
Abstract
Ghrelin and its synthetic analog hexarelin are specific ligands of growth hormone secretagogue (GHS) receptor. GHS have strong growth hormone-releasing effect and other neuroendocrine activities such as stimulatory effects on prolactin and adrenocorticotropic hormone secretion. Recently, several studies have reported other beneficial functions of GHS that are independent of GH. Ghrelin and hexarelin, for examples, have been shown to exert GH-independent cardiovascular activity. Hexarelin has been reported to regulate peroxisome proliferator-activated receptor gamma (PPAR-γ) in macrophages and adipocytes. PPAR-γ is an important regulator of adipogenesis, lipid metabolism, and insulin sensitization. Ghrelin also shows protective effects on beta cells against lipotoxicity through activation of phosphatidylinositol-3 kinase/protein kinase B, c-Jun N-terminal kinase (JNK) inhibition, and nuclear exclusion of forkhead box protein O1. Acylated ghrelin (AG) and unacylated ghrelin (UAG) administration reduces glucose levels and increases insulin-producing beta cell number, and insulin secretion in pancreatectomized rats and in newborn rats treated with streptozotocin, suggesting a possible role of GHS in pancreatic regeneration. Therefore, the discovery of GHS has opened many new perspectives in endocrine, metabolic, and cardiovascular research areas, suggesting the possible therapeutic application in diabetes and diabetic complications especially diabetic cardiomyopathy. Here, we review the physiological roles of ghrelin and hexarelin in the protection and regeneration of beta cells and their roles in the regulation of insulin release, glucose, and fat metabolism and present their potential therapeutic effects in the treatment of diabetes and diabetic-associated heart diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Diabetes mellitus is one of the leading causes of morbidity and mortality worldwide. Currently, about 382 million people suffer from this disease and by 2035, this will rise to 592 million [1]. Diabetes is classified as type 1 diabetes (T1D), characterized by absolute loss of insulin production, and type 2 diabetes (T2D), which is a result of both chronic insulin resistance and loss of pancreatic cell mass and function [2]. T2D is the most common form, accounting for about 95 % of diabetes [3]. The pathogenesis of T2D involves both genetic and environmental factors. Obesity is the most common cause of developing T2D as it represents about 75 % of the causes [4]. Obesity can lead to insulin resistance and impairment in energy metabolism of most tissues such as skeletal muscles, liver, adipose tissue, and pancreatic islets. However, insulin resistance does not lead to T2D unless it is accompanied by pancreatic beta cell dysfunction [5–7].
What are still unknown are the time points when beta cell dysfunction starts and the relative contribution of beta cell dysfunction in the development of T2D. Current evidence suggested that islet function was about 50 % of normal at the time of diagnosis [8]. The reduction of beta cell function most probably starts early about 10–12 years before diagnosis of diabetes [9]. The major factors for causing progressive decrease in beta cell structure and function during the course of the disease are glucotoxicity, lipotoxicity, proinflammatory cytokines, and islet cell amyloidosis [10]. The combined increased flux of free fatty acids and glucose into the beta cell has detrimental consequences on beta cells [11]. The excess free fatty acid entering beta cells inhibits proper glucose utilization in the mitochondria. Additionally, it is reported that these lipids are metabolized in a different way forming lipid intermediates that cause abnormal signaling and beta cell dysfunction. The glucotoxicity and lipotoxicity also may cause an increase in reactive oxygen species (ROS) which can damage the cell and then lead to beta cell death [12]. The role of the proinflammatory cytokines in causing beta cell apoptosis had been demonstrated [13, 14]. Macrophages may infiltrate beta cells when they start becoming damaged. This causes cytokine release from the macrophages, which may alter the ability of the beta cell to proliferate. Impaired beta cell function and mass could be reversible at early stages of the disease [15].
Insulin and its downstream signaling pathway play important roles in the homeostasis of blood levels of glucose, and the disruption of the signaling pathway plays an important role in the pathophysiology of T2D. Insulin reduces blood glucose levels through increasing glucose uptake in muscle and fat and decreasing hepatic glucose production. Skeletal muscle consumes most glucose, accounting for about 75 % of insulin-dependent glucose uptake while adipose tissue accounts for only a small fraction [16]. The adipose tissue, however, is crucial in the normal regulation of the insulin action all over the body. Adipocytes can store excess lipids in obesity but when they become saturated, lipids begin to accumulate inside other organs, and tissues making them insulin resistant. Adipocytes also can produce adipokines such as leptin and adiponectin which have been proved as insulin sensitizers due to their ability to decrease triglycerides (TG) synthesis, to stimulate beta oxidation of fatty acids, and thus to enhance insulin action in both skeletal muscle and liver [17–19].
Genetically modified animals deficient in white adipose tissue usually have severe insulin resistance in liver and muscle [20]. Transplantation of normal fat tissue into white adipose tissue deficient mice restores the insulin sensitivity [21]. Mice with a knockout of the insulin receptor in muscle have normal glucose tolerance [22], whereas those with a knockout of the insulin-sensitive GLUT4 glucose transporter in adipose tissue have impaired glucose tolerance, apparently due to insulin resistance being induced in muscle and liver [23]. Other recent Studies reported that obesity promotes inflammatory signals especially in the adipose tissues that disrupt insulin action and mediate insulin resistance [24]. Accordingly, the therapeutic targets of T2D will be preservation of beta cell structure and function, enhancing metabolic activity especially of the adipose tissues and decreasing lipid content in different tissues. In this review, we will focus on the possible roles of ghrelin, a natural ligand of the growth hormone secretagogue receptor type 1a (GHS-R1a), and hexarelin, a synthetic peptidyl GHS in the treatment of diabetes and diabetic-associated heart diseases.
Growth Hormone Secretagogues (GHS)
GHS are substances which stimulate the release of GH from the pituitary. They include the natural endogenous one, ghrelin, which is predominantly produced by A-like cells of the stomach from its precursor proghrelin [25] and synthetic peptidyl and non-peptidyl molecules. The ghrelin peptide is acylated to acylated ghrelin (AG) by the enzyme ghrelin-O-acyltransferase (GOAT) [26]. AG is present in serum at a 2.5-fold lower concentration than unacylated ghrelin (UAG) [27]. UAG has been thought for a long time to be an inactive metabolite of AG [25, 27]. However, it is now recognized that UAG may exert physiologically relevant effects through an unidentified receptor. UAG can antagonize the effect of AG [28, 29]. However, in some cases UAG acts synergistically with AG [30] or have AG-independent effects [31]. UAG seems to oppose the effect of AG at the metabolic levels such as insulin secretion and food intake but cannot counteract it at the neuroendocrine levels like GH release, prolactin or adrenocorticotrophic response [27]. Most of the effects of AG appear to be mediated through activation of GHS-R1a, as the specific receptor antagonist, [d-Lys3]-GHRP-6 completely blocks the protective effects of ghrelin against oxygen–glucose deprivation insult [32]. In contrast, Baldanzi et al. reported that in cardiomyocytes ghrelin exhibits an antiapoptotic effect through binding to a novel, unidentified receptor that is distinct from GHS-R1a [33].
The GHS-R is a G protein-coupled seven-transmembrane domain receptor and was initially identified as a receptor for small synthetic molecules GHS, such as hexarelin, L-692,429, GHRP-6, and MK-0677, all of which stimulate GH secretion from the pituitary [34, 35]. The GHS-R has two forms GHS-R1a and GHS-R1b. The GHS-R1a is the functionally active and signal transducing form of the GHS-R, whereas the GHS-R1b is devoid of high-affinity ligand-binding and signal transduction activities [36]. Expression of the GHS-R1a receptor was shown in the hypothalamus and anterior pituitary gland which is consistent with its role in regulating GH release [35, 37]. GHS-R1a receptor is also expressed in many peripheral organs. GHS-R1a receptor mRNA was shown in the stomach and intestine [38], pancreas [34], kidney [39], heart, and aorta [40], as well as in different human pituitary adenoma and various endocrine neoplasms of different organs [41–43]. These findings indicate that ghrelin and synthetic GHS have many functions other than the control of GH release. Ghrelin and synthetic GHS are potent stimulators of GH release [44–46]. However, the activity of both ghrelin and synthetic GHS is not fully specific for GH and includes stimulatory effect on both lactotroph and corticotroph secretion [46, 47]. Ghrelin enhances Appetite and increases food intake in humans [48] and animals [49] either injected centrally [50, 51] or peripherally [48, 52]. Ghrelin has been shown to have many protective effects on the cardiovascular system [53–56]. Ghrelin also has been proved to be a potent anti-inflammatory mediator both in vitro and in vivo in lymphocytes, monocytes, and dendritic cells and promoting IL-10 expression and cell migration [57].
The synthetic, peptide GHS are also known as growth hormone-releasing peptides (GHRPs). GHRP-6 was the first hexapeptide shown to release GH in vivo, especially in humans after oral administration with low bioavailability and short-lasting effect [58, 59]. Recently, heptapeptide, GHRP-1, and two other hexapeptides, hexarelin and GHRP-2, have been now synthesized [60, 61]. Hexarelin (His-d-2MeTrp-Ala-Trp-d-Phe-Lys-NH2) differs from growth hormone-releasing peptides GHRP-6 by having a methyl group in position 2 of the DTrp [62]. This minor modification was beneficial, making hexarelin more stable and longer acting in vivo [63]. Hexarelin is more potent than GHRP-6 as a GH releaser [64–66]. Hexarelin has many physiological actions independent of growth hormone such as the protection against cardiac ischemia and impairment of vascular endothelium function in the hearts of GH-deficient rats [67, 68]. GHS-R1a seems to mediate the action of hexarelin. However, some studies have reported that the CD36 receptor mediates the action of hexarelin in the heart and mouse 3T3-L1 preadipocytes [69, 70]. Our review will concern on the functions of ghrelin and hexarelin in the protection, regeneration of beta cells, regulation of insulin release, glucose metabolism, and their possible uses in treatment of diabetes and heart diseases.
Effects of ghrelin on pancreatic beta cells
Ghrelin has been shown to promote survival and inhibit apoptosis in many cell types. Ghrelin protected human endothelial cells against apoptosis caused by high glucose [71]. Treatment of 3T3-L1 preadipocytes with ghrelin inhibited adipocyte apoptosis induced by serum deprivation, induced cellular proliferation and differentiation to mature adipocytes. Ghrelin also increased the basal and insulin-stimulated glucose transport in theses adipocytes [72]. Ghrelin has been confirmed to have protective effects against different stressful factors including TNF-alpha [73] and doxorubicin [33]. Hexarelin is able to both increase cell proliferation and protect adult rat hippocampal progenitor (AHP) cells from apoptosis and necrosis induced by growth factor deprivation [74]. Interestingly, ghrelin is recently shown to protect beta cells from apoptotic effect of saturated fatty acids [75]. It is obvious in many studies that prolonged exposure of isolated islets or beta cell lines to saturated fatty acids is associated with beta cell apoptosis [76–78]. The signaling pathways that regulate pancreatic beta cell apoptosis during lipotoxicity or the oxidative stress could be inactivation of phosphatidylinositol-3 kinase/protein kinase B (PI3K/AKT), sustained c-Jun N-terminal kinase (JNK) activation, phosphorylation of forkhead box protein O1 (Foxo1) at serine and threonine sites distinct from those phosphorylated by AKT, enhancing the nuclear retention of Foxo1 [79, 80]. However, it is unclear whether inhibition of AKT or activation of JNK precedes inhibition of Foxo1 nuclear translocation or if there is a crosstalk between PI3K/AKT activation and JNK inhibition. Therefore, further studies are needed to clarify the mechanisms of these signaling molecules in the protection of beta cells from apoptosis. Ghrelin has been shown to exert the antiapoptotic effects on beta cells via G protein-coupled receptor/CAMP/PKA, activation of PI3K/AKT, and extracellular signal-regulated kinase (ERK) [81, 82] (See Fig. 1). Wang et al. has demonstrated that ghrelin inhibits the apoptosis and promotes beta cell proliferation through nuclear exclusion of Foxo1 and inhibition of endoplasmic reticulum (ER) stress pathway [83].

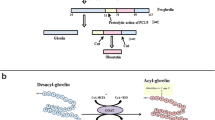
The antiapoptotic, proliferative, and survival effects of AG, UAG, and hexarelin on beta cells. AG, UAG, and hexarelin bind to GHS-R1a and/or unknown other receptor, which are involved in the activation of PI3K/AKT and MAPK (ERK1/2) pathways, JNK inhibition and nuclear extrusion of Foxo1 through activation of the GHS-R1a or other receptor. Ghrelin and hexarelin increase PDX1 mRNA, which is essential for insulin transcription and maintenance of beta cell mass. Furthermore, AG and UAG decrease NO, downregulated active caspase-9 expression, and proapoptotic protein BAX, which have been associated with beta cell dysfunction and death. All these effects contribute to reduced apoptosis, increased beta cell, and islet cell proliferation and survival (GHS-R1a GH secretagogue receptor type 1a, PI3K phosphatidylinositol 3-kinase, MAPK Mitogen-activated protein kinases (MAPK), ERK1/2 extracellular signal-regulated kinase 1/2, NO nitric oxide, PDX1 pancreatic and duodenal homeobox-1, JNK Jun-N-terminal kinase, Foxo1 Forkhead transcription factors of the FoxO family)
AG increased insulin expression and secretion and PDX1 mRNA in the pancreas of STZ-treated rat [84]. PDX1 is a transcriptional factor which is essential for insulin transcription and maintenance of beta cell mass [85]. Similarly, UAG and obstatin, which is encoded in the same gene as ghrelin, increased islet area, islet number, and beta cell mass and insulin and PDX1 mRNA compared to the pancreas of STZ-treated rat and also increased the expression of antiapoptotic gene, BCL-2 [86]. Both UAG and AG induced cell survival and protection against apoptosis in isolated human islets of Langerhans [82, 87]. The insulin-positive beta cells in these islets expressed GHS-R1a, explaining the mechanism of action for AG. However, the radiolabelled binding studies for UAG suggest that these islets also express UAG and AG binding sites that are probably not the classical GHS-R1a [82]. Therefore, the effects of UAG and AG may also occur via mechanisms independent of the GHS-R1a, likely mediated by specific AG and UAG binding sites. Taken together, these findings indicate that the binding sites of AG and UAG are different or may be some GHS-R subtypes could recognize and bind ghrelin independently of its acylation. However, the downstream signaling mechanisms in protecting beta cells may be the same for both peptides via the activation of PI3K/AKT and extracellular signal-regulated kinase (ERK) (as shown in Fig. 1).
Moreover, some studies have reported the potential roles of ghrelin in the regeneration of beta cells. Beta cell regeneration is an alternative strategy to replace islets transplantation especially with the shortage of pancreatic donors [88]. Pancreatic regeneration requires expansion of beta cells through beta cell neogenesis, dedifferentiation of acinar cell to beta cells, and proliferation of progenitor beta cells [89, 90]. Various agents have been studied to increase pancreatic regenerative capacity; the most important ones are glucagon-like peptide 1, actin, members of the transforming growth factor beta family, and the polypeptide growth factor beta [91–93]. Interestingly, ghrelin has been reported to influence the embryologic development of the pancreas and to regulate insulin secretion [94, 95]. Rats received 90 % pancreatectomy (surgical removal of 90 % of pancreas) were shown to have severe hyperglycemia and decreased beta cell mass and insulin levels. AG administration strongly reduced glucose levels, increased insulin-producing beta cell number, and insulin secretion. On the contrary, ghrelin receptor antagonist [d-Lys3]-GHRP-6 administration in these pancreatectomized rats worsens glucose levels and decreases beta cell mass [96]. AG, UAG, and obstatin counteracted the hyperglycaemia and improved plasma and pancreatic insulin levels, which were reduced by the STZ compound in newborn rats treated with STZ [84]. UAG and Obstatin increased islet area, islet number, and beta cell mass with respect to STZ treatment alone [86]. These findings strongly suggest that beta cells damaged by STZ administration during the neonatal stage or reduced by pancreatectomy can be regenerated or replicated following ghrelin treatment. This ability of GHS to regenerate beta cells may be supported by previous studies that have shown the regeneration effect of GH itself on the pancreas [97]. In conclusion, these findings provide evidence that GHS may function as a survival and regenerative factor for beta cells and offer a new perspective on the potential role of these peptides in diabetes.
Effects of ghrelin and hexarelin on insulin secretion and glucose homeostasis
The role of the ghrelin in the regulation of insulin secretion and insulin action remains a controversial topic. Ghrelin is reported to either inhibit or stimulate insulin secretion in animals (as shown in Table 1) and humans (as shown in Table 2). The discrepancies of ghrelin effects in different studies may be due to the following reasons: (1) the implications of ghrelin on insulin secretion and glucose metabolism could be different in different energy states like fasting, fed, obese, and diabetic, (2) Ghrelin, GHS-R, and GOAT are all expressed in pancreatic islets, suggesting a role for locally expressed ghrelin that could be different from that of the systemic ghrelin in the regulation of insulin release, (3) some of effects of ghrelin are independent of GHS-R1a and this highlight the complexity of the ghrelin-signaling pathway. This can be indicated by the paradoxical effects of ghrelin ablation and GHS-R ablation in ob/ob mice [98], (4) the controversy could be also due to differences in experimental design (different doses, different times of observation, mode of injection, in vitro versus in vivo experiments, etc.). This is obvious in one study that tested the effect of different concentration of ghrelin on insulin release. The study showed that ghrelin concentrations in the physiological range had no effect on glucose-stimulated insulin secretion (GSIS), while low ghrelin concentrations inhibited and high stimulated it [99]. The insulin response to glucose was enhanced in the presence of a high ghrelin concentration. Another study showed that the subcutaneous administration of ghrelin induced relatively specific GH release and enhanced cardiac performance in humans without significant adverse effects [99], and (5) long-term versus short-term actions of the peptide. In one study, the stimulatory action of ghrelin on insulin levels in rats was observed at 15 min but not at 5 min after ghrelin administration [100]. Therefore, the net effect on insulin sensitivity of manipulation of the ghrelin system still remains to be determined, although the overall systemic effects suggest promoting insulin resistance.
AG levels may be an important predictor of the glucose control under physiological conditions [101]. Ghrelin clearly stimulated insulin-mediated glucose disposal, an observation that was consistent with enhanced 2-deoxy-d-[3H]glucose (2DG) uptake in muscle and adipose tissue during hyperinsulinemia in ghrelin-treated animals [102]. In contrast, ghrelin hampered inhibition of endogenous glucose production by insulin. Furthermore, simultaneous administration of UAG abolishes the inhibitory effect of ghrelin on hepatic insulin action [102]. AG and UAG have been proposed to have opposite effects on glucose metabolism, which suggests a potentially significant influence of them on glucose metabolism [103]. Transgenic mice overexpressing ghrelin under the rat insulin II promoter had a pancreatic UAG content that was 1,000 times higher than the control mice. Such mice showed reduced GSIS and lower blood glucose levels and triglyceride levels during insulin tolerance test [104]. The suppression of insulin secretion is likely due to the enhancing effect of UAG on insulin sensitivity. Insulin secretion from isolated islets was instead indistinguishable from that of non-transgenic mice. These results imply that UAG, opposite to AG, increases insulin sensitivity. Gauna et al. reported that administration of AG in humans reduces insulin sensitivity, whereas the combination of AG plus UAG strongly improves insulin sensitivity [105]. Another studies reported that the dysregulation of AG/UAG ratio could play a role in pathogenesis of insulin resistance and diabetes [106, 107].
Ghrelin influences insulin and glucose homeostasis through a range of central and peripheral GHS-R-mediated mechanisms. Glucose and insulin also influence ghrelin secretion [108, 109]. Ghrelin influences glucose homeostasis centrally through medial hypothalamus. Re-expression of GHS-R in hypothalamic neurons expressing agouti-related peptide (AgRP) of adult GHS-R-null mice restores the orexigenic response to administered ghrelin and lowered blood glucose levels during caloric restriction [110]. This normalized glucoregulatory response was associated with glucagon rises and hepatic gluconeogenesis, indicating that hypothalamic arcuate-expressing agouti-related peptide (AGRP) neurons are responsible for the orexigenic effects and the central effects of ghrelin on glucose homeostasis. The effect of hexarelin on blood insulin, glucose, and in general on type 2 diabetes mellitus has not been studied well yet. In one study, obese and lean male rats of the Zucker strain were treated with hexarelin (80 μg/kg, b.i.d., s.c.) or saline (1 ml/kg, b.i.d., s.c.) for 30 days [111]. The results showed an increase in plasma insulin concentration in the obese rats treated with hexarelin. Obese rats treated with saline had plasma glucose concentrations similar to those found in lean animals. Treatment with hexarelin increased plasma glucose concentrations in obese rats but not in lean rats. One possibility of these findings may be related to the ability of hexarelin to stimulate the hypothalamic–pituitary–adrenal (HPA) axis [112, 113]. However, the mechanism by which GHS stimulate the HPA axis is unknown.
Effects of ghrelin and hexarelin on adipose tissue
The effect of ghrelin on adipose tissue has also been reported. Ghrelin increased the insulin-stimulated deoxyglucose uptake in isolated white adipose tissue from epididymal fat at concentration of 1,000 nM compared to 0.1 nM insulin alone. Ghrelin increased slightly the glucose uptake at 100 nM without statistical significance [133]. AG did not increase the deoxyglucose uptake in peri-renal adipocytes, which does not express GSH-R1a. UAG had no effect on insulin-stimulated glucose uptake. The dose of ghrelin in this study was above the physiological plasma concentration of ghrelin which could be explained by the difference in the effect and the dose of local and systemic ghrelin. The local ghrelin concentrations in adipose tissues may be considerably higher than those in the circulation and systemic ghrelin could be less important in the modulation of glucose uptake than those found locally. Ghrelin enhanced insulin-stimulated glucose uptake in adipocyte cell line, 3T3-L1 [72]. On the contrary, Ott et al. reported that ghrelin pre-treatment had no effect on insulin-stimulated glucose uptake in an immortalized brown adipocyte cell line [134]. Consequently, ghrelin appears to directly potentiate adipocyte insulin-stimulated glucose uptake in selective adipocyte populations and may play a role in adipocyte regulation of glucose homeostasis. Also, ghrelin regulate cholesterol efflux in macrophages through a GHS-R1a/PPAR-γ-dependent pathway [135]. Interestingly, these studies showed that PI3K/AKT pathway is mediating PPAR-γ activation by ghrelin which is the same pathway that mediates the antiapoptotic effects of ghrelin on various cells.
The potential effects of hexarelin on lipid metabolism have been studied. Hexarelin through binding to CD36 receptor induced an increase in thermogenic coactivator PGC-1α and uncoupling protein-1 (UCP-1) in 3T3-L1 adipocytes as well as in epididymal fat of treated mice, indicating increased fatty acids metabolism in the white fat in response to hexarelin [70]. Hexarelin treatment in obese rats significantly decreased plasma cholesterol but not triglyceride levels [111]. In another study, apolipoprotein E-null mice maintained on a long-term, high-fat, and high-cholesterol diet, a condition known to cause atherosclerosis, showed a significant regression in atherosclerotic lesions when treated with hexarelin compared to saline-treated controls [136]. This study demonstrated the activation of the PPAR-γ- liver X receptor (LXRα)-ATP-binding cassette transporters (ABC) metabolic cascade could be the signaling pathway for the action of hexarelin to cause the macrophages to mobilize excess cholesterol into the HDL cholesterol reverse pathway. These beneficial effects of hexarelin were observed under conditions in which GH was not upregulated, and similar effects observed in mice treated by EP80317, a hexarelin derivative with no GH release activity, supporting a GH-independent role for hexarelin [137].
These effects of hexarelin derived from enhanced expression of PPAR target genes, resulting in a thermogenic-like profile. PPAR-γ is considered a master regulator of fatty acid metabolism in fat through its direct role in regulating the expression of a broad range of genes involved in fatty acid and glucose metabolism [138]. Among the genes upregulated by PPAR-γ, genes related to fatty acid uptake (fatty acid transport protein FATP, CD36), glucose uptake (GLUT4), β-oxidation (acyl-CoA dehydrogenase, carnitine palmitoyltransferase CPT-1, acyl-CoA oxidase), gluconeogenesis (phosphoenolpyruvate carboxykinase PEPCK), and lipid storage (adipophilin). PPAR-γ also plays a central role in coordinating the macrophage response to lipid loading through a transcriptional cascade involving LXRα and the ABC transporters A1 and G1 [139, 140]. Similarly, PPAR-γ agonists of the thiazolidinedione family were shown to exert beneficial anti-diabetic and anti-atherosclerotic actions by promoting activation of the PPAR-γ-LXRα-ABC pathway and cholesterol efflux [141, 142]. Consequently, we suppose that ghrelin or hexarelin by modulation of PPAR-γ activity could enhance insulin sensitivity, regulate cholesterol metabolism in macrophages, and prevent atherosclerotic vascular lesion progression. Recently, hexarelin also inhibited cholesterol synthesis in liver cells through the activation of the adenine monophosphate-activated protein kinase (AMPK) pathway, reduction of 3-hydroxy-3-methylglutaryl-CoA reductase (HMGR), and increase recruitment of the anchor proteins insulin-induced genes (Insig-1 and Insig-2) in the liver cells [143]. HMGR is the rate-limiting enzyme in the cholesterol biosynthesis pathway. These findings support that CD36 and its ligands such as hexarelin play a role in regulating AMPK activity fatty acid metabolism and reverse cholesterol transport.
Ghrelin and hexarelin promote mitochondrial activity and biogenesis
The cause of apoptosis and failure of beta cell that occur during T2D are still unknown. However, mitochondria may play a role in regulating apoptosis and insulin secretion [144, 145]. Many studies have reported the beta cells in T2D do not sense glucose properly and consequently do not release enough amounts of insulin. The increased glucose and fatty acids could cause beta cell mitochondrial damage, leading to decreased ATP due to increase of mitochondrial membrane potential and production of ROS [146] (as shown in Fig. 2). Beta cells are susceptible to oxidative stress due to the weak expression of antioxidative enzymatic defenses, e.g., catalase and superoxide dismutase (SOD). Increased ROS can cause leakage of protons into the mitochondrial matrix, leading to decreased ATP production, and thus reduced GSIS [147]. Uncoupling protein 2 (UCP-2) modulates coupling efficiency and may regulate GSIS [148]. A small reduction in the mitochondrial membrane potential induced by mild uncoupling has a significant effect in attenuating ROS production [149]. However, the increase mitochondrial UCP2 decreases GSIS by decreasing intracellular ATP/ADP ratio [148].

The role of mitochondria in T2D. High concentration of glucose and fatty acids can lead to mitochondrial stress, which in turn results in increased production of reactive oxygen species (ROS). Increased ROS can decrease ATP production and reduced glucose-stimulated insulin secretion
It was interesting to observe that hexarelin and ghrelin enhanced mitochondrial structure and metabolism by upregulating many genes related to the mitochondrial metabolism of glucose and fatty acids [70]. More specifically, the upregulation of the genes which encode acetyl CoA acyl transferase, CPT-1, and several subunits of the ATP synthase and cytochrome c oxidase complexes may suggest an increased fatty acid metabolism through the mitochondrial oxidative phosphorylation pathway. The mitochondria of hexarelin-treated 3T3-L1 adipocytes showed an intense and highly organized cristae formation that spanned the entire width of mitochondria compared to that in untreated cells as shown by the electron microscopy [70]. Ghrelin also exerts a protective effect against H2O2-induced apoptosis in H9c2 rat cardiomyocytes by reducing the proapoptotic protein Bax and increasing Bcl-2 expression, accompanied by decreasing activation of caspase-9 and caspase-3 [150]. These results demonstrate that the antiapoptotic effect of ghrelin is probably caused by decrease H2O2-induced mitochondrial stress and by blocking activation of NF-κB. The transcription factor NF-κB is a critical signaling molecule in oxidant stress responses. UCP-2 mRNA expression was downregulated in INS-1 beta cell by ghrelin in the presence of 26.4 mM glucose [151]. Other studies showed that ghrelin decreased oxidative injury in the stomach [152], brain [153], blood vessels [154], and liver [155].
Cardioprotective effects of ghrelin and hexarelin
Individuals with diabetes are at a significantly greater risk of developing cardiovascular diseases including cardiomyopathy (CM) and heart failure [156]. Insulin resistance has been recognized as an important risk factor in the development of cardiovascular diseases [157, 158]. The diabetes-associated cardiovascular disease is responsible for 70 % of diabetes-related deaths. Diabetes can cause direct injury to myocardium, resulting in a distinct disease called “diabetic CM.” Data from experimental, epidemiologic, and clinical studies have shown that diabetes results in structural and functional cardiac changes. The structural changes of diabetic CM include higher left ventricle (LV) mass, wall thickness, and arterial stiffness [159]. These changes could be caused by deposition of advanced glycation end-products (AGEs) and collagen [160, 161]. Functionally, there are diastolic abnormalities which have been suggested as early functional abnormalities of diabetic CM [162, 163]. The cardioprotective effects of hexarelin and ghrelin have been well investigated. Many studies have shown their beneficial effects on the cardiovascular system such as protection against cardiac ischemia, cardiac fibrosis, improving cardiac function, and decreasing peripheral resistance after myocardial infarction (MI) in animals [164–166] and humans [167]. Our group showed that treatment of spontaneously hypertensive rats (SHRs) with hexarelin for 5 weeks from an age of 16 weeks significantly reduced cardiac fibrosis in SHRs by decreasing interstitial and perivascular myocardial collagen deposition and myocardial hydroxyproline content and reducing mRNA and protein expression of collagen I and III in SHR hearts [168]. In addition, hexarelin treatment significantly attenuated left ventricular (LV) hypertrophy, LV diastolic dysfunction, and high blood pressure in SHRs. So, our data demonstrate that hexarelin reduces cardiac fibrosis in SHRs, perhaps by decreasing collagen synthesis and accelerating collagen degradation [168].
Ghrelin and hexarelin showed protective effects on cardiac ischaemia. Administration of ghrelin in vitro to ischaemia/reperfusion (I/R) rat hearts was shown to reduce the infarct size through the activation of PKC, and these effects were likely initiated by the binding of ghrelin to its receptor, GHS-R1a [169]. Pre-administration of ghrelin in vivo greatly ameliorated the damaged heart function, attenuated myocardial injury, and apoptosis through inhibition of ER stress [170]. The role of hexarelin on the protection against cardiac ischemia has been reported earlier [67, 68, 171]. Rats pre-treated with ghrelin or hexarelin for 7 days in vivo prior to in vitro I/R injury showed an improvement in cardiac function [172]. A single dose of oral hexarelin treatment at the very acute phase after MI has protective effects on chronic cardiac function [173]. Aragno et al. also showed that obestatin, which is encoded by the same gene for ghrelin, protected STZ-caused myocardial dysfunction [174]. Our group has confirmed the protective effect of hexarelin and ghrelin on cardiac ischaemia [164]. Ghrelin (10 nM) or hexarelin (1 nM) was administered in the perfusion solution of isolated hearts before or after ischemia for 10 min. It was found that relative sarcomere shortening was significantly reduced after I/R and exhibited a reduction in both the amplitude and rising rate of intracellular calcium ([Ca2+]i) transients but increases in cytoplasmic [Ca2+]. The alternation in [Ca2+]i transients and cytoplasmic [Ca2+] which occurs during the cardiac ischemia and reperfusion is due to reduction of sarcoplasmic reticulum (SR) Ca2+ ATPase and Na/K ATPase activities or due to increased oxidative stress which occurs during the reperfusion which leads to subsequent damage to the plasma and SR membranes and further increases in cytoplasmic Ca2+. Pre- and post-treatments with ghrelin and hexarelin protected freshly isolated mouse ventricular cardiomyocytes against these negative effects of I/R. They resulted in normal cardiomyocytes contractility, normal amplitude, and rising rate of [Ca2+]i transients in the cardiomyocytes during an in vitro I/R injury, which could be attributed to normalized SR Ca2+ ATPase activity and SR Ca2+ content [164].
To compare the effect of ghrelin and hexarelin on cardiac function after infarction MI, ghrelin-knockout mice were used to show whether hexarelin could compensate for the ghrelin deficiency [175]. The results showed that hexarelin-treated mice had better cardiac function than ghrelin-treated mice, as indicated by the higher absolute values of percent of the ejection fraction (EF), peak rate of pressure rise (dP/dt max) and dP/dt min, fractional shortening (FS), preload-adjusted maximal power (PAMP), and maximal elastance (Emax). Hexarelin and ghrelin treatments also significantly reduced collagen volume fraction in the non-infarcted LV walls and plasma atrial natriuretic peptide levels than vehicle administration [175]. These stronger effects of hexarelin was also confirmed by another study in which hypophysectomized rats were treated in vivo for 7 days with either ghrelin (320 g/kg) or hexarelin (80 g/kg), and then their hearts were subjected in vitro to the ischemia and reperfusion procedure [172]. Ghrelin was far less effective than hexarelin in preventing increases in LV end-diastolic pressure (15 and 60 % protection for ghrelin and hexarelin, respectively), coronary perfusion pressure (10 and 45 % reduction), and release of creatine kinase in the heart perfusate (15 and 55 % reduction). The effects of hexarelin were mediated largely by interactions with CD36 in the heart and to a lesser extent by GHS-R. However, other studies reported that ghrelin and hexarelin had similar effects [176].
In addition, AG and UAG also exert antiapoptotic effects on mouse and rat cardiomyocytes by acting on MAPK and PI3K/AKT pathways [33]. Similarly, our studies have confirmed the cardioprotective role of hexarelin in I/R models of hearts from mice [164]. GHS can preserve the electrophysiological properties of cardiomyocytes after I/R and inhibit cardiomyocyte apoptosis and promote cell survival by modification of MAPK pathways through activating GHS-R1a [176]. Hexarelin also showed a protective role on angiotensin II (ANG II)-induced apoptosis of isolated cardiomyocytes from neonatal rats [177]. Administration of hexarelin significantly decreased ANG II-induced apoptosis and DNA fragmentation and increased cardiomyocyte viability via inhibiting the increased caspase-3 activity and BAX expression and by increasing the expression of BCL-2. GHS-R mRNA was abundantly expressed in cardiomyocytes and was upregulated after administration of hexarelin.
Recently, some studies have reported that the beneficial effect of ghrelin on the cardiovascular system might be mediated by modulation of cardiac autonomic nerve activity. Ghrelin has been shown to suppress sympathetic activity and to decrease blood pressure through mechanisms involving the central nervous system [178, 179]. The beneficial effects of ghrelin were accompanied by the suppression of MI-induced increase of heart rate and plasma norepinephrine concentration [180]. These effects of ghrelin decreased with atropine pre-treatment or vagotomy. Ghrelin exerted antiarrhythmic effects in rats during acute MI via modulation of vagal activity by showing increased high-frequency (HF) component as an index of parasympathetic activity and decreased low-frequency (LF)/HF ratio as an index of sympathetic activity and restoration of phosphorylated connexin-43 protein levels [181].
As connexin-43-deficient mice have been reported to be markedly susceptible to ischemia-induced ventricular arrhythmias [182]. Other studies have been proposed to explain the reduction in blood pressure after ghrelin administration, including vasodilation via endothelium activation or a direct effect on vascular smooth muscle cells [183, 184]. Ghrelin also inhibited post-infarct myocardial remodeling, while improving cardiac function through its anti-inflammatory effects as shown by decreased mRNA and protein levels of interleukin-1beta and tumor necrosis factor-alpha [185].
Clinical studies in human with growth hormone deficiency (GHD) suggested that patients with either ischemic or non-ischemic CM might benefit from GHS therapy, mainly from reduction of LV maladaptive remodeling and cardiomyocyte loss. In healthy volunteers and in patients with GHD, an acute administration of hexarelin (2 µg/kg) elicited a short-term increase in contractility and LV ejection fraction in a GH-independent manner [186]. Similar results were observed in patients with ischemic CM but not with dilated CM [187]. Studies with ghrelin showed an improved cardiac function following acute administration, although this effect was associated with a dose-dependent increase in GH [188]. Also, a short-term infusion of ghrelin (0.1 µg/kg/min60 min) decreased mean arterial pressure and increased cardiac and stroke volume index, which may be related to a reduced systemic vascular resistance in healthy volunteers and in patients with heart failure. The cardiovascular effects of GHS could be through GHS-R1a in the heart and the blood vessels or through CD36 that is a specific cardiac receptor for hexarelin [189].
Conclusions
The exact mechanisms by which GHS promote their metabolic response are not fully understood. However, it becomes clear that interacting with GHS-R1a and/or other receptors induces profound changes in metabolic activities of target tissues, especially regarding PPAR-γ-downregulated events. These metabolic effects besides the proliferative, regenerative, and antiapoptotic effects of GHS on beta cells represent a promising avenue in the treatment of diabetes and diabetic related heart diseases.
References
L. Guariguata, D.R. Whiting, I. Hambleton, J. Beagley, U. Linnenkamp, J.E. Shaw, Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Res. Clin. Pract. 103(2), 137–149 (2014). doi:10.1016/j.diabres.2013.11.002
D.A. Stoffers, The development of beta-cell mass: recent progress and potential role of GLP-1. Hormone Metab. Res. 36(11–12), 811–821 (2004). doi:10.1055/s-2004-826168
R. Colagiuri, R. Short, A. Buckley, The status of national diabetes programmes: a global survey of IDF member associations. Diabetes Res. Clin. Pract. 87(2), 137–142 (2010). doi:10.1016/j.diabres.2009.10.005
J.A. Wali, H.E. Thomas, A.P. Sutherland, Linking obesity with type 2 diabetes: the role of T-bet. Diabetes Metabol. Syndr. Obes. 7, 331–340 (2014). doi:10.2147/DMSO.S51432
H. Sakuraba, H. Mizukami, N. Yagihashi, R. Wada, C. Hanyu, S. Yagihashi, Reduced beta-cell mass and expression of oxidative stress-related DNA damage in the islet of Japanese Type II diabetic patients. Diabetologia 45(1), 85–96 (2002). doi:10.1007/s001250200009
P. Marchetti, S. Del Guerra, L. Marselli, R. Lupi, M. Masini, M. Pollera, M. Bugliani, U. Boggi, F. Vistoli, F. Mosca, S. Del Prato, Pancreatic islets from type 2 diabetic patients have functional defects and increased apoptosis that are ameliorated by metformin. J. Clin. Endocrinol. Metab. 89(11), 5535–5541 (2004). doi:10.1210/jc.2004-0150
I. Cozar-Castellano, N. Fiaschi-Taesch, T.A. Bigatel, K.K. Takane, A. Garcia-Ocana, R. Vasavada, A.F. Stewart, Molecular control of cell cycle progression in the pancreatic beta-cell. Endocr. Rev. 27(4), 356–370 (2006). doi:10.1210/er.2006-0004
D.R. Matthews, The natural history of diabetes-related complications: the UKPDS experience. United Kingdom Prospective Diabetes Study. Diabetes Obes. Metab. 1(Suppl 2), S7–S13 (1999)
R.R. Holman, Assessing the potential for alpha-glucosidase inhibitors in prediabetic states. Diabetes Res. Clin. Pract. 40(Suppl), S21–S25 (1998)
M. Prentki, C.J. Nolan, Islet beta cell failure in type 2 diabetes. J. Clin. Investig. 116(7), 1802–1812 (2006). doi:10.1172/JCI29103
B.L. Wajchenberg, Beta-cell failure in diabetes and preservation by clinical treatment. Endocr. Rev. 28(2), 187–218 (2007). doi:10.1210/10.1210/er.2006-0038
P.A. Halban, K.S. Polonsky, D.W. Bowden, M.A. Hawkins, C. Ling, K.J. Mather, A.C. Powers, C.J. Rhodes, L. Sussel, G.C. Weir, Beta-cell failure in type 2 diabetes: postulated mechanisms and prospects for prevention and treatment. Diabetes Care 37(6), 1751–1758 (2014). doi:10.2337/dc14-0396
W. Quan, E.K. Jo, M.S. Lee, Role of pancreatic beta-cell death and inflammation in diabetes. Diabetes Obes. Metab. 15(Suppl 3), 141–151 (2013). doi:10.1111/dom.12153
M.S. Akash, K. Rehman, S. Chen, Role of inflammatory mechanisms in pathogenesis of type 2 diabetes mellitus. J. Cell. Biochem. 114(3), 525–531 (2013). doi:10.1002/jcb.24402
R.P. Robertson, J. Harmon, P.O. Tran, Y. Tanaka, H. Takahashi, Glucose toxicity in beta-cells: type 2 diabetes, good radicals gone bad, and the glutathione connection. Diabetes 52(3), 581–587 (2003)
A. Klip, M.R. Paquet, Glucose transport and glucose transporters in muscle and their metabolic regulation. Diabetes Care 13(3), 228–243 (1990)
I. Shimomura, R.E. Hammer, S. Ikemoto, M.S. Brown, J.L. Goldstein, Leptin reverses insulin resistance and diabetes mellitus in mice with congenital lipodystrophy. Nature 401(6748), 73–76 (1999). doi:10.1038/43448
K. Ebihara, Y. Ogawa, H. Masuzaki, M. Shintani, F. Miyanaga, M. Aizawa-Abe, T. Hayashi, K. Hosoda, G. Inoue, Y. Yoshimasa, O. Gavrilova, M.L. Reitman, K. Nakao, Transgenic overexpression of leptin rescues insulin resistance and diabetes in a mouse model of lipoatrophic diabetes. Diabetes 50(6), 1440–1448 (2001)
K. Hotta, T. Funahashi, Y. Arita, M. Takahashi, M. Matsuda, Y. Okamoto, H. Iwahashi, H. Kuriyama, N. Ouchi, K. Maeda, M. Nishida, S. Kihara, N. Sakai, T. Nakajima, K. Hasegawa, M. Muraguchi, Y. Ohmoto, T. Nakamura, S. Yamashita, T. Hanafusa, Y. Matsuzawa, Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arterioscler. Thromb. Vasc. Biol. 20(6), 1595–1599 (2000)
M.L. Reitman, O. Gavrilova, A-ZIP/F-1 mice lacking white fat: a model for understanding lipoatrophic diabetes. Int. J. Obes. Relat. Metabol. Disord. 24(Suppl 4), S11–S14 (2000)
O. Gavrilova, B. Marcus-Samuels, D. Graham, J.K. Kim, G.I. Shulman, A.L. Castle, C. Vinson, M. Eckhaus, M.L. Reitman, Surgical implantation of adipose tissue reverses diabetes in lipoatrophic mice. J. Clin. Investig. 105(3), 271–278 (2000). doi:10.1172/JCI7901
J.C. Bruning, M.D. Michael, J.N. Winnay, T. Hayashi, D. Horsch, D. Accili, L.J. Goodyear, C.R. Kahn, A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol. Cell 2(5), 559–569 (1998)
E.D. Abel, O. Peroni, J.K. Kim, Y.B. Kim, O. Boss, E. Hadro, T. Minnemann, G.I. Shulman, B.B. Kahn, Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature 409(6821), 729–733 (2001). doi:10.1038/35055575
C.J. Shu, C. Benoist, D. Mathis, The immune system’s involvement in obesity-driven type 2 diabetes. Semin. Immunol. 24(6), 436–442 (2012). doi:10.1016/j.smim.2012.12.001
M. Kojima, K. Kangawa, Ghrelin: structure and function. Physiol. Rev. 85(2), 495–522 (2005). doi:10.1152/physrev.00012.2004
C.Y. Chen, A. Asakawa, M. Fujimiya, S.D. Lee, A. Inui, Ghrelin gene products and the regulation of food intake and gut motility. Pharmacol. Rev. 61(4), 430–481 (2009). doi:10.1124/pr.109.001958
F. Broglio, C. Gottero, F. Prodam, C. Gauna, G. Muccioli, M. Papotti, T. Abribat, A.J. Van Der Lely, E. Ghigo, Non-acylated ghrelin counteracts the metabolic but not the neuroendocrine response to acylated ghrelin in humans. J. Clin. Endocrinol. Metab. 89(6), 3062–3065 (2004). doi:10.1210/jc.2003-031964
C. Gauna, P.J. Delhanty, L.J. Hofland, J.A. Janssen, F. Broglio, R.J. Ross, E. Ghigo, A.J. van der Lely, Ghrelin stimulates, whereas des-octanoyl ghrelin inhibits, glucose output by primary hepatocytes. J. Clin. Endocrinol. Metab. 90(2), 1055–1060 (2005). doi:10.1210/jc.2004-1069
R. Kumar, A. Salehi, J.F. Rehfeld, P. Hoglund, E. Lindstrom, R. Hakanson, Proghrelin peptides: desacyl ghrelin is a powerful inhibitor of acylated ghrelin, likely to impair physiological effects of acyl ghrelin but not of obestatin A study of pancreatic polypeptide secretion from mouse islets. Regul. Pept. 164(2–3), 65–70 (2010). doi:10.1016/j.regpep.2010.06.005
G. Muccioli, N. Pons, C. Ghe, F. Catapano, R. Granata, E. Ghigo, Ghrelin and des-acyl ghrelin both inhibit isoproterenol-induced lipolysis in rat adipocytes via a non-type 1a growth hormone secretagogue receptor. Eur. J. Pharmacol. 498(1–3), 27–35 (2004). doi:10.1016/j.ejphar.2004.07.066
K. Toshinai, H. Yamaguchi, Y. Sun, R.G. Smith, A. Yamanaka, T. Sakurai, Y. Date, M.S. Mondal, T. Shimbara, T. Kawagoe, N. Murakami, M. Miyazato, K. Kangawa, M. Nakazato, Des-acyl ghrelin induces food intake by a mechanism independent of the growth hormone secretagogue receptor. Endocrinology 147(5), 2306–2314 (2006). doi:10.1210/en.2005-1357
H. Chung, S. Seo, M. Moon, S. Park, Phosphatidylinositol-3-kinase/Akt/glycogen synthase kinase-3 beta and ERK1/2 pathways mediate protective effects of acylated and unacylated ghrelin against oxygen-glucose deprivation-induced apoptosis in primary rat cortical neuronal cells. J. Endocrinol. 198(3), 511–521 (2008). doi:10.1677/JOE-08-0160
G. Baldanzi, N. Filigheddu, S. Cutrupi, F. Catapano, S. Bonissoni, A. Fubini, D. Malan, G. Baj, R. Granata, F. Broglio, M. Papotti, N. Surico, F. Bussolino, J. Isgaard, R. Deghenghi, F. Sinigaglia, M. Prat, G. Muccioli, E. Ghigo, A. Graziani, Ghrelin and des-acyl ghrelin inhibit cell death in cardiomyocytes and endothelial cells through ERK1/2 and PI 3-kinase/AKT. J. Cell Biol. 159(6), 1029–1037 (2002). doi:10.1083/jcb.200207165
X.M. Guan, H. Yu, O.C. Palyha, K.K. McKee, S.D. Feighner, D.J. Sirinathsinghji, R.G. Smith, L.H. Van der Ploeg, A.D. Howard, Distribution of mRNA encoding the growth hormone secretagogue receptor in brain and peripheral tissues. Brain Res. Mol. Brain Res. 48(1), 23–29 (1997)
A.D. Howard, S.D. Feighner, D.F. Cully, J.P. Arena, P.A. Liberator, C.I. Rosenblum, M. Hamelin, D.L. Hreniuk, O.C. Palyha, J. Anderson, P.S. Paress, C. Diaz, M. Chou, K.K. Liu, K.K. McKee, S.S. Pong, L.Y. Chaung, A. Elbrecht, M. Dashkevicz, R. Heavens, M. Rigby, D.J. Sirinathsinghji, D.C. Dean, D.G. Melillo, A.A. Patchett, R. Nargund, P.R. Griffin, J.A. DeMartino, S.K. Gupta, J.M. Schaeffer, R.G. Smith, L.H. Van der Ploeg, A receptor in pituitary and hypothalamus that functions in growth hormone release. Science 273(5277), 974–977 (1996)
G. Muccioli, A. Baragli, R. Granata, M. Papotti, E. Ghigo, Heterogeneity of ghrelin/growth hormone secretagogue receptors. Toward the understanding of the molecular identity of novel ghrelin/GHS receptors. Neuroendocrinology 86(3), 147–164 (2007). doi:10.1159/000105141
Y. Shuto, T. Shibasaki, K. Wada, I. Parhar, J. Kamegai, H. Sugihara, S. Oikawa, I. Wakabayashi, Generation of polyclonal antiserum against the growth hormone secretagogue receptor (GHS-R): evidence that the GHS-R exists in the hypothalamus, pituitary and stomach of rats. Life Sci. 68(9), 991–996 (2001)
Y. Date, M. Kojima, H. Hosoda, A. Sawaguchi, M.S. Mondal, T. Suganuma, S. Matsukura, K. Kangawa, M. Nakazato, Ghrelin, a novel growth hormone-releasing acylated peptide, is synthesized in a distinct endocrine cell type in the gastrointestinal tracts of rats and humans. Endocrinology 141(11), 4255–4261 (2000). doi:10.1210/endo.141.11.7757
K. Mori, A. Yoshimoto, K. Takaya, K. Hosoda, H. Ariyasu, K. Yahata, M. Mukoyama, A. Sugawara, H. Hosoda, M. Kojima, K. Kangawa, K. Nakao, Kidney produces a novel acylated peptide, ghrelin. FEBS Lett. 486(3), 213–216 (2000)
N. Nagaya, M. Kojima, M. Uematsu, M. Yamagishi, H. Hosoda, H. Oya, Y. Hayashi, K. Kangawa, Hemodynamic and hormonal effects of human ghrelin in healthy volunteers. Am. J. Physiol. Regul. Integr. Comp. Physiol. 280(5), R1483–R1487 (2001)
M. Korbonits, R.A. Jacobs, S.J. Aylwin, J.M. Burrin, P.L. Dahia, J.P. Monson, J. Honegger, R. Fahlbush, P.J. Trainer, S.L. Chew, G.M. Besser, A.B. Grossman, Expression of the growth hormone secretagogue receptor in pituitary adenomas and other neuroendocrine tumors. J. Clin. Endocrinol. Metab. 83(10), 3624–3630 (1998). doi:10.1210/jcem.83.10.5210
K. Kim, K. Arai, N. Sanno, R.Y. Osamura, A. Teramoto, T. Shibasaki, Ghrelin and growth hormone (GH) secretagogue receptor (GHSR) mRNA expression in human pituitary adenomas. Clin. Endocrinol. 54(6), 759–768 (2001)
M. Papotti, P. Cassoni, M. Volante, R. Deghenghi, G. Muccioli, E. Ghigo, Ghrelin-producing endocrine tumors of the stomach and intestine. J. Clin. Endocrinol. Metab. 86(10), 5052–5059 (2001). doi:10.1210/jcem.86.10.7918
M. Kojima, H. Hosoda, Y. Date, M. Nakazato, H. Matsuo, K. Kangawa, Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 402(6762), 656–660 (1999). doi:10.1038/45230
E. Arvat, L. Di Vito, F. Broglio, M. Papotti, G. Muccioli, C. Dieguez, F.F. Casanueva, R. Deghenghi, F. Camanni, E. Ghigo, Preliminary evidence that Ghrelin, the natural GH secretagogue (GHS)-receptor ligand, strongly stimulates GH secretion in humans. J. Endocrinol. Invest. 23(8), 493–495 (2000)
E. Arvat, M. Maccario, L. Di Vito, F. Broglio, A. Benso, C. Gottero, M. Papotti, G. Muccioli, C. Dieguez, F.F. Casanueva, R. Deghenghi, F. Camanni, E. Ghigo, Endocrine activities of ghrelin, a natural growth hormone secretagogue (GHS), in humans: comparison and interactions with hexarelin, a nonnatural peptidyl GHS, and GH-releasing hormone. J. Clin. Endocrinol. Metab. 86(3), 1169–1174 (2001). doi:10.1210/jcem.86.3.7314
F. Broglio, A. Benso, C. Castiglioni, C. Gottero, F. Prodam, S. Destefanis, C. Gauna, A.J. van der Lely, R. Deghenghi, M. Bo, E. Arvat, E. Ghigo, The endocrine response to ghrelin as a function of gender in humans in young and elderly subjects. J. Clin. Endocrinol. Metab. 88(4), 1537–1542 (2003). doi:10.1210/jc.2002-021504
A.M. Wren, L.J. Seal, M.A. Cohen, A.E. Brynes, G.S. Frost, K.G. Murphy, W.S. Dhillo, M.A. Ghatei, S.R. Bloom, Ghrelin enhances appetite and increases food intake in humans. J. Clin. Endocrinol. Metab. 86(12), 5992 (2001). doi:10.1210/jcem.86.12.8111
D.E. Cummings, J. Overduin, Gastrointestinal regulation of food intake. J. Clin. Investig. 117(1), 13–23 (2007). doi:10.1172/JCI30227
J. Kamegai, H. Tamura, T. Shimizu, S. Ishii, H. Sugihara, I. Wakabayashi, Chronic central infusion of ghrelin increases hypothalamic neuropeptide Y and Agouti-related protein mRNA levels and body weight in rats. Diabetes 50(11), 2438–2443 (2001)
M. Shintani, Y. Ogawa, K. Ebihara, M. Aizawa-Abe, F. Miyanaga, K. Takaya, T. Hayashi, G. Inoue, K. Hosoda, M. Kojima, K. Kangawa, K. Nakao, Ghrelin, an endogenous growth hormone secretagogue, is a novel orexigenic peptide that antagonizes leptin action through the activation of hypothalamic neuropeptide Y/Y1 receptor pathway. Diabetes 50(2), 227–232 (2001)
M. Tschop, D.L. Smiley, M.L. Heiman, Ghrelin induces adiposity in rodents. Nature 407(6806), 908–913 (2000). doi:10.1038/35038090
N. Nagaya, M. Uematsu, M. Kojima, Y. Ikeda, F. Yoshihara, W. Shimizu, H. Hosoda, Y. Hirota, H. Ishida, H. Mori, K. Kangawa, Chronic administration of ghrelin improves left ventricular dysfunction and attenuates development of cardiac cachexia in rats with heart failure. Circulation 104(12), 1430–1435 (2001)
N. Nagaya, K. Kangawa, Ghrelin improves left ventricular dysfunction and cardiac cachexia in heart failure. Curr. Opin. Pharmacol. 3(2), 146–151 (2003)
N. Nagaya, K. Kangawa, Ghrelin, a novel growth hormone-releasing peptide, in the treatment of chronic heart failure. Regul. Pept. 114(2–3), 71–77 (2003)
Y. Lin, K. Matsumura, M. Fukuhara, S. Kagiyama, K. Fujii, M. Iida, Ghrelin acts at the nucleus of the solitary tract to decrease arterial pressure in rats. Hypertension 43(5), 977–982 (2004). doi:10.1161/01.HYP.0000122803.91559.55
D. Baatar, K. Patel, D.D. Taub, The effects of ghrelin on inflammation and the immune system. Mol. Cell. Endocrinol. 340(1), 44–58 (2011). doi:10.1016/j.mce.2011.04.019
N. Pandya, R. DeMott-Friberg, C.Y. Bowers, A.L. Barkan, C.A. Jaffe, Growth hormone (GH)-releasing peptide-6 requires endogenous hypothalamic GH-releasing hormone for maximal GH stimulation. J. Clin. Endocrinol. Metab. 83(4), 1186–1189 (1998). doi:10.1210/jcem.83.4.4711
E. Ghigo, G. Aimaretti, E. Arvat, F. Camanni, Growth hormone-releasing hormone combined with arginine or growth hormone secretagogues for the diagnosis of growth hormone deficiency in adults. Endocrine 15(1), 29–38 (2001). doi:10.1385/ENDO:15:1:029
E. Ghigo, E. Arvat, G. Muccioli, F. Camanni, Growth hormone-releasing peptides. Eur. J. Endocrinol. 136(5), 445–460 (1997)
V. Guerlavais, D. Boeglin, D. Mousseaux, C. Oiry, A. Heitz, R. Deghenghi, V. Locatelli, A. Torsello, C. Ghe, F. Catapano, G. Muccioli, J.C. Galleyrand, J.A. Fehrentz, J. Martinez, New active series of growth hormone secretagogues. J. Med. Chem. 46(7), 1191–1203 (2003). doi:10.1021/jm020985q
R. Deghenghi, M.M. Cananzi, A. Torsello, C. Battisti, E.E. Muller, V. Locatelli, GH-releasing activity of Hexarelin, a new growth hormone releasing peptide, in infant and adult rats. Life Sci. 54(18), 1321–1328 (1994)
A.V. Kaisary, W.G. Bowsher, D.A. Gillatt, J.B. Anderson, P.R. Malone, B.P. Imbimbo, Pharmacodynamics of a long acting depot preparation of avorelin in patients with prostate cancer. Avorelin Study Group. J. Urol. 162(6), 2019–2023 (1999)
M. Korbonits, G. Kaltsas, L.A. Perry, A.B. Grossman, J.P. Monson, G.M. Besser, P.J. Trainer, Hexarelin as a test of pituitary reserve in patients with pituitary disease. Clin. Endocrinol. 51(3), 369–375 (1999)
M. Gasperi, G. Aimaretti, G. Scarcello, G. Corneli, C. Cosci, E. Arvat, E. Martino, E. Ghigo, Low dose hexarelin and growth hormone (GH)-releasing hormone as a diagnostic tool for the diagnosis of GH deficiency in adults: comparison with insulin-induced hypoglycemia test. J. Clin. Endocrinol. Metab. 84(8), 2633–2637 (1999). doi:10.1210/jcem.84.8.5904
Z. Laron, J. Frenkel, R. Deghenghi, S. Anin, B. Klinger, A. Silbergeld, Intranasal administration of the GHRP hexarelin accelerates growth in short children. Clin. Endocrinol. 43(5), 631–635 (1995)
V. Locatelli, G. Rossoni, F. Schweiger, A. Torsello, V. De Gennaro Colonna, M. Bernareggi, R. Deghenghi, E.E. Muller, F. Berti, Growth hormone-independent cardioprotective effects of hexarelin in the rat. Endocrinology 140(9), 4024–4031 (1999). doi:10.1210/endo.140.9.6948
G. Rossoni, V. Locatelli, V. De GennaroColonna, A. Torsello, F. Schweiger, M. Boghen, M. Nilsson, M. Bernareggi, E.E. Muller, F. Berti, Growth hormone and hexarelin prevent endothelial vasodilator dysfunction in aortic rings of the hypophysectomized rat. J. Cardiovasc. Pharmacol. 34(3), 454–460 (1999)
V. Bodart, M. Febbraio, A. Demers, N. McNicoll, P. Pohankova, A. Perreault, T. Sejlitz, E. Escher, R.L. Silverstein, D. Lamontagne, H. Ong, CD36 mediates the cardiovascular action of growth hormone-releasing peptides in the heart. Circ. Res. 90(8), 844–849 (2002)
A. Rodrigue-Way, A. Demers, H. Ong, A. Tremblay, A growth hormone-releasing peptide promotes mitochondrial biogenesis and a fat burning-like phenotype through scavenger receptor CD36 in white adipocytes. Endocrinology 148(3), 1009–1018 (2007). doi:10.1210/en.2006-0975
H. Zhao, G. Liu, Q. Wang, L. Ding, H. Cai, H. Jiang, Z. Xin, Effect of ghrelin on human endothelial cells apoptosis induced by high glucose. Biochem. Biophys. Res. Commun. 362(3), 677–681 (2007). doi:10.1016/j.bbrc.2007.08.021
M.S. Kim, C.Y. Yoon, P.G. Jang, Y.J. Park, C.S. Shin, H.S. Park, J.W. Ryu, Y.K. Pak, J.Y. Park, K.U. Lee, S.Y. Kim, H.K. Lee, Y.B. Kim, K.S. Park, The mitogenic and antiapoptotic actions of ghrelin in 3T3-L1 adipocytes. Mol. Endocrinol. 18(9), 2291–2301 (2004). doi:10.1210/me.2003-0459
W.G. Li, D. Gavrila, X. Liu, L. Wang, S. Gunnlaugsson, L.L. Stoll, M.L. McCormick, C.D. Sigmund, C. Tang, N.L. Weintraub, Ghrelin inhibits proinflammatory responses and nuclear factor-kappaB activation in human endothelial cells. Circulation 109(18), 2221–2226 (2004). doi:10.1161/01.CIR.0000127956.43874.F2
I. Johansson, S. Destefanis, N.D. Aberg, M.A. Aberg, K. Blomgren, C. Zhu, C. Ghe, R. Granata, E. Ghigo, G. Muccioli, P.S. Eriksson, J. Isgaard, Proliferative and protective effects of growth hormone secretagogues on adult rat hippocampal progenitor cells. Endocrinology 149(5), 2191–2199 (2008). doi:10.1210/en.2007-0733
W. Wang, D. Zhang, H. Zhao, Y. Chen, Y. Liu, C. Cao, L. Han, G. Liu, Ghrelin inhibits cell apoptosis induced by lipotoxicity in pancreatic beta-cell line. Regul. Pept. 161(1–3), 43–50 (2010). doi:10.1016/j.regpep.2009.12.017
C.E. Wrede, L.M. Dickson, M.K. Lingohr, I. Briaud, C.J. Rhodes, Protein kinase B/Akt prevents fatty acid-induced apoptosis in pancreatic beta-cells (INS-1). J. Biol. Chem. 277(51), 49676–49684 (2002). doi:10.1074/jbc.M208756200
J. Buteau, W. El-Assaad, C.J. Rhodes, L. Rosenberg, E. Joly, M. Prentki, Glucagon-like peptide-1 prevents beta cell glucolipotoxicity. Diabetologia 47(5), 806–815 (2004). doi:10.1007/s00125-004-1379-6
E. Karaskov, C. Scott, L. Zhang, T. Teodoro, M. Ravazzola, A. Volchuk, Chronic palmitate but not oleate exposure induces endoplasmic reticulum stress, which may contribute to INS-1 pancreatic beta-cell apoptosis. Endocrinology 147(7), 3398–3407 (2006). doi:10.1210/en.2005-1494
D. Kawamori, H. Kaneto, Y. Nakatani, T.A. Matsuoka, M. Matsuhisa, M. Hori, Y. Yamasaki, The forkhead transcription factor Foxo1 bridges the JNK pathway and the transcription factor PDX-1 through its intracellular translocation. J. Biol. Chem. 281(2), 1091–1098 (2006). doi:10.1074/jbc.M508510200
G. Solinas, W. Naugler, F. Galimi, M.S. Lee, M. Karin, Saturated fatty acids inhibit induction of insulin gene transcription by JNK-mediated phosphorylation of insulin-receptor substrates. Proc. Natl. Acad. Sci. USA 103(44), 16454–16459 (2006). doi:10.1073/pnas.0607626103
Y. Zhang, B. Ying, L. Shi, H. Fan, D. Yang, D. Xu, Y. Wei, X. Hu, Y. Zhang, X. Zhang, T. Wang, D. Liu, L. Dou, G. Chen, F. Jiang, F. Wen, Ghrelin inhibit cell apoptosis in pancreatic beta cell line HIT-T15 via mitogen-activated protein kinase/phosphoinositide 3-kinase pathways. Toxicology 237(1–3), 194–202 (2007). doi:10.1016/j.tox.2007.05.013
R. Granata, F. Settanni, L. Biancone, L. Trovato, R. Nano, F. Bertuzzi, S. Destefanis, M. Annunziata, M. Martinetti, F. Catapano, C. Ghe, J. Isgaard, M. Papotti, E. Ghigo, G. Muccioli, Acylated and unacylated ghrelin promote proliferation and inhibit apoptosis of pancreatic beta-cells and human islets: involvement of 3′,5′-cyclic adenosine monophosphate/protein kinase A, extracellular signal-regulated kinase 1/2, and phosphatidyl inositol 3-Kinase/Akt signaling. Endocrinology 148(2), 512–529 (2007). doi:10.1210/en.2006-0266
W. Wang, Y. Liu, Y. Chen, C. Cao, Y. Xiang, D. Zhang, L. Han, H. Zhao, G. Liu, Inhibition of Foxo1 mediates protective effects of ghrelin against lipotoxicity in MIN6 pancreatic beta-cells. Peptides 31(2), 307–314 (2010). doi:10.1016/j.peptides.2009.11.011
T. Irako, T. Akamizu, H. Hosoda, H. Iwakura, H. Ariyasu, K. Tojo, N. Tajima, K. Kangawa, Ghrelin prevents development of diabetes at adult age in streptozotocin-treated newborn rats. Diabetologia 49(6), 1264–1273 (2006). doi:10.1007/s00125-006-0226-3
A.M. Ackermann, M. Gannon, Molecular regulation of pancreatic beta-cell mass development, maintenance, and expansion. J. Mol. Endocrinol. 38(1–2), 193–206 (2007). doi:10.1677/JME-06-0053
R. Granata, M. Volante, F. Settanni, C. Gauna, C. Ghe, M. Annunziata, B. Deidda, I. Gesmundo, T. Abribat, A.J. van der Lely, G. Muccioli, E. Ghigo, M. Papotti, Unacylated ghrelin and obestatin increase islet cell mass and prevent diabetes in streptozotocin-treated newborn rats. J. Mol. Endocrinol. 45(1), 9–17 (2010). doi:10.1677/JME-09-0141
R. Granata, F. Settanni, M. Julien, R. Nano, G. Togliatto, A. Trombetta, D. Gallo, L. Piemonti, M.F. Brizzi, T. Abribat, A.J. van Der Lely, E. Ghigo, Des-acyl ghrelin fragments and analogues promote survival of pancreatic beta-cells and human pancreatic islets and prevent diabetes in streptozotocin-treated rats. J. Med. Chem. 55(6), 2585–2596 (2012). doi:10.1021/jm201223m
S. Bonner-Weir, D.F. Trent, G.C. Weir, Partial pancreatectomy in the rat and subsequent defect in glucose-induced insulin release. J. Clin. Investig. 71(6), 1544–1553 (1983)
R. Rafaeloff, G.L. Pittenger, S.W. Barlow, X.F. Qin, B. Yan, L. Rosenberg, W.P. Duguid, A.I. Vinik, Cloning and sequencing of the pancreatic islet neogenesis associated protein (INGAP) gene and its expression in islet neogenesis in hamsters. J. Clin. Investig. 99(9), 2100–2109 (1997). doi:10.1172/JCI119383
D. Gu, N. Sarvetnick, Epithelial cell proliferation and islet neogenesis in IFN-g transgenic mice. Development 118(1), 33–46 (1993)
J.F. List, J.F. Habener, Glucagon-like peptide 1 agonists and the development and growth of pancreatic beta-cells. Am. J. Physiol. Endocrinol. Metab. 286(6), E875–E881 (2004). doi:10.1152/ajpendo.00007.2004
F. Sanvito, P.L. Herrera, J. Huarte, A. Nichols, R. Montesano, L. Orci, J.D. Vassalli, TGF-beta 1 influences the relative development of the exocrine and endocrine pancreas in vitro. Development 120(12), 3451–3462 (1994)
Y.Q. Zhang, H. Zhang, A. Maeshima, H. Kurihara, J. Miyagawa, T. Takeuchi, I. Kojima, Up-regulation of the expression of activins in the pancreatic duct by reduction of the beta-cell mass. Endocrinology 143(9), 3540–3547 (2002). doi:10.1210/en.2002-220089
J.T. Hill, T.L. Mastracci, C. Vinton, M.L. Doyle, K.R. Anderson, Z.L. Loomis, J.M. Schrunk, A.D. Minic, K.R. Prabakar, A. Pugliese, Y. Sun, R.G. Smith, L. Sussel, Ghrelin is dispensable for embryonic pancreatic islet development and differentiation. Regul. Pept. 157(1–3), 51–56 (2009). doi:10.1016/j.regpep.2009.02.013
N. Wierup, S. Yang, R.J. McEvilly, H. Mulder, F. Sundler, Ghrelin is expressed in a novel endocrine cell type in developing rat islets and inhibits insulin secretion from INS-1 (832/13) cells. J. Histochem. Cytochem. 52(3), 301–310 (2004)
M. Kerem, B. Salman, S. Ozsoy, H. Pasaoglu, A. Bedirli, R. Haziroglu, T.U. Yilmaz, Exogenous ghrelin enhances endocrine and exocrine regeneration in pancreatectomized rats. J. Gastrointest. Surg. 13(4), 775–783 (2009). doi:10.1007/s11605-008-0778-2
J.H. Nielsen, E.D. Galsgaard, A. Moldrup, B.N. Friedrichsen, N. Billestrup, J.A. Hansen, Y.C. Lee, C. Carlsson, Regulation of beta-cell mass by hormones and growth factors. Diabetes 50(Suppl 1), S25–S29 (2001)
X. Ma, Y. Lin, L. Lin, G. Qin, F.A. Pereira, M.W. Haymond, N.F. Butte, Y. Sun, Ablation of ghrelin receptor in leptin-deficient ob/ob mice has paradoxical effects on glucose homeostasis when compared with ablation of ghrelin in ob/ob mice. Am. J. Physiol. Endocrinol. Metab. 303(3), E422–E431 (2012). doi:10.1152/ajpendo.00576.2011
A. Salehi, C. de la Dornonville Cour, R. Hakanson, I. Lundquist, Effects of ghrelin on insulin and glucagon secretion: a study of isolated pancreatic islets and intact mice. Regul. Pept. 118(3), 143–150 (2004). doi:10.1016/j.regpep.2003.12.001
H.M. Lee, G. Wang, E.W. Englander, M. Kojima, G.H. Greeley Jr, Ghrelin, a new gastrointestinal endocrine peptide that stimulates insulin secretion: enteric distribution, ontogeny, influence of endocrine, and dietary manipulations. Endocrinology 143(1), 185–190 (2002). doi:10.1210/endo.143.1.8602
K. Spiegel, E. Tasali, R. Leproult, N. Scherberg, E. Van Cauter, Twenty-four-hour profiles of acylated and total ghrelin: relationship with glucose levels and impact of time of day and sleep. J. Clin. Endocrinol. Metab. 96(2), 486–493 (2011). doi:10.1210/jc.2010-1978
A.C. Heijboer, A.M. van den Hoek, E.T. Parlevliet, L.M. Havekes, J.A. Romijn, H. Pijl, E.P. Corssmit, Ghrelin differentially affects hepatic and peripheral insulin sensitivity in mice. Diabetologia 49(4), 732–738 (2006). doi:10.1007/s00125-006-0138-2
K.M. Heppner, J. Tong, H. Kirchner, R. Nass, M.H. Tschop, The ghrelin O-acyltransferase-ghrelin system: a novel regulator of glucose metabolism. Curr. Opin. Endocrinol. Diabetes Obes. 18(1), 50–55 (2011). doi:10.1097/MED.0b013e328341e1d3
H. Iwakura, K. Hosoda, C. Son, J. Fujikura, T. Tomita, M. Noguchi, H. Ariyasu, K. Takaya, H. Masuzaki, Y. Ogawa, T. Hayashi, G. Inoue, T. Akamizu, H. Hosoda, M. Kojima, H. Itoh, S. Toyokuni, K. Kangawa, K. Nakao, Analysis of rat insulin II promoter-ghrelin transgenic mice and rat glucagon promoter-ghrelin transgenic mice. J. Biol. Chem. 280(15), 15247–15256 (2005). doi:10.1074/jbc.M411358200
C. Gauna, F.M. Meyler, J.A. Janssen, P.J. Delhanty, T. Abribat, P. van Koetsveld, L.J. Hofland, F. Broglio, E. Ghigo, A.J. van der Lely, Administration of acylated ghrelin reduces insulin sensitivity, whereas the combination of acylated plus unacylated ghrelin strongly improves insulin sensitivity. J. Clin. Endocrinol. Metab. 89(10), 5035–5042 (2004). doi:10.1210/jc.2004-0363
L. Pacifico, E. Poggiogalle, F. Costantino, C. Anania, F. Ferraro, F. Chiarelli, C. Chiesa, Acylated and nonacylated ghrelin levels and their associations with insulin resistance in obese and normal weight children with metabolic syndrome. Eur. J. Endocrinol. 161(6), 861–870 (2009). doi:10.1530/EJE-09-0375
L. Huang, Y. Tong, F. Zhang, Q. Yang, D. Li, S. Xie, Y. Li, H. Cao, L. Tang, X. Zhang, N. Tong, Increased acyl ghrelin but decreased total ghrelin and unacyl ghrelin in Chinese Han people with impaired fasting glucose combined with impaired glucose tolerance. Peptides 60, 86–94 (2014). doi:10.1016/j.peptides.2014.07.022
P. Lucidi, G. Murdolo, C. Di Loreto, A. De Cicco, N. Parlanti, C. Fanelli, F. Santeusanio, G.B. Bolli, P. De Feo, Ghrelin is not necessary for adequate hormonal counterregulation of insulin-induced hypoglycemia. Diabetes 51(10), 2911–2914 (2002)
M.F. Saad, B. Bernaba, C.M. Hwu, S. Jinagouda, S. Fahmi, E. Kogosov, R. Boyadjian, Insulin regulates plasma ghrelin concentration. J. Clin. Endocrinol. Metab. 87(8), 3997–4000 (2002). doi:10.1210/jcem.87.8.8879
Q. Wang, C. Liu, A. Uchida, J.C. Chuang, A. Walker, T. Liu, S. Osborne-Lawrence, B.L. Mason, C. Mosher, E.D. Berglund, J.K. Elmquist, J.M. Zigman, Arcuate AgRP neurons mediate orexigenic and glucoregulatory actions of ghrelin. Mol. Metab. 3(1), 64–72 (2014). doi:10.1016/j.molmet.2013.10.001
V. De Gennaro-Colonna, G. Rossoni, D. Cocchi, A.E. Rigamonti, F. Berti, E.E. Muller, Endocrine, metabolic and cardioprotective effects of hexarelin in obese Zucker rats. J. Endocrinol. 166(3), 529–536 (2000)
R.M. Frieboes, H. Murck, P. Maier, T. Schier, F. Holsboer, A. Steiger, Growth hormone-releasing peptide-6 stimulates sleep, growth hormone, ACTH and cortisol release in normal man. Neuroendocrinology 61(5), 584–589 (1995)
R.G. Clark, G.B. Thomas, D.L. Mortensen, W.B. Won, Y.H. Ma, E.E. Tomlinson, K.M. Fairhall, I.C. Robinson, Growth hormone secretagogues stimulate the hypothalamic-pituitary-adrenal axis and are diabetogenic in the Zucker diabetic fatty rat. Endocrinology 138(10), 4316–4323 (1997). doi:10.1210/endo.138.10.5424
Y. Date, M. Nakazato, S. Hashiguchi, K. Dezaki, M.S. Mondal, H. Hosoda, M. Kojima, K. Kangawa, T. Arima, H. Matsuo, T. Yada, S. Matsukura, Ghrelin is present in pancreatic alpha-cells of humans and rats and stimulates insulin secretion. Diabetes 51(1), 124–129 (2002)
E. Adeghate, A.S. Ponery, Ghrelin stimulates insulin secretion from the pancreas of normal and diabetic rats. J. Neuroendocrinol. 14(7), 555–560 (2002)
K. Dezaki, H. Hosoda, M. Kakei, S. Hashiguchi, M. Watanabe, K. Kangawa, T. Yada, Endogenous ghrelin in pancreatic islets restricts insulin release by attenuating Ca2 + signaling in beta-cells: implication in the glycemic control in rodents. Diabetes 53(12), 3142–3151 (2004)
K. Dezaki, H. Sone, M. Koizumi, M. Nakata, M. Kakei, H. Nagai, H. Hosoda, K. Kangawa, T. Yada, Blockade of pancreatic islet-derived ghrelin enhances insulin secretion to prevent high-fat diet-induced glucose intolerance. Diabetes 55(12), 3486–3493 (2006). doi:10.2337/db06-0878
Y. Wang, M. Nishi, A. Doi, T. Shono, Y. Furukawa, T. Shimada, H. Furuta, H. Sasaki, K. Nanjo, Ghrelin inhibits insulin secretion through the AMPK-UCP2 pathway in beta cells. FEBS Lett. 584(8), 1503–1508 (2010). doi:10.1016/j.febslet.2010.02.069
E.M. Egido, J. Rodriguez-Gallardo, R.A. Silvestre, J. Marco, Inhibitory effect of ghrelin on insulin and pancreatic somatostatin secretion. Eur. J. Endocrinol. 146(2), 241–244 (2002)
M. Colombo, S. Gregersen, J. Xiao, K. Hermansen, Effects of ghrelin and other neuropeptides (CART, MCH, orexin A and B, and GLP-1) on the release of insulin from isolated rat islets. Pancreas 27(2), 161–166 (2003)
C. Gauna, P.J. Delhanty, M.O. van Aken, J.A. Janssen, A.P. Themmen, L.J. Hofland, M. Culler, F. Broglio, E. Ghigo, A.J. van der Lely, Unacylated ghrelin is active on the INS-1E rat insulinoma cell line independently of the growth hormone secretagogue receptor type 1a and the corticotropin releasing factor 2 receptor. Mol. Cell. Endocrinol. 251(1–2), 103–111 (2006). doi:10.1016/j.mce.2006.03.040
M.K. Reimer, G. Pacini, B. Ahren, Dose-dependent inhibition by ghrelin of insulin secretion in the mouse. Endocrinology 144(3), 916–921 (2003). doi:10.1210/en.2002-220819
S.K. Chacko, M.W. Haymond, Y. Sun, J.C. Marini, P.J. Sauer, X. Ma, A.L. Sunehag, Effect of ghrelin on glucose regulation in mice. Am. J. Physiol. Endocrinol. Metab. 302(9), E1055–E1062 (2012). doi:10.1152/ajpendo.00445.2011
R.M. Kiewiet, M.O. van Aken, K. van der Weerd, P. Uitterlinden, A.P. Themmen, L.J. Hofland, Y.B. de Rijke, P.J. Delhanty, E. Ghigo, T. Abribat, A.J. van der Lely, Effects of acute administration of acylated and unacylated ghrelin on glucose and insulin concentrations in morbidly obese subjects without overt diabetes. Eur. J. Endocrinol. 161(4), 567–573 (2009). doi:10.1530/EJE-09-0339
J. Tong, R.L. Prigeon, H.W. Davis, M. Bidlingmaier, S.E. Kahn, D.E. Cummings, M.H. Tschop, D. D’Alessio, Ghrelin suppresses glucose-stimulated insulin secretion and deteriorates glucose tolerance in healthy humans. Diabetes 59(9), 2145–2151 (2010). doi:10.2337/db10-0504
F. Tassone, F. Broglio, S. Destefanis, S. Rovere, A. Benso, C. Gottero, F. Prodam, R. Rossetto, C. Gauna, A.J. van der Lely, E. Ghigo, M. Maccario, Neuroendocrine and metabolic effects of acute ghrelin administration in human obesity. J. Clin. Endocrinol. Metab. 88(11), 5478–5483 (2003). doi:10.1210/jc.2003-030564
F. Broglio, C. Gottero, A. Benso, F. Prodam, S. Destefanis, C. Gauna, M. Maccario, R. Deghenghi, A.J. van der Lely, E. Ghigo, Effects of ghrelin on the insulin and glycemic responses to glucose, arginine, or free fatty acids load in humans. J. Clin. Endocrinol. Metab. 88(9), 4268–4272 (2003). doi:10.1210/jc.2002-021940
F. Broglio, A. Benso, C. Gottero, F. Prodam, C. Gauna, L. Filtri, E. Arvat, A.J. van der Lely, R. Deghenghi, E. Ghigo, Non-acylated ghrelin does not possess the pituitaric and pancreatic endocrine activity of acylated ghrelin in humans. J. Endocrinol. Invest. 26(3), 192–196 (2003)
E.T. Vestergaard, C.B. Djurhuus, J. Gjedsted, S. Nielsen, N. Moller, J.J. Holst, J.O. Jorgensen, O. Schmitz, Acute effects of ghrelin administration on glucose and lipid metabolism. J. Clin. Endocrinol. Metab. 93(2), 438–444 (2008). doi:10.1210/jc.2007-2018
E.T. Vestergaard, L.C. Gormsen, N. Jessen, S. Lund, T.K. Hansen, N. Moller, J.O. Jorgensen, Ghrelin infusion in humans induces acute insulin resistance and lipolysis independent of growth hormone signaling. Diabetes 57(12), 3205–3210 (2008). doi:10.2337/db08-0025
S.S. Damjanovic, N.M. Lalic, P.M. Pesko, M.S. Petakov, A. Jotic, D. Miljic, K.S. Lalic, L. Lukic, M. Djurovic, V.B. Djukic, Acute effects of ghrelin on insulin secretion and glucose disposal rate in gastrectomized patients. J. Clin. Endocrinol. Metab. 91(7), 2574–2581 (2006). doi:10.1210/jc.2005-1482
E.T. Vestergaard, N. Moller, J.O. Jorgensen, Acute peripheral tissue effects of ghrelin on interstitial levels of glucose, glycerol, and lactate: a microdialysis study in healthy human subjects. Am. J. Physiol. Endocrinol. Metab. 304(12), E1273–E1280 (2013). doi:10.1152/ajpendo.00662.2012
A.D. Patel, S.A. Stanley, K.G. Murphy, G.S. Frost, J.V. Gardiner, A.S. Kent, N.E. White, M.A. Ghatei, S.R. Bloom, Ghrelin stimulates insulin-induced glucose uptake in adipocytes. Regul. Pept. 134(1), 17–22 (2006). doi:10.1016/j.regpep.2005.11.001
V. Ott, M. Fasshauer, A. Dalski, B. Meier, N. Perwitz, H.H. Klein, M. Tschop, J. Klein, Direct peripheral effects of ghrelin include suppression of adiponectin expression. Hormone Metab. Res. 34(11–12), 640–645 (2002). doi:10.1055/s-2002-38261
A. Demers, V. Caron, A. Rodrigue-Way, W. Wahli, H. Ong, A. Tremblay, A concerted kinase interplay identifies PPARgamma as a molecular target of ghrelin signaling in macrophages. PLoS ONE 4(11), e7728 (2009). doi:10.1371/journal.pone.0007728
R. Avallone, A. Demers, A. Rodrigue-Way, K. Bujold, D. Harb, S. Anghel, W. Wahli, S. Marleau, H. Ong, A. Tremblay, A growth hormone-releasing peptide that binds scavenger receptor CD36 and ghrelin receptor up-regulates sterol transporters and cholesterol efflux in macrophages through a peroxisome proliferator-activated receptor gamma-dependent pathway. Mol. Endocrinol. 20(12), 3165–3178 (2006). doi:10.1210/me.2006-0146
S. Marleau, D. Harb, K. Bujold, R. Avallone, K. Iken, Y. Wang, A. Demers, M.G. Sirois, M. Febbraio, R.L. Silverstein, A. Tremblay, H. Ong, EP 80317, a ligand of the CD36 scavenger receptor, protects apolipoprotein E-deficient mice from developing atherosclerotic lesions. FASEB J. 19(13), 1869–1871 (2005). doi:10.1096/fj.04-3253fje
M. Lehrke, M.A. Lazar, The many faces of PPARgamma. Cell 123(6), 993–999 (2005). doi:10.1016/j.cell.2005.11.026
A. Chawla, W.A. Boisvert, C.H. Lee, B.A. Laffitte, Y. Barak, S.B. Joseph, D. Liao, L. Nagy, P.A. Edwards, L.K. Curtiss, R.M. Evans, P. Tontonoz, A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol. Cell 7(1), 161–171 (2001)
A. Castrillo, P. Tontonoz, Nuclear receptors in macrophage biology: at the crossroads of lipid metabolism and inflammation. Annu. Rev. Cell Dev. Biol. 20, 455–480 (2004). doi:10.1146/annurev.cellbio.20.012103.134432
A.C. Li, K.K. Brown, M.J. Silvestre, T.M. Willson, W. Palinski, C.K. Glass, Peroxisome proliferator-activated receptor gamma ligands inhibit development of atherosclerosis in LDL receptor-deficient mice. J. Clin. Investig. 106(4), 523–531 (2000). doi:10.1172/JCI10370
A.R. Collins, W.P. Meehan, U. Kintscher, S. Jackson, S. Wakino, G. Noh, W. Palinski, W.A. Hsueh, R.E. Law, Troglitazone inhibits formation of early atherosclerotic lesions in diabetic and nondiabetic low density lipoprotein receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 21(3), 365–371 (2001)
A. Rodrigue-Way, V. Caron, S. Bilodeau, S. Keil, M. Hassan, E. Levy, G.A. Mitchell, A. Tremblay, Scavenger receptor CD36 mediates inhibition of cholesterol synthesis via activation of the PPARgamma/PGC-1alpha pathway and Insig1/2 expression in hepatocytes. FASEB J. 28(4), 1910–1923 (2014). doi:10.1096/fj.13-240168
H. Hui, F. Dotta, U. Di Mario, R. Perfetti, Role of caspases in the regulation of apoptotic pancreatic islet beta-cells death. J. Cell. Physiol. 200(2), 177–200 (2004). doi:10.1002/jcp.20021
P. Maechler, C.B. Wollheim, Mitochondrial function in normal and diabetic beta-cells. Nature 414(6865), 807–812 (2001). doi:10.1038/414807a
S. Krauss, C.Y. Zhang, L. Scorrano, L.T. Dalgaard, J. St-Pierre, S.T. Grey, B.B. Lowell, Superoxide-mediated activation of uncoupling protein 2 causes pancreatic beta cell dysfunction. J. Clin. Investig. 112(12), 1831–1842 (2003). doi:10.1172/JCI19774
M. Anello, R. Lupi, D. Spampinato, S. Piro, M. Masini, U. Boggi, S. Del Prato, A.M. Rabuazzo, F. Purrello, P. Marchetti, Functional and morphological alterations of mitochondria in pancreatic beta cells from type 2 diabetic patients. Diabetologia 48(2), 282–289 (2005). doi:10.1007/s00125-004-1627-9
C.Y. Zhang, G. Baffy, P. Perret, S. Krauss, O. Peroni, D. Grujic, T. Hagen, A.J. Vidal-Puig, O. Boss, Y.B. Kim, X.X. Zheng, M.B. Wheeler, G.I. Shulman, C.B. Chan, B.B. Lowell, Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, beta cell dysfunction, and type 2 diabetes. Cell 105(6), 745–755 (2001)
Y. Teshima, M. Akao, S.P. Jones, E. Marban, Uncoupling protein-2 overexpression inhibits mitochondrial death pathway in cardiomyocytes. Circ. Res. 93(3), 192–200 (2003). doi:10.1161/01.RES.0000085581.60197.4D
Q. Zhang, W.D. Huang, X.Y. Lv, Y.M. Yang, Ghrelin protects H9c2 cells from hydrogen peroxide-induced apoptosis through NF-kappaB and mitochondria-mediated signaling. Eur. J. Pharmacol. 654(2), 142–149 (2011). doi:10.1016/j.ejphar.2010.12.011
J. Chmielewska, D. Szczepankiewicz, M. Skrzypski, D. Kregielska, M.Z. Strowski, K.W. Nowak, Ghrelin but not obestatin regulates insulin secretion from INS1 beta cell line via UCP2-dependent mechanism. J. Biol. Regul. Homeost. Agents 24(4), 397–402 (2010)
E. ElEter, A. AlTuwaijiri, H. Hagar, M. Arafa, In vivo and in vitro antioxidant activity of ghrelin: attenuation of gastric ischemic injury in the rat. J. Gastroenterol. Hepatol. 22(11), 1791–1799 (2007). doi:10.1111/j.1440-1746.2006.04696.x
B.D. Obay, E. Tasdemir, C. Tumer, H. Bilgin, M. Atmaca, Dose dependent effects of ghrelin on pentylenetetrazole-induced oxidative stress in a rat seizure model. Peptides 29(3), 448–455 (2008). doi:10.1016/j.peptides.2007.11.020
A. Kawczynska-Drozdz, R. Olszanecki, J. Jawien, T. Brzozowski, W.W. Pawlik, R. Korbut, T.J. Guzik, Ghrelin inhibits vascular superoxide production in spontaneously hypertensive rats. Am. J. Hypertens. 19(7), 764–767 (2006). doi:10.1016/j.amjhyper.2006.01.022
S.O. Iseri, G. Sener, B. Saglam, F. Ercan, N. Gedik, B.C. Yegen, Ghrelin alleviates biliary obstruction-induced chronic hepatic injury in rats. Regul. Pept. 146(1–3), 73–79 (2008). doi:10.1016/j.regpep.2007.08.014
X. Zhang, C. Chen, A new insight of mechanisms, diagnosis and treatment of diabetic cardiomyopathy. Endocrine 41(3), 398–409 (2012). doi:10.1007/s12020-012-9623-1
S.N. Verhagen, A.M. Wassink, Y. van der Graaf, Y. Gorter, P.M. Visseren, F.L. Visseren, S.S. Group, Insulin resistance increases the occurrence of new cardiovascular events in patients with manifest arterial disease without known diabetes. The SMART study. Cardiovasc. Diabetol. 10, 100 (2011). doi:10.1186/1475-2840-10-100
E. Bonora, G. Targher, G. Formentini, F. Calcaterra, S. Lombardi, F. Marini, L. Zenari, F. Saggiani, M. Poli, S. Perbellini, A. Raffaelli, L. Gemma, L. Santi, R.C. Bonadonna, M. Muggeo, The metabolic syndrome is an independent predictor of cardiovascular disease in Type 2 diabetic subjects. Prospective data from the Verona Diabetes Complications Study. Diabetic Med. 21(1), 52–58 (2004)
A. Dei Cas, V. Spigoni, V. Ridolfi, M. Metra, Diabetes and chronic heart failure: from diabetic cardiomyopathy to therapeutic approach. Endocr. Metab. Immune Disord. Drug Targets 13(1), 38–50 (2013)
L. van Heerebeek, N. Hamdani, M.L. Handoko, I. Falcao-Pires, R.J. Musters, K. Kupreishvili, A.J. Ijsselmuiden, C.G. Schalkwijk, J.G. Bronzwaer, M. Diamant, A. Borbely, J. van der Velden, G.J. Stienen, G.J. Laarman, H.W. Niessen, W.J. Paulus, Diastolic stiffness of the failing diabetic heart: importance of fibrosis, advanced glycation end products, and myocyte resting tension. Circulation 117(1), 43–51 (2008). doi:10.1161/CIRCULATIONAHA.107.728550
W.D. Gao, Y. Liu, E. Marban, Selective effects of oxygen free radicals on excitation-contraction coupling in ventricular muscle. Implications for the mechanism of stunned myocardium. Circulation 94(10), 2597–2604 (1996)
M. Zabalgoitia, M.F. Ismaeil, L. Anderson, F.A. Maklady, Prevalence of diastolic dysfunction in normotensive, asymptomatic patients with well-controlled type 2 diabetes mellitus. Am. J. Cardiol. 87(3), 320–323 (2001)
J.K. Boyer, S. Thanigaraj, K.B. Schechtman, J.E. Perez, Prevalence of ventricular diastolic dysfunction in asymptomatic, normotensive patients with diabetes mellitus. Am. J. Cardiol. 93(7), 870–875 (2004). doi:10.1016/j.amjcard.2003.12.026
Y. Ma, L. Zhang, J.N. Edwards, B.S. Launikonis, C. Chen, Growth hormone secretagogues protect mouse cardiomyocytes from in vitro ischemia/reperfusion injury through regulation of intracellular calcium. PLoS ONE 7(4), e35265 (2012). doi:10.1371/journal.pone.0035265
F. Berti, E. Muller, V. De GennaroColonna, G. Rossoni, Hexarelin exhibits protective activity against cardiac ischaemia in hearts from growth hormone-deficient rats. Growth Hormone IGF Res. 8(Suppl B), 149–152 (1998)
S. Frascarelli, S. Ghelardoni, S. Ronca-Testoni, R. Zucchi, Effect of ghrelin and synthetic growth hormone secretagogues in normal and ischemic rat heart. Basic Res. Cardiol. 98(6), 401–405 (2003). doi:10.1007/s00395-003-0434-7
G. Bisi, V. Podio, M.R. Valetto, F. Broglio, G. Bertuccio, G. Del Rio, E. Arvat, M.F. Boghen, R. Deghenghi, G. Muccioli, H. Ong, E. Ghigo, Acute cardiovascular and hormonal effects of GH and hexarelin, a synthetic GH-releasing peptide, in humans. J. Endocrinol. Invest. 22(4), 266–272 (1999)
X. Xu, F. Ding, J. Pang, X. Gao, R.K. Xu, W. Hao, J.M. Cao, C. Chen, Chronic administration of hexarelin attenuates cardiac fibrosis in the spontaneously hypertensive rat. Am. J. Physiol. Heart Circ. Physiol. 303(6), H703–H711 (2012). doi:10.1152/ajpheart.00257.2011
L. Chang, Y. Ren, X. Liu, W.G. Li, J. Yang, B. Geng, N.L. Weintraub, C. Tang, Protective effects of ghrelin on ischemia/reperfusion injury in the isolated rat heart. J. Cardiovasc. Pharmacol. 43(2), 165–170 (2004)
G.G. Zhang, X. Teng, Y. Liu, Y. Cai, Y.B. Zhou, X.H. Duan, J.Q. Song, Y. Shi, C.S. Tang, X.H. Yin, Y.F. Qi, Inhibition of endoplasm reticulum stress by ghrelin protects against ischemia/reperfusion injury in rat heart. Peptides 30(6), 1109–1116 (2009). doi:10.1016/j.peptides.2009.03.024
V. De Gennaro Colonna, G. Rossoni, M. Bernareggi, E.E. Muller, F. Berti, Cardiac ischemia and impairment of vascular endothelium function in hearts from growth hormone-deficient rats: protection by hexarelin. Eur. J. Pharmacol. 334(2–3), 201–207 (1997)
A. Torsello, E. Bresciani, G. Rossoni, R. Avallone, G. Tulipano, D. Cocchi, I. Bulgarelli, R. Deghenghi, F. Berti, V. Locatelli, Ghrelin plays a minor role in the physiological control of cardiac function in the rat. Endocrinology 144(5), 1787–1792 (2003). doi:10.1210/en.2002-221048
Y. Mao, T. Tokudome, I. Kishimoto, K. Otani, M. Miyazato, K. Kangawa, One dose of oral hexarelin protects chronic cardiac function after myocardial infarction. Peptides 56, 156–162 (2014). doi:10.1016/j.peptides.2014.04.004
M. Aragno, R. Mastrocola, C. Ghe, E. Arnoletti, E. Bassino, G. Alloatti, G. Muccioli, Obestatin induced recovery of myocardial dysfunction in type 1 diabetic rats: underlying mechanisms. Cardiovasc. Diabetol. 11, 129 (2012). doi:10.1186/1475-2840-11-129
Y. Mao, T. Tokudome, I. Kishimoto, K. Otani, H. Hosoda, C. Nagai, N. Minamino, M. Miyazato, K. Kangawa, Hexarelin treatment in male ghrelin knockout mice after myocardial infarction. Endocrinology 154(10), 3847–3854 (2013). doi:10.1210/en.2013-1291
Y. Ma, L. Zhang, B.S. Launikonis, C. Chen, Growth hormone secretagogues preserve the electrophysiological properties of mouse cardiomyocytes isolated from in vitro ischemia/reperfusion heart. Endocrinology 153(11), 5480–5490 (2012). doi:10.1210/en.2012-1404
J.J. Pang, R.K. Xu, X.B. Xu, J.M. Cao, C. Ni, W.L. Zhu, K. Asotra, M.C. Chen, C. Chen, Hexarelin protects rat cardiomyocytes from angiotensin II-induced apoptosis in vitro. Am. J. Physiol. Heart Circ. Physiol. 286(3), H1063–H1069 (2004). doi:10.1152/ajpheart.00648.2003
T. Soeki, K. Koshiba, T. Niki, K. Kusunose, K. Yamaguchi, H. Yamada, T. Wakatsuki, M. Shimabukuro, K. Minakuchi, I. Kishimoto, K. Kangawa, M. Sata, Effect of ghrelin on autonomic activity in healthy volunteers. Peptides 62C, 1–5 (2014). doi:10.1016/j.peptides.2014.09.015
J. Isgaard, Ghrelin and the cardiovascular system. Endocrinol. Dev. 25, 83–90 (2013). doi:10.1159/000346056
T. Soeki, I. Kishimoto, D.O. Schwenke, T. Tokudome, T. Horio, M. Yoshida, H. Hosoda, K. Kangawa, Ghrelin suppresses cardiac sympathetic activity and prevents early left ventricular remodeling in rats with myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 294(1), H426–H432 (2008). doi:10.1152/ajpheart.00643.2007
T. Soeki, T. Niki, E. Uematsu, S. Bando, T. Matsuura, K. Kusunose, T. Ise, Y. Ueda, N. Tomita, K. Yamaguchi, K. Koshiba, S. Yagi, D. Fukuda, Y. Taketani, T. Iwase, H. Yamada, T. Wakatsuki, M. Akaike, M. Shimabukuro, I. Kishimoto, K. Kangawa, M. Sata, Ghrelin protects the heart against ischemia-induced arrhythmias by preserving connexin-43 protein. Heart Vessel. 28(6), 795–801 (2013). doi:10.1007/s00380-013-0333-2
D.L. Lerner, K.A. Yamada, R.B. Schuessler, J.E. Saffitz, Accelerated onset and increased incidence of ventricular arrhythmias induced by ischemia in Cx43-deficient mice. Circulation 101(5), 547–552 (2000)
K.E. Wiley, A.P. Davenport, Comparison of vasodilators in human internal mammary artery: ghrelin is a potent physiological antagonist of endothelin-1. Br. J. Pharmacol. 136(8), 1146–1152 (2002). doi:10.1038/sj.bjp.0704815
N. Nagaya, K. Miyatake, M. Uematsu, H. Oya, W. Shimizu, H. Hosoda, M. Kojima, N. Nakanishi, H. Mori, K. Kangawa, Hemodynamic, renal, and hormonal effects of ghrelin infusion in patients with chronic heart failure. J. Clin. Endocrinol. Metab. 86(12), 5854–5859 (2001). doi:10.1210/jcem.86.12.8115
C.X. Huang, M.J. Yuan, H. Huang, G. Wu, Y. Liu, S.B. Yu, H.T. Li, T. Wang, Ghrelin inhibits post-infarct myocardial remodeling and improves cardiac function through anti-inflammation effect. Peptides 30(12), 2286–2291 (2009). doi:10.1016/j.peptides.2009.09.004
G. Bisi, V. Podio, M.R. Valetto, F. Broglio, G. Bertuccio, G. Aimaretti, E. Pelosi, G. Del Rio, G. Muccioli, H. Ong, M.F. Boghen, R. Deghenghi, E. Ghigo, Cardiac effects of hexarelin in hypopituitary adults. Eur. J. Pharmacol. 381(1), 31–38 (1999)
M. Imazio, M. Bobbio, F. Broglio, A. Benso, V. Podio, M.R. Valetto, G. Bisi, E. Ghigo, G.P. Trevi, GH-independent cardiotropic activities of hexarelin in patients with severe left ventricular dysfunction due to dilated and ischemic cardiomyopathy. Eur. J. Heart Fail. 4(2), 185–191 (2002)
M. Enomoto, N. Nagaya, M. Uematsu, H. Okumura, E. Nakagawa, F. Ono, H. Hosoda, H. Oya, M. Kojima, K. Kanmatsuse, K. Kangawa, Cardiovascular and hormonal effects of subcutaneous administration of ghrelin, a novel growth hormone-releasing peptide, in healthy humans. Clin. Sci. 105(4), 431–435 (2003). doi:10.1042/CS20030184
Y. Mao, T. Tokudome, I. Kishimoto, The cardiovascular action of hexarelin. J. Geriatr. Cardiol. 11(3), 253–258 (2014). doi:10.11909/j.issn.1671-5411.2014.03.007
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Mosa, R.M.H., Zhang, Z., Shao, R. et al. Implications of ghrelin and hexarelin in diabetes and diabetes-associated heart diseases. Endocrine 49, 307–323 (2015). https://doi.org/10.1007/s12020-015-0531-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-015-0531-z