged at 3 days and 3 months after surgery, but then increased significantly at 12 months after surgery (+61%, p < 0.0001). Mean (SD) 25OHD levels were 59.6 (8.3) at baseline, 51.5 (8.7) at 3 days, 67.8 (20.0) at 3 months and 94.4 (33.8) at 12 months after surgery. Besides, parathyroid hormone was within the normal range for all participants at baseline. It increased significantly at 3 days after surgery (+87%, p < 0.0001) but then remained within normal limits for all participants at 3 and 12 months after surgery [20]. The IUCPQ ethical review board approved the protocol. Consent form was signed by all participants before entering the study.
Outcome measurements
Outcome measurements were assessed before surgery (45 ± 8 days before surgery) and at 3 days, 3 months and 12 months after BPD.
Uncarboxylated osteocalcin and bone turnover markers
At each timepoint, 12-h fasting early morning frozen EDTA plasma was analyzed in a single batch to determine levels of CTX (intra-assay CV 1.8–4.5% and inter-assay CV 2.5–6.5%) and 1–43/49 (N-terminal and intact) OCN (intra-assay CV 0.7–2.4% and inter-assay CV 2.0–4.1%) with an automated electrochemiluminescence assay (Elecsys, Roche Diagnostics). ucOCN was assessed with a recently developed Sandwich Enzyme-Linked Immunosorbent Assay recognizing human ucOCN, but not carboxylated OCN (intra-assay CV 3.1–4.1% and inter-assay CV 7.1–8.1%) [21].
Glucose homeostasis markers
Fasting concentrations of glucose (hexokinase method, Roche Glucose Analyzer) and insulin (Luminex LX200 multiplex immunoassays, Millipore, Billerica, MA) were assessed. Details of the methods to assess glucose homeostasis markers have been described in the original paper and summarized here [6]. HOMA-IR, a marker of hepatic insulin resistance, was calculated by multiplying fasting plasma insulin (U/L) by fasting plasma glucose (mmol/L)/22.5 [24]. Each participant completed a 6-h stable isotopic tracer infusion study in which a euglycemic-hyperinsulinemic clamp was maintained for 4 h. The first 2 h was an equilibration period during which subjects received a continuous perfusion of glycerol and palmitate, as previously described [6]. High-dose insulin perfusion (1.2 mUI/kg/min) started 120 min later, and patients received variable glucose infusion to achieve and maintain euglycemia (5 mmol/L) over 4 h. Peripheral insulin sensitivity index (SI), which represents muscle sensitivity to insulin, was determined during the 4-h euglycemic-hyperinsulinemic clamp. Adipose tissue insulin resistance index (ADIPO-IR), which represents adipose tissue resistance to insulin [25], was calculated by multiplying whole-body rate of appearance of glycerol by fasting insulin levels, during the euglycemic-hyperinsulinemic clamp with and without intravenous fat loading [6, 26]. ISR was determined during a hyperglycemic clamp where patients received graded intravenous glucose infusion, reaching and sustaining for 2 h a peak plasma glucose of 10 mmol/L [6, 27]. Insulin disposition index (DI), an indicator of pancreatic β-cell function [28, 29], was assessed by multiplying SI by ISR. Bodyweight was measured using a bioimpedance scale (InBody520, body composition analyzer, Biospace, Los Angeles, California) calibrated for subjects with severe obesity at baseline, 3 months and 12 months. Measurements were obtained in light clothing to the nearest 0.1 kg. Height was measured with a wall-mounted stadiometer to the nearest 0.1 cm. BMI was calculated and reported in kg/m2.
Statistical analyses
Statistical analyses were performed using JMP Pro version 12.1.0 and SAS version 9.4 (SAS Institute Inc, Cary, NC, USA). A general linear mixed model with a variance component covariance structure for repeated measures was used to compare concentrations as well as their changes from baseline in ucOCN, bone remodeling markers and glucose homeostasis indices at each timepoint. The chosen matrix was based on minimal Akaike information criterion. The subject-specific intercept was considered a random effect, and time was considered a fixed effect. When necessary, log transformation of the variables was performed to ensure normality of distribution. The Tukey–Kramer adjustment was used for multiple comparison tests, comparing all timepoints versus all other timepoints. Due to the small sample size, spearman correlations were performed to determine the associations between changes in ucOCN, bone remodeling markers and glucose homeostasis indices, with or without adjustment for weight loss at the specified time of measurement. For all statistical analyses, changes were similar between patients with or without a diagnosis of type 2 diabetes at baseline (data not shown). Data are presented as mean ± SD, unless stated otherwise. A two-tailed p value < 0.05 was considered statistically significant for all analyses.
Results
Study population
Study population included eleven men and five women with a mean age of 41.6 ± 8.8 years and a mean BMI of 49.4 ± 5.6 kg/m2. At baseline, 40% of the women (n = 2) were postmenopausal. Besides, 11 individuals were diagnosed with type 2 diabetes and treated with diet, metformin or a sulfonylurea, as monotherapy or in combination, and 5 did not have diabetes but were insulin-resistant based on HOMA-IR.
Changes in total bodyweight, ucOCN and bone remodeling markers after BPD
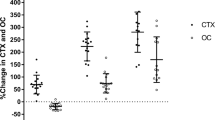
Details of the changes in total bodyweight, CTX and OCN (not ucOCN) have been published previously [20]. Briefly, total bodyweight was not measured at 3 days after BPD but was presumably stable. However, it decreased by 26 ± 7 kg (−19%, p < 0.0001) at 3 months and 59 ± 12 kg (−43%, p < 0.0001) at 12 months. Change in CTX was inversely associated with weight loss at 3 months (r = −0.63, p = 0.009) and 12 months (r = −0.58, p = 0.039). Changes in ucOCN and OCN did not correlate with weight loss. Descriptive analyses of ucOCN and the bone remodeling markers are detailed in Table 1. As shown in Fig. 1, while the individual change in ucOCN at 3 days after BPD was variable (8% reduction on average, p = 0.986), the increase in ucOCN was universal and impressive at 3 months (+257%, p < 0.0001) and 12 months (+498%, p < 0.0001). Similarly, OCN first declined (−19%, p = 0.012) 3 days after BPD, and then rose to a lesser extent than ucOCN at 3 months (+69%, p < 0.0001) and 12 months (+164%, p < 0.0001). CTX increased significantly at 3 days (+66%, p < 0.0001) after BPD, and continued to rise at 3 months (+219%, p < 0.0001) and 12 months (+295%, p < 0.0001) after surgery.

Change in ucOCN at 3 days, 3 months and 12 months after BPD. ucOCN uncarboxylated osteocalcin
Changes in glucose homeostasis indices after BPD
Descriptive analyses of the glucose homeostasis indices are shown in Table 2. Fasting plasma glucose decreased 3 days after BPD (−15%, p < 0.0001) and continued to decrease at 3 months (−25%, p < 0.0001) and 12 months (−32%, p < 0.0001). In parallel, insulin concentrations fell as early as 3 days after BPD (−53%, p < 0.0001), and continued to decline throughout follow-up (−71% at 3 months; −84% at 12 months, all p < 0.0001). Both HOMA-IR (−61% at 3 days; −81% at 3 months; −92% at 12 months; all p < 0.0001) and ADIPO-IR (−80% at 3 days; −82% at 3 months; −83% at 12 months; all p < 0.0001) declined at every timepoint after BPD. SI and DI were unchanged at 3 days but increased at 3 months (SI + 194%; DI + 213%; all p < 0.0001) and remained elevated 12 months after surgery (SI + 242%; DI + 120%; all p < 0.0001). While ISR did not change significantly at either 3 days or 3 months after BPD, it decreased significantly at 12 months (−45%, p = 0.003).
Correlations between changes in ucOCN, bone remodeling markers and glucose homeostasis indices after BPD
Significant correlations between changes in ucOCN, bone turnover markers and glucose homeostasis indices before and after adjustment for weight loss are shown in Table 3. Change in ucOCN was positively associated with change in CTX at 3 months and 12 months (Fig. 2). Moreover, change in ucOCN correlated negatively with change in fasting insulin and HOMA-IR, and positively with change in SI at 3 days after BPD (Fig. 3). At 3 months, change in ucOCN was negatively associated with ADIPO-IR and HbA1c (Fig. 3). Furthermore, change in CTX correlated negatively with change in ADIPO-IR at 3 months and at 12 months, and positively with change in DI at 3 months. None of the glucose homeostasis indices correlated significantly with OCN. Adjustment for weight loss at the specified time of measurement did not affect the results.

Non-adjusted Spearman correlations between change in ucOCN and change in CTX at 3 months (a) and 12 months (b) after BPD. ucOCN uncarboxylated osteocalcin; CTX C-terminal telopeptide

Non-adjusted significant Spearman correlations between change in ucOCN and a change in fasting insulin, b change in HOMA-IR and c change in SI, at 3 days after BPD, and d change in HbA1c and e change in ADIPO-IR at 3 months after BPD. ucOCN uncarboxylated osteocalcin; HOMA-IR Homeostasis Model Assessment of insulin resistance; SI Insulin sensitivity index; HbA1c glycated hemoglobin; ADIPO-IR adipose tissue insulin resistance index
Discussion
In the present study, we explored for the first time the association between changes in ucOCN, bone remodeling markers and glucose homeostasis indices following bariatric surgery. We found that even though mean ucOCN levels did not change significantly 3 days after BPD, individual changes were variable and were associated with changes in hepatic and muscle insulin sensitivity. However, ucOCN increased in all participants at 3 months and continued to rise at 12 months. Furthermore, we observed significant associations between the change in ucOCN and improvement in hepatic and muscle insulin sensitivity indices at 3 days, as well as with adipose tissue insulin sensitivity and HbA1c at 3 months after BPD, independent of weight loss. These findings are consistent with previous studies in mice and humans [12, 15, 30, 31], and support a potential role of ucOCN in the regulation of whole-body insulin sensitivity after BPD. In line with our hypothesis, ucOCN correlated positively with CTX, suggesting that bone resorption is required to decarboxylate OCN [14, 30]. Besides, the increase in CTX correlated with the improvement in adipose tissue insulin resistance and pancreatic β-cell function. Altogether, these findings suggest a potential implication of ucOCN in the improvement of glucose homeostasis after BPD, that may be driven by bone resorption.
We could find four cross-sectional studies [16, 17, 32, 33] and one prospective study [31] that examined associations between ucOCN levels and glucose homeostasis in humans. However, the assay used to measure ucOCN in two of these studies [16, 31] tends to overestimate ucOCN levels [15] while the assays in the other studies have not been validated. Nevertheless, the first study showed that lower ucOCN levels were associated with increased fasting insulin and HOMA-IR in 98 obese patients [16] while the second study found a positive correlation between ucOCN levels and insulin secretion indices in 50 Japanese patients with type 2 diabetes [17]. Conversely, the other cross-sectional studies found no correlations between ucOCN levels and insulin resistance indices in 129 patients with type 2 diabetes assessed with a euglycemic-hyperinsulinemic clamp [32], as well as in women with gestational diabetes [33]. Finally, low baseline concentrations of ucOCN were significantly associated with an increased risk of developing type 2 diabetes at 5 years [31]. Furthermore, in line with our results, Ferron et al. found that the metabolically inactive gamma-carboxylated form of OCN/intact OCN ratio correlated negatively with insulin sensitivity assessed by the euglycemic-hyperinsulinemic clamp or SI assessed by the oral glucose tolerance test [15]. Despite some inconsistencies among studies, which could be attributed to different assay methods and study populations, most analyses support that increased ucOCN, or reduced gamma-carboxylated OCN, is associated with greater insulin sensitivity, insulin secretion and possibly lower type 2 diabetes risk.
To our knowledge, no study addressed specifically whether changes in ucOCN are associated with glucose homeostasis markers after bariatric surgery or medical weight loss. Our findings support the existence of a bone-pancreas-adipose tissue endocrine loop regulating whole-body glucose homeostasis in humans, as previously described in animals. Indeed, Ferron et al. demonstrated that insulin signaling in osteoblasts decreases the expression of osteoprotegerin [34]. This reduction in osteoprotegerin activates bone resorption, which creates a highly acid environment in the bone milieu that facilitates OCN decarboxylation and the release of ucOCN. Activation of bone resorption is thus a necessary step to increase bioactive ucOCN, which then stimulates insulin secretion in pancreatic β-cells and enhances adiponectin expression in adipose tissue, a well-known insulin-sensitizing hormone in muscle and liver [35,36,37]. Indeed, we found a significant correlation between changes in ucOCN and changes in high molecular weight adiponectin at 3 days after BPD (r = 0.57, p = 0.021), but correlations were not significant at 3 months and 12 months. Our hypothesis is also supported by the earlier increase in CTX, a marker of bone resorption, and by the correlation we found between CTX and ucOCN at 3 and 12 months, as well as with several indices of insulin sensitivity. It is not possible to determine from our study the sequence of events leading to increased unOCN and insulin sensitivity. Indeed, the increase in bone resorption may activate decarboxylation of available OCN, increase unOCN and then improve insulin sensitivity. Concomitantly, osteoblasts may secrete more OCN as they become more sensitive to insulin after BPD, similar to what is observed in other insulin-sensitive organs [38,39,40,41]. Hence, it has been shown in animal studies that bone is a site of insulin resistance [42]. Thus, one of the possible mechanisms responsible for the improvement in whole-body insulin sensitivity after BPD may be the increase in ucOCN levels resulting from the activation of bone resorption and the increased bone sensitivity to insulin. Therefore, while the increase in bone resorption resulting from BPD might be detrimental to bone health [43, 44], it may be favorable at the metabolic level by enhancing systemic insulin sensitivity through decarboxylation of OCN.
Regarding the associations that we found between the change in CTX and glucose homeostasis markers, we could find only one study reporting a correlation between the increase in CTX and the reduction in HbA1c 1 year after Roux-in-Y gastric bypass (r = −0.74, p = 0.023) [45]. Moreover, CTX levels were found to correlate positively with insulin sensitivity indices derived from a euglycemic-hyperinsulinemic clamp in 14 overweight and obese individuals, with or without type 2 diabetes [46]. Those results are in line with a few studies suggesting that bone may influence insulin sensitivity, independently of OCN [47, 48]. Moreover, mice studies have shown that bone resorption is modulated by insulin sensitivity in the bone cells, and insulin secretion by the pancreas [34, 42]. Therefore, the findings that changes in CTX correlate with improvements in adipose tissue insulin resistance and DI may reflect those associations. Nonetheless, it also supports our general hypothesis that the increase in bone resorption enhances the bioavailability of ucOCN, which then improves whole-body insulin sensitivity. Besides, it is worth mentioning that we did not find any association between the change in OCN and glucose homeostasis markers after BPD, further supporting that ucOCN is the metabolically-active form of OCN and the importance of accurately measuring it. Our results are in contrast with another study in which higher total OCN levels were significantly associated with improved glucose tolerance, higher SI and lower HOMA-IR in overweight and obese postmenopausal women during an oral glucose tolerance test [15]. The lack of correlation with OCN in our study may be explained by the fact that the assay we used to measure OCN does not detect ucOCN, or by a lack of statistical power compared with the latter study (n = 16 vs. n = 132) [15].
This study explored prospectively for the first time the associations between short- and medium-term changes in ucOCN, bone remodeling markers and glucose homeostasis indices after BPD, measured precisely using gold standard methods. There are limitations that need to be highlighted including the small and heterogeneous study population comprising men as well as pre- and postmenopausal women with and without T2D. Indeed, bone turnover and glucose homeostasis are affected by both diabetes and prediabetes [49] as well as menopausal status [50]. Moreover, the observational nature of our study does not enable us to establish a causal link between ucOCN, bone remodeling markers and glucose homeostasis after BPD.
In conclusion, the increase in ucOCN may be associated with the improvement in insulin sensitivity and glucose homeostasis observed after BPD, independently of weight loss. We hypothesized that this relationship may be driven by bone resorption. Our study lends further support to the existence of endocrine communication between bone and organs involved in glucose metabolism in humans. It also provides additional novel hypotheses that could explain the improvement in glucose homeostasis after bariatric surgery. Our exploratory findings however need to be confirmed in a larger and less heterogenous study population. Moreover, interventional studies should investigate specifically the involvement of ucOCN and bone remodeling in the regulation of glucose homeostasis in the context of bariatric surgery, weight loss, obesity, and diabetes.
Data availability
The datasets generated during and/or analyzed during the current study are not publicly available but are available from the corresponding author on reasonable request.
References
G. Mingrone, S. Panunzi, A. De Gaetano, C. Guidone, A. Iaconelli, L. Leccesi, G. Nanni, A. Pomp, M. Castagneto, G. Ghirlanda, F. Rubino, Bariatric surgery versus conventional medical therapy for type 2 diabetes. N. Engl. J. Med. 366(17), 1577–1585 (2012). https://doi.org/10.1056/NEJMoa1200111
H. Buchwald, R. Estok, K. Fahrbach, D. Banel, M.D. Jensen, W.J. Pories, J.P. Bantle, I. Sledge, Weight and type 2 diabetes after bariatric surgery: systematic review and meta-analysis. Am. J. Med. 122(3), 248–256.e245 (2009). https://doi.org/10.1016/j.amjmed.2008.09.041
H. Buchwald, Y. Avidor, E. Braunwald, M.D. Jensen, W. Pories, K. Fahrbach, K. Schoelles, Bariatric surgery: a systematic review and meta-analysis. JAMA 292(14), 1724–1737 (2004). https://doi.org/10.1001/jama.292.14.1724
C. Guidone, M. Manco, E. Valera-Mora, A. Iaconelli, D. Gniuli, A. Mari, G. Nanni, M. Castagneto, M. Calvani, G. Mingrone, Mechanisms of recovery from type 2 diabetes after malabsorptive bariatric surgery. Diabetes 55(7), 2025–2031 (2006). https://doi.org/10.2337/db06-0068
A. Michaud, T. Grenier-Larouche, D. Caron-Dorval, S. Marceau, L. Biertho, S. Simard, D. Richard, A. Tchernof, A.C. Carpentier, Biliopancreatic diversion with duodenal switch leads to better postprandial glucose level and beta cell function than sleeve gastrectomy in individuals with type 2 diabetes very early after surgery. Metab.: Clin. Exp. 74, 10–21 (2017). https://doi.org/10.1016/j.metabol.2017.06.005
T. Grenier-Larouche, A.M. Carreau, A. Geloen, F. Frisch, L. Biertho, S. Marceau, S. Lebel, F.S. Hould, D. Richard, A. Tchernof, A.C. Carpentier, Fatty acid metabolic remodeling during type 2 diabetes remission after bariatric surgery. Diabetes 66(11), 2743–2755 (2017). https://doi.org/10.2337/db17-0414
T. Grenier-Larouche, A.M. Carreau, A.C. Carpentier, Early metabolic improvement after bariatric surgery: the first steps toward remission of type 2 diabetes. Can. J. Diabetes 41(4), 418–425 (2017). https://doi.org/10.1016/j.jcjd.2016.10.013
C.E. Plourde, T. Grenier-Larouche, D. Caron-Dorval, S. Biron, S. Marceau, S. Lebel, L. Biertho, A. Tchernof, D. Richard, A.C. Carpentier, Biliopancreatic diversion with duodenal switch improves insulin sensitivity and secretion through caloric restriction. Obesity 22(8), 1838–1846 (2014). https://doi.org/10.1002/oby.20771
C. Koliaki, S. Liatis, C.W. le Roux, A. Kokkinos, The role of bariatric surgery to treat diabetes: current challenges and perspectives. BMC Endocr. Disord. 17(1), 50 (2017). https://doi.org/10.1186/s12902-017-0202-6
G. Karsenty, M. Ferron, The contribution of bone to whole-organism physiology. Nature 481(7381), 314–320 (2012). https://doi.org/10.1038/nature10763
P. Mera, M. Ferron, I. Mosialou, Regulation of energy metabolism by bone-derived hormones. Cold Spring Harbor Perspect. Med. (2017). https://doi.org/10.1101/cshperspect.a031666
N.K. Lee, H. Sowa, E. Hinoi, M. Ferron, J.D. Ahn, C. Confavreux, R. Dacquin, P.J. Mee, M.D. McKee, D.Y. Jung, Z. Zhang, J.K. Kim, F. Mauvais-Jarvis, P. Ducy, G. Karsenty, Endocrine regulation of energy metabolism by the skeleton. Cell 130(3), 456–469 (2007). https://doi.org/10.1016/j.cell.2007.05.047
M. Ferron, E. Hinoi, G. Karsenty, P. Ducy, Osteocalcin differentially regulates beta cell and adipocyte gene expression and affects the development of metabolic diseases in wild-type mice. Proc. Natl Acad. Sci. USA 105(13), 5266–5270 (2008). https://doi.org/10.1073/pnas.0711119105
J. Lacombe, G. Karsenty, M. Ferron, In vivo analysis of the contribution of bone resorption to the control of glucose metabolism in mice. Mol. Metab. 2(4), 498–504 (2013). https://doi.org/10.1016/j.molmet.2013.08.004
J. Bonneau, G. Ferland, A.D. Karelis, E. Doucet, M. Faraj, R. Rabasa-Lhoret, M. Ferron, Association between osteocalcin gamma-carboxylation and insulin resistance in overweight and obese postmenopausal women. J. Diabetes Complicat. 31(6), 1027–1034 (2017). https://doi.org/10.1016/j.jdiacomp.2017.01.023
U. Razny, D. Fedak, B. Kiec-Wilk, J. Goralska, A. Gruca, A. Zdzienicka, M. Kiec-Klimczak, B. Solnica, A. Hubalewska-Dydejczyk, M. Malczewska-Malec, Carboxylated and undercarboxylated osteocalcin in metabolic complications of human obesity and prediabetes. Diabetes/Metab. Res. Rev. 33(3), (2017). https://doi.org/10.1002/dmrr.2862
Y. Takashi, M. Koga, Y. Matsuzawa, J. Saito, M. Omura, T. Nishikawa, Undercarboxylated osteocalcin can predict insulin secretion ability in type 2 diabetes. J. Diabetes Investig. 8(4), 471–474 (2017). https://doi.org/10.1111/jdi.12601
Q. Guo, H. Li, L. Xu, S. Wu, H. Sun, B. Zhou, Undercarboxylated osteocalcin reverts insulin resistance induced by endoplasmic reticulum stress in human umbilical vein endothelial cells. Sci. Rep. 7(1), 46 (2017). https://doi.org/10.1038/s41598-017-00163-2
D.M. Liu, X.Z. Guo, H.J. Tong, B. Tao, L.H. Sun, H.Y. Zhao, G. Ning, J.M. Liu, Association between osteocalcin and glucose metabolism: a meta-analysis. Osteoporos. Int. 26(12), 2823–2833 (2015). https://doi.org/10.1007/s00198-015-3197-8
A.F. Turcotte, T. Grenier-Larouche, R.V. Ung, D. Simonyan, A.M. Carreau, A.C. Carpentier, F. Mac-Way, L. Michou, A. Tchernof, L. Biertho, S. Lebel, S. Marceau, C. Gagnon, Effects of biliopancreatic diversion on bone turnover markers and association with hormonal factors in patients with severe obesity. Obes. Surg. 29(3), 990–998 (2019). https://doi.org/10.1007/s11695-018-3617-x
J. Lacombe, O. Al Rifai, L. Loter, T. Moran, A.F. Turcotte, T. Grenier-Larouche, A. Tchernof, L. Biertho, A.C. Carpentier, D. Prud'homme, R. Rabasa-Lhoret, G. Karsenty, C. Gagnon, W. Jiang, M. Ferron, Measurement of bioactive osteocalcin in humans using a novel immunoassay reveals association with glucose metabolism and beta-cell function. Am. J. Physiol. Endocrinol. Metab. 318(3), E381–E391 (2020). https://doi.org/10.1152/ajpendo.00321.2019
Standards of Medical Care in Diabetes, Abridged for Primary Care Providers. Clin. Diabetes Publ. Am. Diabetes Assoc. 35(1), 5–26 (2017). https://doi.org/10.2337/cd16-0067
L. Biertho, S. Lebel, S. Marceau, F.S. Hould, F. Julien, S. Biron, Biliopancreatic diversion with duodenal switch: surgical technique and perioperative care. Surg. Clin. N. Am. 96(4), 815–826 (2016). https://doi.org/10.1016/j.suc.2016.03.012
D.R. Matthews, J.P. Hosker, A.S. Rudenski, B.A. Naylor, D.F. Treacher, R.C. Turner, Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28(7), 412–419 (1985)
E. Sondergaard, A.E. Espinosa De Ycaza, M. Morgan-Bathke, M.D. Jensen, How to measure adipose tissue insulin sensitivity. J. Clin. Endocrinol. Metab. 102(4), 1193–1199 (2017). https://doi.org/10.1210/jc.2017-00047
A.C. Carpentier, F. Frisch, D. Cyr, P. Genereux, B.W. Patterson, R. Giguere, J.P. Baillargeon, On the suppression of plasma nonesterified fatty acids by insulin during enhanced intravascular lipolysis in humans. Am. J. Physiol. Endocrinol. Metab. 289(5), E849–E856 (2005). https://doi.org/10.1152/ajpendo.00073.2005
A. Carpentier, S.D. Mittelman, B. Lamarche, R.N. Bergman, A. Giacca, G.F. Lewis, Acute enhancement of insulin secretion by FFA in humans is lost with prolonged FFA elevation. Am. J. Physiol. 276(6 Pt 1), E1055–E1066 (1999)
R.N. Bergman, M. Ader, K. Huecking, G. Van Citters, Accurate assessment of beta-cell function: the hyperbolic correction. Diabetes 51(Suppl 1), S212–S220 (2002)
S.E. Kahn, The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of Type 2 diabetes. Diabetologia 46(1), 3–19 (2003). https://doi.org/10.1007/s00125-002-1009-0
M. Ferron, J. Lacombe, Regulation of energy metabolism by the skeleton: osteocalcin and beyond. Arch. Biochem. Biophysics 561, 137–146 (2014). https://doi.org/10.1016/j.abb.2014.05.022
A. Diaz-Lopez, M. Bullo, M. Juanola-Falgarona, M.A. Martinez-Gonzalez, R. Estruch, M.I. Covas, F. Aros, J. Salas-Salvado, Reduced serum concentrations of carboxylated and undercarboxylated osteocalcin are associated with risk of developing type 2 diabetes mellitus in a high cardiovascular risk population: a nested case-control study. J. Clin. Endocrinol. Metab. 98(11), 4524–4531 (2013). https://doi.org/10.1210/jc.2013-2472
K. Mori, M. Emoto, K. Motoyama, E. Lee, S. Yamada, T. Morioka, Y. Imanishi, T. Shoji, M. Inaba, Undercarboxylated osteocalcin does not correlate with insulin resistance as assessed by euglycemic hyperinsulinemic clamp technique in patients with type 2 diabetes mellitus. Diabetol. Metab. Syndr. 4(1), 53 (2012). https://doi.org/10.1186/1758-5996-4-53
R. Saucedo, G. Rico, G. Vega, L. Basurto, L. Cordova, R. Galvan, M. Hernandez, E. Puello, A. Zarate, Osteocalcin, under-carboxylated osteocalcin and osteopontin are not associated with gestational diabetes mellitus but are inversely associated with leptin in non-diabetic women. J. Endocrinol. Investig. 38(5), 519–526 (2015). https://doi.org/10.1007/s40618-014-0220-4
M. Ferron, J. Wei, T. Yoshizawa, A. Del Fattore, R.A. DePinho, A. Teti, P. Ducy, G. Karsenty, Insulin signaling in osteoblasts integrates bone remodeling and energy metabolism. Cell 142(2), 296–308 (2010). https://doi.org/10.1016/j.cell.2010.06.003
B. Lee, J. Shao, Adiponectin and energy homeostasis. Rev. Endocr. Metab. Disord. 15(2), 149–156 (2014). https://doi.org/10.1007/s11154-013-9283-3
R. Ye, P.E. Scherer, Adiponectin, driver or passenger on the road to insulin sensitivity? Mol. Metab. 2(3), 133–141 (2013). https://doi.org/10.1016/j.molmet.2013.04.001
X. Wu, H. Motoshima, K. Mahadev, T.J. Stalker, R. Scalia, B.J. Goldstein, Involvement of AMP-activated protein kinase in glucose uptake stimulated by the globular domain of adiponectin in primary rat adipocytes. Diabetes 52(6), 1355–1363 (2003)
J.J. Cao, Effects of obesity on bone metabolism. J. Orthop. Surg. Res. 6, 30 (2011). https://doi.org/10.1186/1749-799x-6-30
B.A. Gower, K. Casazza, Divergent effects of obesity on bone health. J. Clin. Densitom. 16(4), 450–454 (2013). https://doi.org/10.1016/j.jocd.2013.08.010
A.L. Evans, M.A. Paggiosi, R. Eastell, J.S. Walsh, Bone density, microstructure and strength in obese and normal weight men and women in younger and older adulthood. J. Bone Miner. Res. 30(5), 920–928 (2015). https://doi.org/10.1002/jbmr.2407
J.S. Walsh, T. Vilaca, Obesity, type 2 diabetes and bone in adults. Calcif. Tissue Int. 100(5), 528–535 (2017). https://doi.org/10.1007/s00223-016-0229-0
J. Wei, M. Ferron, C.J. Clarke, Y.A. Hannun, H. Jiang, W.S. Blaner, G. Karsenty, Bone-specific insulin resistance disrupts whole-body glucose homeostasis via decreased osteocalcin activation. J. Clin. Investig. 124(4), 1–13 (2014). https://doi.org/10.1172/jci72323
C. Gagnon, A.L. Schafer, Bone health after bariatric surgery. JBMR 2(3), 121–133 (2018). https://doi.org/10.1002/jbm4.10048
C. Rousseau, S. Jean, P. Gamache, S. Lebel, F. Mac-Way, L. Biertho, L. Michou, C. Gagnon, Change in fracture risk and fracture pattern after bariatric surgery: nested case-control study. BMJ (Clin. Res. ed.) 354, i3794 (2016). https://doi.org/10.1136/bmj.i3794
E.W. Yu, M. Wewalka, S.A. Ding, D.C. Simonson, K. Foster, J.J. Holst, A. Vernon, A.B. Goldfine, F. Halperin, Effects of gastric bypass and gastric banding on bone remodeling in obese patients with type 2 diabetes. J. Clin. Endocrinol. Metab. 101(2), 714–722 (2016). https://doi.org/10.1210/jc.2015-3437
R. Basu, J. Peterson, R. Rizza, S. Khosla, Effects of physiological variations in circulating insulin levels on bone turnover in humans. J. Clin. Endocrinol. Metab. 96(5), 1450–1455 (2011). https://doi.org/10.1210/jc.2010-2877
M.M. Weivoda, C.K. Chew, D.G. Monroe, J.N. Farr, E.J. Atkinson, J.R. Geske, B. Eckhardt, B. Thicke, M. Ruan, A.J. Tweed, L.K. McCready, R.A. Rizza, A. Matveyenko, M. Kassem, T.L. Andersen, A. Vella, M.T. Drake, B.L. Clarke, M.J. Oursler, S. Khosla, Identification of osteoclast-osteoblast coupling factors in humans reveals links between bone and energy metabolism. Nat. Commun. 11(1), 87 (2020). https://doi.org/10.1038/s41467-019-14003-6
I. Mosialou, S. Shikhel, J.M. Liu, A. Maurizi, N. Luo, Z. He, Y. Huang, H. Zong, R.A. Friedman, J. Barasch, P. Lanzano, L. Deng, R.L. Leibel, M. Rubin, T. Nickolas, W. Chung, L.M. Zeltser, K.W. Williams, J.E. Pessin, S. Kousteni, MC4R-dependent suppression of appetite by bone-derived lipocalin 2. Nature 543(7645), 385–390 (2017). https://doi.org/10.1038/nature21697
S. Costantini, C. Conte, Bone health in diabetes and prediabetes. World J. Diabetes 10(8), 421–445 (2019). https://doi.org/10.4239/wjd.v10.i8.421
F. Gossiel, H. Altaher, D.M. Reid, C. Roux, D. Felsenberg, C.C. Gluer, R. Eastell, Bone turnover markers after the menopause: T-score approach. Bone 111, 44–48 (2018). https://doi.org/10.1016/j.bone.2018.03.016
Acknowledgements
The authors would like to thank M. David Simonyan for his help with statistical analyses.
Funding
The Canadian Institutes of Health Research (MOP 97947 & MOP 133652), Canadian Diabetes Association (NC-3-17-5232-CG) and CHU de Québec-Université Laval Foundation provided funding for this research. AMC is the recipient of Fonds de recherche du Québec-Santé (FRQ-S) and Diabetes Canada scholarships. MF holds the Canada Research Chair in Bone and Energy Metabolism. FM has a scholarship from FRQ-S and is a co-Chair of the Amgen Research Chair in Nephrology from Université Laval Foundation. CG is a clinical research scholar of the FRQ-S and the recipient of a Diabetes Canada New Investigator Award.
Author information
Authors and Affiliations
Contributions
A.F.T. and C.G. contributed to the study conception and design. Material preparation, data collection and analysis were performed by A.F.T., T.G.L., J.L., A.C.C., M.F. and C.G. The first draft of the paper was written by A.F.T. and all authors commented on previous versions of the paper. All authors read and approved the final paper.
Corresponding author
Ethics declarations
Conflict of interest
A.M.C. receives consultation honoraria from Pfizer. A.C.C. is the recipient of the Canada Research Chair in Molecular Imaging of Diabetes. A.T. and L.B. receive funding from Johnson Johnson Medical Companies and Medtronic for research studies on bariatric surgery. M.F. receives royalties from BioLegend for the development of the ucOCN ELISA assay. T.G.L. is now a full-time employee of Boehringer Ingelheim Canada. F.M. received speaker honoraria from Amgen and Sanofi, and participated in advisory committee for Otsuka. C.G. received research funding from Shire and speaker honoraria from Amgen, Eli Lilly and Janssen.
Ethical approval
This study was performed in line with the principles of the Declaration of Helsinki. Approval was granted by the Institut Universitaire de cardiologie et de pneumologie de Québec (IUCPQ) ethical review board.
Informed consent
Informed consent was obtained for all participants before entering the study.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Turcotte, AF., Grenier-Larouche, T., Lacombe, J. et al. Association between changes in bioactive osteocalcin and glucose homeostasis after biliopancreatic diversion. Endocrine 69, 526–535 (2020). https://doi.org/10.1007/s12020-020-02340-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-020-02340-2