
Abstract
Purpose of Review
Canonical growth hormone (GH)-dependent signaling is essential for growth and counterregulatory responses to hypoglycemia, but also may contribute to glucose homeostasis (even in the absence of hypoglycemia) via its impact on metabolism of carbohydrates, lipids and proteins, body composition, and cardiovascular risk profile. The aim of this review is to summarize recent data implicating GH action in metabolic control, including both IGF-1-dependent and -independent pathways, and its potential role as target for T2D therapy.
Recent Findings
Experimental blockade of the GHR can modulate glucose metabolism. Moreover, the soluble form of the GH receptor (GHR, or GHBP) was recently identified as a mediator of improvement in glycemic control in patients with T2D randomized to bariatric surgery vs. medical therapy. Reductions in GHR were accompanied by increases in plasma GH, but unchanged levels of both total and free IGF-1. Likewise, hepatic GHR expression is reduced following both RYGB and VSG in rodents.
Summary
Emerging data indicate that GH signaling is important for regulation of long-term glucose metabolism in T2D. Future studies will be required to dissect tissue-specific GH signaling and sensitivity and their contributions to systemic glucose metabolism.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Despite advances in pharmacologic options for T2D [1], achievement of durable and clinically significant diabetes control remains an elusive goal for many patients. Recent results from randomized clinical trials have demonstrated that bariatric/metabolic surgery has potent metabolic effects in patients with T2D, inducing remission and/or sustained metabolic control in patients with T2D [2,3,4,5].
The potent effects of surgically mediated intestinal rearrangement suggest that additional mechanisms contribute to systemic glucose metabolism in T2D. We recently analyzed the plasma proteome and metabolome in the fasting state in individuals randomized to surgical vs. medical therapy within the SLIMM-T2D clinical trial [6•]. Consistent with the large impact of surgery on both body weight and HbA1c, a large number of both weight-dependent and -independent protein and metabolite changes differed between surgical and medical therapy. Mediation analysis determined that the top-ranking mediator of improved glycemic control was plasma growth hormone receptor (GHR); reductions in GHR at 3 months mediated improved HbA1c at 1 year. Strikingly, GHR was more significant as a mediator than any clinical markers including body mass index.
Given these new findings suggesting that GH signaling is a potent mediator of systemic glycemic control in T2D, we now review current information about GH signaling, including both IGF-1-dependent and -independent downstream metabolic effects.
IGF-1 Dependent GH Signaling in Physiology
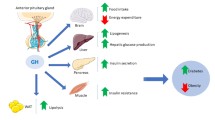
GH interacts with the cell-surface GHR, a member of the cytokine receptor superfamily. GHR is expressed in multiple tissues, including liver, muscle, adipose, heart, kidney, intestine, and bone, with greatest abundance in liver [7]. Binding of GH to its receptor results in the rapid phosphorylation/activation of Janus activating kinase 2 (JAK2) [8] and downstream activation of the signal transducers and activators of transcription (STAT) transcription factor family [9]. Of these, STAT5b is the major isoform activated by GH in the liver and is necessary for GH-stimulated insulin-like growth factor-1 (IGF-1) expression [10]. Consistent with this canonical pathway, mutation or ablation of STAT5b leads to GH insensitivity in both mice and humans [11]. Conversely, negative regulators of GHR/JAK2 signaling are not completely elucidated; however, suppressors of cytokine signaling (SOCS) and protein tyrosine phosphatases (PTP) play an important role in physiological states [12].
While IGF-1 is well-recognized as the effector of GH signaling which is critical for growth-related phenotypes, GH and IGF-1 are very distinct in terms of effects on glucose and lipid metabolism. IGF-1, which has 48% amino acid sequence identity with proinsulin, enhances insulin sensitivity in humans [13, 14]. GH (but not IGF-1) stimulates lipolysis [15], providing free fatty acids (FFA) and glycerol as substrates for energy metabolism and inhibiting insulin action to suppress hepatic gluconeogenesis [16].
In this review, we will focus primarily on IGF-1-independent GH action; we suggest that readers may wish to refer to other reviews of IGF-1-dependent effects [7, 12, 17, 18].
GH Action in Metabolism
Several lines of evidence demonstrate that GH is a potent regulator of carbohydrate metabolism in humans. Firstly, GH excess due to either acromegaly or pharmacologic use of GH in adults is associated with increased glucose levels and diabetes [19], despite increases in IGF-1. Secondly, the GHR antagonist pegvisomant improves systemic insulin action during fasting, predominantly via suppression of endogenous glucose production [20•].
Additional support is provided from animal studies. For example, mice with liver-specific IGF-1 deletion (LID) [21] have a 75% reduction in circulating IGF-1 levels and a corresponding fourfold increase in GH levels, attributed to lack of normal feedback of IGF-1. At 16 weeks of age, LID mice are severely insulin resistant in muscle, liver, and fat, with a fourfold increase in serum insulin levels. This insulin resistance was reversed by suppressing GH via GH-releasing hormone antagonist [22] or by simultaneous transgenic overexpression of GH antagonist (LID + GHa transgenic mice) [23]. In contrast, mice with deletion of the acid-labile subunit (ALSKO) demonstrate a 65% reduction in circulating IGF-1 levels, with normal GH levels and no signs of insulin resistance [22]. These studies suggest that whereas GH plays a major role in inducing insulin resistance, IGF-1 may play a modulatory role. Together, these data are consistent with the ability of GH to affect various tissues via direct, and IGF-1-independent, regulation of systemic glucose homeostasis and insulin action.
The cellular mechanisms and tissues responsible for IGF-1-independent GH action on glucose metabolism remain ill-defined. While JAK2 is the canonical kinase mediating GHR action to IGF-1 transcription, GHR is constitutively associated with another non-receptor tyrosine kinase, SRC kinase. Activated SRC phosphorylates protein kinase C (PKC) and activates extracellular signal regulated kinase (ERK1/2) signaling [23]. GHR can also activate the PI3K-AKT pathway via phosphorylation of IRS1/2, as demonstrated in mice [24]. However, studies in humans have demonstrated that GH-induced insulin resistance is independent of effects on ERK and PI3K signaling [25]. For example, a study from Denmark [26] observed that a bolus or short-term infusion of GH to achieve levels similar to either normal or twice-normal daily GH production, respectively, caused insulin resistance in skeletal muscle of healthy males; however, they were unable to detect any changes in insulin-stimulated signaling to ERK, PI3K, or AKT in skeletal muscle [27]. Moreover, insulin-stimulated AKT phosphorylation did not differ with coadministration of either GH or pegvisomant. The same group demonstrated no effect of a 0.5-mg GH bolus on PI3K activity or AKT phosphorylation in muscle or adipose tissue 30 and 60 min after GH exposure in the fasting state [28].
GH action on systemic metabolism may be mediated via adipose tissue lipolysis [29]; GH activation of the MEK–ERK pathway results in phosphorylation and inactivation of PPARγ and downregulation of FSP27. Decreased FSP27 expression, together with GH-mediated increased expression of HSL, leads to increased lipolysis and higher plasma FFA levels, a hallmark of insulin resistance and T2D. While GH also activates STAT5, a positive regulator of PPARγ, the MEK–ERK activation predominates in adipose tissue to yield net inactivation of PPARγ [30].
Other mechanisms by which GH may impact carbohydrate metabolism include modulation of IGF binding proteins (IGFBPs). While named for their ability to bind IGF-1, the six IGFBPs also impact IGF-1 tissue delivery and clearance (together with the acid-labile subunit ALS), and have independent metabolic effects as well. The effects of IGFBP2 are particularly striking. IGFBP2 circulates at high levels, largely reflecting liver-derived synthesis; however, it is also expressed in multiple tissues, where it may exert paracrine effects via IGF-1-independent mechanisms, such as signaling via cell-surface integrin receptor-mediated pathways [31]. IGFBP2 is increased in catabolic states and in response to leptin signaling [32], while it is reduced in T2D and obesity [33], and is potently suppressed by GH [34]. Experimental increases in IGFBP2 yield resistance to diet-induced obesity [35], and diabetes remission in several models, in parallel with improved hepatic insulin sensitivity. Recent data also implicate IGFBP2 as a contributor to improved metabolism after bariatric surgery [36, 37]. These changes are also concordant with reduced GH effect within IGF-1-independent pathways regulating hepatic and systemic metabolism.
Like other cytokines, GH is produced locally in many tissues (brain, many immune cells, mammary tissue, teeth, pineal gland, and placenta) [38]. The presence of GHR in virtually every cell of the body has led to the realization that GH acts not only through its classical mediator IGF-1 but also through additional intracellular signaling pathways [12]. Approximately 20% of GH localizes to the nucleus [39] in a JAK2-independent mode [40]; GH is also specifically imported into mitochondria, where has a direct metabolic effect, independent of cell surface receptors and signal transduction [41].
Analysis of liver gene expression in mutant mice with ablation of specific pathways downstream of GHR has been informative. Mice lacking JAK2 signaling (GHRBox1−/−), mice lacking STAT5 activation but with retained JAK2 and Src activation by GH (391-GHR mutants), and mice with global GHR knockout (GHR−/−) have distinct expression patterns. Of the 90 GH-regulated hepatic genes identified at high stringency, 52% were regulated specifically by the distal part of the GHR at the site of STAT5 signaling initiation, 14% were regulated by JAK2, and 27% by Src/ERK signaling. IGF-I was equally decreased in GHRBox1−/− and GHR−/− mice. Thus, the GHRBox1−/− demonstrates a set of hepatic transcripts that can be regulated by GH independently of JAK2, including the GHR itself. Likewise, some genes are regulated directly by JAK2 even in the absence of STAT5 activation, including those with roles in metabolic processes and DNA–protein/chromatin interactions. Additional studies will be required to discern the complex mechanisms mediating IGF-1-independent GH action in both physiologic and pathophysiologic states.
Another level of regulation of GH action arises from the extracellular domain of the GHR. Circulating GHBP (soluble form of GHR), cell surface GHR expression in the specific cell/tissue, and post-receptor signaling can all contribute to net GH signaling effects. Human GHBP, first described in 1986 [42], is derived from cleavage of the extracellular domain of the GHR [43]. Approximately 50% of GH circulates in a complex with GHBP, which prolongs half-life and affects bioavailability of GH. Methods to measure GHBP have evolved from chromatographic, activity-based procedures to direct immunoassays. Mutations in the extracellular domain of the GHR contribute to very low or absent GHBP levels in classical Laron syndrome [44], but normal or high levels of GHBP have been found in other forms of GH insensitivity [45]. In vitro studies have shown that GHBP inhibits multiple effects of GH, including GH binding to GHR, lipolysis, proliferation, and IGF-I expression [46, 47]. GHBP itself may even contribute to transcriptional regulation of the GHR gene, with high levels of GHBP reducing expression [48]. While the exact physiologic role of GHBP is not fully understand, it is likely that GHBP contributes to the complexity of tissue-specific GH sensitivity. Additionally, measurement of GHBP can help to differentiate between GH deficiency and GH insensitivity [49, 50].
Impact of GH Excess in Metabolism
Patients with acromegaly often have insulin resistance, hyperinsulinemia, and impaired glucose intolerance [51]. When secreted in excess, GH acts directly to block insulin action at the level of IRS-1 and PI3K [52], contributing to tissue insulin resistance. Moreover, the known lipolytic effects of GH increase flux of FFA from adipose tissue to both muscle and liver, with potential effects to reduce glucose uptake and utilization in muscle and increase lipid accumulation in both liver and muscle [16, 53]. Interestingly, administration of a GHR antagonist to individuals with acromegaly reduces IGF-1 dramatically and improves, but does not normalize, insulin sensitivity [54].
GH transgenic mice (bGH), which overexpress bovine GH, are a commonly studied model of acromegaly. Relative to controls, bGH mice are giant and lean (with ≈1.5-fold greater lean mass at both 6 weeks and 1 year of age), have increased IGF-I levels, and are insulin resistant [55]. Furthermore, they show a 12–18-month decrease in lifespan, dying primarily from kidney, heart, and liver disease (vs. wild type, with average lifespan ~ 24 months) [56].
Impact of GH Deficiency in Metabolism
An important question is what is the relative role of GH in maintaining carbohydrate and lipid homeostasis under normal conditions as compared with conditions with excessive GH secretion [57]. In contrast to GH-overexpressing transgenic mice (bGH), mice with global GHR knockout have increased white adipose tissue mass and decreased lean mass [58•]. Despite increased adiposity, these mice have low-normal glucose, low insulin levels, and increased lifespan (30–36 months) — holding the record for the longest-lived laboratory mouse! [58•]. These phenotypes are similar to those of patients with Laron syndrome, who have deletions or mutations in the GHR gene or post-receptor pathways, IGF-1 deficiency, short stature, obesity, and longevity [59]. To determine the role of GHR in adult life, Kopchick and colleagues generated an inducible GHR mutant mouse (aGHRKO) [60]. Disruption of the GHR gene at 6 weeks of age, after growth is complete, yielded an 2748% increase in GH by 9 months of age in males, with lower magnitude increases in females, but expected suppression of IGF-1 levels to approximately 9% of controls in both sexes and reduction in tissue IGF-1 levels (up to 99% in the liver, 29–57% in adipose) [60]. aGHRKO mice had significantly lower body weight and increased total adiposity starting at 8–10 weeks of age. Moreover, aGHRKO mice had reduced glucose tolerance at both 5 and 13 months of age, despite greater insulin sensitivity measured via insulin tolerance tests.
Creation of multiple tissue-specific GHR knockout mice, including liver [61, 62], fat [63, 64], muscle [65], and bone, as well as in specific cell types, such as pancreatic β-cells [66], macrophages [67], and intestinal epithelial cells [68], has underscored the distinct tissue-specific metabolic effects of GHR signaling. For example, GHR knockout in liver (LiGHRKO) [62] and in muscle (mGHRKO and MuGHRKO [65, 69], both using creatine kinase to drive Cre expression) have decreased body weight and decreased fat mass. However, another liver-specific knockout GHRLD [61] did not impact body weight, and an additional muscle line (using the Mef-2c-73 k promoter, also expressed during adulthood specifically in muscle but also a regulator of brain, bone, lymphocyte, and vascular development) [60] have increased body weight and adipose mass. Fat-specific knockouts including FaGHRKO (Fabp4 promoter) and AdGHRKO (adiponectin promoter) have increased fat mass [63, 64]. Intestinal GHRKO mice have no persistent changes in body weight or fat mass [68]. The reasons behind these model-specific effects on body composition remain uncertain.
Tissue-specific effects of GHR disruption also have divergent effects on glucose homeostasis. Liver-specific disruption of GHR (> 90% in GHRLD and > 99% in LiGHRKO) increased fasting glucose, insulin, and C-peptide, and reduced insulin sensitivity and glucose tolerance. Conversely, insulin sensitivity is increased in AdGHRKO mice and in mGHRKO/MuGHRKO males, but is reduced in mice with GHR disruption in cardiac, intestinal epithelium, macrophage, and skeletal muscle (Mef-2c-73 k). Glucose metabolism in intestinal GHRKO mice ranges from normal (in males) to impaired (in females). When placed on a high-fat diet (HFD), β-cell GHR knockout mice had decreased β-cell proliferation and mass and impaired glucose tolerance, but these differences were not present on standard chow [66]. Weak effects on glucose metabolism were observed in macrophage GHR knockouts, with impaired glucose tolerance only on a high-fat diet.
The Kopchick laboratory also disrupted cardiac GHR in 4-month-old iC-GHRKO mice [70] to avoid developmental effects of perinatal GHR gene ablation. Interestingly, these iC-GHRKO mice had decreased fat mass and improved insulin sensitivity as adults. By 12.5 months of age, however, iC-GHRKO mice no longer had significant decreases in fat mass and had developed glucose intolerance and insulin resistance. Furthermore, they had decreased insulin-stimulated Akt phosphorylation, specifically in heart and liver, but not in epididymal white adipose tissue, indicating complex tissue cross-talk.
GHR is known to be expressed in neurons that express leptin receptor, agouti-related protein (AgRP), kisspeptin receptor, proopiomelanocortin (POMC) prohormone, and steroidogenic factor-1 (SF1), all of which are involved in metabolic adaptations during food restriction. Four brain-specific GHR knockout lines (2 in leptin receptor neurons, and one each in AgRP neurons and whole brain) were created to probe GH action in the brain [71]. While body weight was reduced in AgRP GHRKO mice when subjected to food restriction [72], both body weight and length were increased in leptin receptor and whole-brain GHRKO mice [72, 73]. Leptin receptor GHRKO mice had impaired glucose homeostasis with both ad libitum and high-fat feeding. In contrast, AgRP GHRKO mice had normal glucose tolerance and insulin sensitivity, while GH action in POMC neurons was not required for glucose homeostasis or energy balance. Taken together, these results suggest that GHR in leptin receptor neurons is important for maintaining glucose homeostasis [73] while GHR in AgRP neurons is important for energy conservation during food restriction [71, 72].
The most important phenotypic changes in mice with organ/cell-specific disruption of GHR signaling are summarized in Table 1. It is important to note that the disparate results seen in knockout models can be reflective of the different time points for disruption of gene expression and metabolic measurements, and the unanticipated expression of Cre in tissues other than the target [74]. The complex tissue cross-talk of GH signaling also could be a major confounder. Future studies are still needed to discover the key tissue or cells responsible for IGF-1-independent GH action.
The GH Axis in Obesity in Humans
GH secretion, either spontaneous or evoked by provocative stimuli (insulin-induced hypoglycemia, arginine, galanin, L-dopa, clonidine, acute glucocorticoid administration, or exogenous growth hormone-releasing hormone), is markedly blunted in obesity [75] (Table 2). Individuals with obesity display reduced frequency of GH secretory bursts, a modest reduction in half-life, and a fourfold reduction in daily production rate [76, 77]. These alterations are correlated with the magnitude of obesity; daily GH secretion has been estimated to fall by 6% for each unit increase in BMI. GH responsiveness to physiological challenges such as physical exercise and sleep is also reduced in patients with obesity [78, 79]. Abdominal adiposity, particularly visceral adipose mass, is a strong negative determinant of GH secretion, with effect greater in magnitude than age, sex, or generalized obesity [80, 81].
Circulating levels of the high-affinity GH binding protein (GHBP), corresponding to the extracellular domain of the GHR, are also increased in patients with obesity [82] with positive correlations with both BMI [83] and percent body fat [84]. Reductions in GHBP after weight loss have been related to changes in fat mass rather than to insulin levels [85]; additionally, insulin secretion and sensitivity and leptin levels can also influence plasma GHBP [86].
Tissue GHR expression is also influenced by obesity. GHR mRNA expression is 2–threefold lower in omental and subcutaneous adipose of obese vs. lean women [87]. Moreover, between-depot differences in GHR seen in lean individuals (visceral > subcutaneous) are not observed in individuals with obesity. Lower GHR expression in adipocytes [87] may provide one possible explanation for the lack of efficacy of GH therapy in individuals with obesity.
Assessment of total IGF-I levels in individuals with obesity has yielded conflicting results, with normal [88, 89], low [82, 90, 91], or high [92, 93] serum concentrations. Likewise, high free IGF-I, low IGFBP-1 and IGFBP-2, and normal or high IGFBP-3 circulating levels have been described in obesity [89, 94, 95]; higher levels of free (bioavailable) IGF-1 may contribute to the normal growth in children with obesity despite markedly impaired GH secretion. On the other hand, other authors have demonstrated decreases in free IGF-1, IGFBP-1, and IGFBP-2 in adults with obesity [85].
It remains unclear whether obesity-related alterations in IGF-1 are related to differences in GH sensitivity. In one study, short-term treatment with low-dose rhGH increased IGF-I levels to a greater extent in individuals with obesity as compared to lean; the authors hypothesized that hyperinsulinism and obesity enhance sensitivity to GH [96]. A double-blind, crossover design study [97] evaluated the impact of 5 weeks of placebo or GH administration in nine women with obesity (BMI 37.0 ± 2.8 kg/m2). Obesity was associated with marked elevations in GHBP, but these were unaffected by GH. Serum IGF-1 levels were subnormal but increased significantly after GH, similar to the previous study. Serum IGFBP3 was in the normal range and increased significantly during GH treatment, resulting in increases in the IGF-1/IGFBP3 molar ratio. Serum IGFBP1, which was low with placebo, became almost completely suppressed during GH treatment. By contrast, IGF2 levels were in the normal range and remained unchanged. Thus, these patterns are complex and may vary according to the population under study; however, a consistent theme is that obesity-linked changes in the GH/IGF-1 axis are distinct from those of patients with classic GH deficiency.
Can GH Signaling via the GHR Be Modulated During Adult Life in Order to Influence Metabolism?
GHR antagonists (GHA) provide a unique tool to determine the impact of GH signaling on metabolism during adult life, after growth has been completed. GHA were initially discovered via expression of mutated GH genes in transgenic mice. Production of a modified GH molecule, containing nine amino acid substitutions (the original substitution of glycine 120 and eight mutations in site 1) and addition of several polyethylene glycol (PEG) molecules, resulted in markedly prolonged half-life of the molecule, from approximately 30 min to more than 100 h. This molecule, known as pegvisomant (PEG-hGH G120K), maintains its GHR binding and antagonistic properties [98], and thus can normalize IGF-1 levels in patients with acromegaly.
Since the GH/IGF-1 axis has been implicated in several metabolic disorders, some clinical studies have investigated the use of pegvisomant as a novel tool and potential therapeutic. Pedersen et al. [20•] recently tested the impact of GHR blockade on insulin resistance in nine obese individuals during 72 h of fasting in the presence or absence of pegvisomant. As expected, endogenous GH levels increased in response to fasting. In the presence of pegvisomant, there was a substantial reduction of serum IGF-I, indicating successful suppression of GH action. Pegvisomant also reduced fasting-induced insulin resistance, predominantly via suppression of endogenous glucose production, and augmented insulin-induced suppression of circulating lipids. Taken together, this study indicated that fasting-induced GH elevations contribute to insulin resistance in obesity and that GHR blockade could improve insulin resistance.
Another small pilot study evaluated the impact of a 4-week treatment with 20 mg pegvisomant daily in four men (ages 18–62, BMI 18–35 kg/m2) with insulin resistance but not diabetes. Insulin sensitivity was measured using hyperinsulinemic euglycemic clamp, and endogenous glucose production and lipolysis were measured using stable isotope tracers. There was a small but statistically significant decrease in appendicular fat mass post-pegvisomant, but no significant effect on insulin sensitivity, measured by hyperinsulinemic euglycemic clamp [99].
The GH/GHR/IGF-1 Axis in Diabetes
Type 1 Diabetes
Prior studies of individuals with type 1 diabetes (T1D) indicate high circulating GH, but low levels of IGF-1, suggesting hepatic GH resistance [100, 101]. To investigate the possibility that GHR/GHBP could contribute to these changes, Mercado et al. (102) measured plasma GHBP in patients with T1D and controls. Plasma GHBP levels were significantly lower in T1D, even when analysis was restricted to lean patients only. No correlations were found between GHBP and HbA1c, duration of diabetes, or plasma GH. The authors concluded that T1D is associated with low GHBP levels, potentially related to insulinopenia. Consistent with this possibility, a subsequent study also showed a significant direct relation between GHBP level and total insulin dose in T1D [103]. Frystyk et al. also demonstrated marked reduction in GHBP, free IGF-1, and IGF-2 in T1D, normal total IGF-1, IGF-2, and IGFBP3, and increased IGFBP1/2, again suggesting a role for insulinopenia [104].
Although most of the alterations in the GH axis in T1D have been described in situations of poor glycemic control, hyperglycemia does not seem to be the predominant mediator of these patterns [105]. Rather, another interesting possibility is that the route of insulin delivery and its hepatic action affects GH action. Peritoneal delivery of insulin for 12 months resulted in a slight and transient improvement in HbA1c, increases in GHBP, near-normalization of IGF-1, and normalization of IGFBP3. Thus, intraperitoneal insulin delivery, allowing primary portal venous absorption, may influence hepatic GH sensitivity.
One small study tested experimental modulation of GH signaling in patients with T1D. In this randomized, placebo-controlled, crossover study [106], 10 young adults with T1D were evaluated at baseline and after 4 weeks of treatment with either 10 mg of pegvisomant or placebo. GHR blockade improved hepatic insulin sensitivity, but had no impact on FFA levels. Future studies will be necessary to dissect the mechanisms responsible for these effect(s).
Type 2 Diabetes
Changes in the GH/GHBP axis are more complex in T2D [102]. GHBP concentrations in patients with T2D were significantly lower in a subgroup of those who were hypoinsulinemic vs. those with hyperinsulinemia, and were lower in those who were insulin-treated vs. those not requiring insulin or controls. Likewise, GHBP was correlated with insulin, proinsulin, and C-peptide and inversely with IGFBP1 in individuals with T2D [103].
In a comparative study of individuals with obesity, with or without T2D [104], IGFBP1 and total and free IGF-1 did not differ in those with or without T2D, and IGFBP3 remained elevated to a similar extent as in simple obesity. Notably, the impact of T2D on the GH/IGF system was clearly different from that of T1D.
To our knowledge, there is lack of experimental modulation of GH signaling in patients with T2D. Studies of GH blockade in more insulin-resistant individuals, such as newly diagnosed patients with T2D, may be more informative.
Impact of Acquired GH Resistance During Adult Life
While classical genetically determined growth hormone resistance causes growth defects and hypoglycemia, mild abnormalities of GH action in the adult (after completion of linear growth) yield a broader range of phenotypes [107]; such acquired GH insensitivity in the adult can result from malnutrition, liver disease, or chronic inflammatory conditions [45]. Collectively, the mechanisms underlying inflammation-induced GH resistance may be related to direct impacts on the GHR through changes in transcription and translation, as well as on downstream signaling intermediates and inflammatory cytokine receptors, potentially via SIRT1-dependent mechanisms [108, 109]. Note that acquired GH resistance can be associated with variable effects on both stature and total IGF-1, depending upon the timing of the GH resistance, additional transcriptional influences, and regulation of free IGF-1 levels. For example, total IGF-1 and IGFBP3 can be variable in the setting of malnutrition or intestinal causes of acquired GH resistance [45]. Our recent findings of reductions in circulating GHR, increases in plasma GH, and increases in IGFBP1/2 also support the possibility of acquired GH resistance as a consequence of bariatric surgery which may contribute to both weight loss and glycemic benefits [6•]. Similar patterns of reductions in GHR and increased SOCS3 expression in rodent models also support this hypothesis [110]. Future studies focusing on general or tissue-specific GH resistance will be helpful to better understand GH signaling and potentially provide novel approaches to improve insulin resistance and glucose metabolism.
Besides whole-body and tissue-specific knockout mice as stated above, multiple mouse lines with disruption of GH downstream signaling, such as JAK2, STAT5b, and SOCS2, were established in the past decade with disruption of GH downstream signaling, in the hope of discerning phenotypes linked to GH resistance [58•]. Although JAK2 and STAT5b comprise the canonical signaling pathway of GH and SOCS2 is a negative regulator of STAT5b signaling, these signaling intermediates are not specific to GH action and may also be activated by other cytokines or growth factors, making phenotypic interpretation challenging [7, 12, 111, 112]. Curiously, different doses of SOCS2 have opposite effects in GHR downstream signaling and activities in vitro [113]. Future studies to exhibit GH resistance by disrupting new and specific GH downstream signaling will be required to address these important questions.
Conclusion
The role of GH signaling as a determinant of systemic glucose metabolism is complex. Changes in GH itself, GHBP, and downstream GHR-dependent signaling via IGF-1 dependent and independent pathways add to a complex network of possible outcomes at each level. These factors are even more complex when considering the distinct tissue-specific effects of GH signaling. Nevertheless, given recent data implicating changes in GH signaling as a key mediator of changes in glucose metabolism after bariatric surgery in both humans and mice, further investigations into these important pathways are sorely needed to address key scientific questions: does potential GH resistance after bariatric surgery reflect induction of a catabolic state? What is the likely tissue responsible? Are there new GH-GHR pathway targets in specific tissues which can explain the impact of surgery on diabetes remission? Can we identify a novel therapeutic target to replace invasive bariatric surgery? These answers will come from further studies in expanded studies, as well as from the rest of the animal kingdom.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Libianto R, Ekinci EI. New agents for the treatment of type 2 diabetes. Crit Care Clin. 2019;35:315–28.
Simonson DC, Halperin F, Foster K, Vernon A, Goldfine AB. Clinical and patient-centered outcomes in obese patients with type 2 diabetes 3 years after randomization to Roux-en-Y gastric bypass surgery versus intensive lifestyle management: the SLIMM-T2D study. Diabetes Care. 2018;41:670–9.
Schauer PR, Bhatt DL, Kirwan JP, Wolski K, Aminian A, Brethauer SA, Navaneethan SD, Singh RP, Pothier CE, Nissen SE, Kashyap SR, Investigators S. Bariatric surgery versus intensive medical therapy for diabetes — 5-year outcomes. N Engl J Med. 2017;376:641–51.
Cummings DE, Arterburn DE, Westbrook EO, Kuzma JN, Stewart SD, Chan CP, Bock SN, Landers JT, Kratz M, Foster-Schubert KE, Flum DR. Gastric bypass surgery vs intensive lifestyle and medical intervention for type 2 diabetes: the CROSSROADS randomised controlled trial. Diabetologia. 2016;59:945–53.
Mingrone G, Panunzi S, De Gaetano A, Guidone C, Iaconelli A, Capristo E, Chamseddine G, Bornstein SR, Rubino F. Metabolic surgery versus conventional medical therapy in patients with type 2 diabetes: 10-year follow-up of an open-label, single-centre, randomised controlled trial. Lancet. 2021;397:293–304.
• Dreyfuss JM, Yuchi Y, Dong X, Efthymiou V, Pan H, Simonson DC, Vernon A, Halperin F, Aryal P, Konkar A, Sebastian Y, Higgs BW, Grimsby J, Rondinone CM, Kasif S, Kahn BB, Foster K, Seeley R, Goldfine A, Djordjilovic V, Patti ME. High-throughput mediation analysis of human proteome and metabolome identifies mediators of post-bariatric surgical diabetes control. Nat Commun. 2021;12:6951. This paper identified reductions in GHR as a mediator of metabolic changes after bariatric surgery in patients with T2D as well as increases in IGFBP1/2. Moreover, experimental modulation of GHR altered metabolism in hepatocytes.
Waters MJ. The growth hormone receptor. Growth Horm IGF Res. 2016;28:6–10.
Argetsinger LS, Campbell GS, Yang X, Witthuhn BA, Silvennoinen O, Ihle JN, Carter-Su C. Identification of JAK2 as a growth hormone receptor-associated tyrosine kinase. Cell. 1993;74:237–44.
Carter-Su C, Schwartz J, Smit LS. Molecular mechanism of growth hormone action. Annu Rev Physiol. 1996;58:187–207.
Chia DJ, Ono M, Woelfle J, Schlesinger-Massart M, Jiang H, Rotwein P. Characterization of distinct Stat5b binding sites that mediate growth hormone-stimulated IGF-I gene transcription. J Biol Chem. 2006;281:3190–7.
Woelfle J, Chia DJ, Rotwein P. Mechanisms of growth hormone (GH) action. Identification of conserved Stat5 binding sites that mediate GH-induced insulin-like growth factor-I gene activation. J Biol Chem. 2003;278:51261–6.
Dehkhoda F, Lee CMM, Medina J, Brooks AJ. The growth hormone receptor: mechanism of receptor activation, cell signaling, and physiological aspects. Front Endocrinol (Lausanne). 2018;9:35.
Moses AC, Young SC, Morrow LA, O’Brien M, Clemmons DR. Recombinant human insulin-like growth factor I increases insulin sensitivity and improves glycemic control in type II diabetes. Diabetes. 1996;45:91–100.
Clemmons DR, Moses AC, McKay MJ, Sommer A, Rosen DM, Ruckle J. The combination of insulin-like growth factor I and insulin-like growth factor-binding protein-3 reduces insulin requirements in insulin-dependent type 1 diabetes: evidence for in vivo biological activity. J Clin Endocrinol Metab. 2000;85:1518–24.
Kofoed EM, Hwa V, Little B, Woods KA, Buckway CK, Tsubaki J, Pratt KL, Bezrodnik L, Jasper H, Tepper A, Heinrich JJ, Rosenfeld RG. Growth hormone insensitivity associated with a STAT5b mutation. N Engl J Med. 2003;349:1139–47.
Moller N, Jorgensen JO. Effects of growth hormone on glucose, lipid, and protein metabolism in human subjects. Endocr Rev. 2009;30:152–77.
Clemmons DR. Metabolic actions of insulin-like growth factor-I in normal physiology and diabetes. Endocrinol Metab Clin N Am. 2012;41:425–43, vii-viii.
Lu M, Flanagan JU, Langley RJ, Hay MP, Perry JK. Targeting growth hormone function: strategies and therapeutic applications. Signal Transduct Target Ther. 2019;4:3.
Vila G, Jorgensen JOL, Luger A, Stalla GK. Insulin resistance in patients with acromegaly. Front Endocrinol (Lausanne). 2019;10:509.
• Pedersen MH, Svart MV, Lebeck J, Bidlingmaier M, Stodkilde-Jorgensen H, Pedersen SB, Moller N, Jessen N, Jorgensen JOL. Substrate metabolism and insulin sensitivity during fasting in obese human subjects: impact of GH blockade. J Clin Endocrinol Metab. 2017;102:1340–9. This clinical study demonstrated the short-term metabolic effects of GHR blockade with pegvisomant in humans with obesity.
Yakar S, Liu JL, Fernandez AM, Wu Y, Schally AV, Frystyk J, Chernausek SD, Mejia W, Le Roith D. Liver-specific igf-1 gene deletion leads to muscle insulin insensitivity. Diabetes. 2001;50:1110–8.
Haluzik M, Yakar S, Gavrilova O, Setser J, Boisclair Y, LeRoith D. Insulin resistance in the liver-specific IGF-1 gene-deleted mouse is abrogated by deletion of the acid-labile subunit of the IGF-binding protein-3 complex: relative roles of growth hormone and IGF-1 in insulin resistance. Diabetes. 2003;52:2483–9.
Yakar S, Setser J, Zhao H, Stannard B, Haluzik M, Glatt V, Bouxsein ML, Kopchick JJ, LeRoith D. Inhibition of growth hormone action improves insulin sensitivity in liver IGF-1-deficient mice. J Clin Invest. 2004;113:96–105.
Brooks AJ, Waters MJ. The growth hormone receptor: mechanism of activation and clinical implications. Nat Rev Endocrinol. 2010;6:515–25.
Zhu T, Ling L, Lobie PE. Identification of a JAK2-independent pathway regulating growth hormone (GH)-stimulated p44/42 mitogen-activated protein kinase activity. GH activation of Ral and phospholipase D is Src-dependent. J Biol Chem. 2002;277:45592–603.
Nielsen C, Gormsen LC, Jessen N, Pedersen SB, Moller N, Lund S, Jorgensen JO. Growth hormone signaling in vivo in human muscle and adipose tissue: impact of insulin, substrate background, and growth hormone receptor blockade. J Clin Endocrinol Metab. 2008;93:2842–50.
Jessen N, Djurhuus CB, Jorgensen JO, Jensen LS, Moller N, Lund S, Schmitz O. Evidence against a role for insulin-signaling proteins PI 3-kinase and Akt in insulin resistance in human skeletal muscle induced by short-term GH infusion. Am J Physiol Endocrinol Metab. 2005;288:E194-199.
Jorgensen JO, Jessen N, Pedersen SB, Vestergaard E, Gormsen L, Lund SA, Billestrup N. GH receptor signaling in skeletal muscle and adipose tissue in human subjects following exposure to an intravenous GH bolus. Am J Physiol Endocrinol Metab. 2006;291:E899-905.
Kopchick JJ, Berryman DE, Puri V, Lee KY, Jorgensen JOL. The effects of growth hormone on adipose tissue: old observations, new mechanisms. Nat Rev Endocrinol. 2020;16:135–46.
Sharma VM, Vestergaard ET, Jessen N, Kolind-Thomsen P, Nellemann B, Nielsen TS, Vendelbo MH, Moller N, Sharma R, Lee KY, Kopchick JJ, Jorgensen JOL, Puri V. Growth hormone acts along the PPARgamma-FSP27 axis to stimulate lipolysis in human adipocytes. Am J Physiol Endocrinol Metab. 2019;316:E34–42.
Wheatcroft SB, Kearney MT. IGF-dependent and IGF-independent actions of IGF-binding protein-1 and -2: implications for metabolic homeostasis. Trends Endocrinol Metab. 2009;20:153–62.
Yakar S, Liu JL, Stannard B, Butler A, Accili D, Sauer B, LeRoith D. Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc Natl Acad Sci U S A. 1999;96:7324–9.
Hedbacker K, Birsoy K, Wysocki RW, Asilmaz E, Ahima RS, Farooqi IS, Friedman JM. Antidiabetic effects of IGFBP2, a leptin-regulated gene. Cell Metab. 2010;11:11–22.
Munzer T, Rosen CJ, Harman SM, Pabst KM, St Clair C, Sorkin JD, Blackman MR. Effects of GH and/or sex steroids on circulating IGF-I and IGFBPs in healthy, aged women and men. Am J Physiol Endocrinol Metab. 2006;290:E1006-1013.
Wheatcroft SB, Kearney MT, Shah AM, Ezzat VA, Miell JR, Modo M, Williams SC, Cawthorn WP, Medina-Gomez G, Vidal-Puig A, Sethi JK, Crossey PA. IGF-binding protein-2 protects against the development of obesity and insulin resistance. Diabetes. 2007;56:285–94.
Al-Regaiey K, Alshubrami S, Al-Beeshi I, Alnasser T, Alwabel A, Al-Beladi H, Al-Tujjar O, Alnasser A, Alfadda AA, Iqbal M. Effects of gastric sleeve surgery on the serum levels of GH, IGF-1 and IGF-binding protein 2 in healthy obese patients. BMC Gastroenterol. 2020;20:199.
Faramia J, Hao Z, Mumphrey MB, Townsend RL, Miard S, Carreau AM, Nadeau M, Frisch F, Baraboi ED, Grenier-Larouche T, Noll C, Li M, Biertho L, Marceau S, Hould FS, Lebel S, Morrison CD, Munzberg H, Richard D, Carpentier AC, Tchernof A, Berthoud HR, Picard F. IGFBP-2 partly mediates the early metabolic improvements caused by bariatric surgery. Cell Rep Med. 2021;2:100248.
Waters MJ, Shang CA, Behncken SN, Tam SP, Li H, Shen B, Lobie PE. Growth hormone as a cytokine. Clin Exp Pharmacol Physiol. 1999;26:760–4.
Lobie PE, Mertani H, Morel G, Morales-Bustos O, Norstedt G, Waters MJ. Receptor-mediated nuclear translocation of growth hormone. J Biol Chem. 1994;269:21330–9.
Mertani HC, Raccurt M, Abbate A, Kindblom J, Tornell J, Billestrup N, Usson Y, Morel G, Lobie PE. Nuclear translocation and retention of growth hormone. Endocrinology. 2003;144:3182–95.
Ardail D, Debon A, Perret-Vivancos C, Biol-N’Garagba MC, Krantic S, Lobie PE, Morel G. Growth hormone internalization in mitochondria decreases respiratory chain activity. Neuroendocrinology. 2010;91:16–26.
Herington AC, Ymer S, Stevenson J. Identification and characterization of specific binding proteins for growth hormone in normal human sera. J Clin Invest. 1986;77:1817–23.
Mannor DA, Winer LM, Shaw MA, Baumann G. Plasma growth hormone (GH)-binding proteins: effect on GH binding to receptors and GH action. J Clin Endocrinol Metab. 1991;73:30–4.
Milward A, Metherell L, Maamra M, Barahona MJ, Wilkinson IR, Camacho-Hubner C, Savage MO, Bidlingmaier M, Clark AJ, Ross RJ, Webb SM. Growth hormone (GH) insensitivity syndrome due to a GH receptor truncated after Box1, resulting in isolated failure of STAT 5 signal transduction. J Clin Endocrinol Metab. 2004;89:1259–66.
Kurtoglu S, Hatipoglu N. Growth hormone insensitivity: diagnostic and therapeutic approaches. J Endocrinol Invest. 2016;39:19–28.
Hansen BS, Hjorth S, Welinder BS, Skriver L, De Meyts P. The growth hormone (GH)-binding protein cloned from human IM-9 lymphocytes modulates the down-regulation of GH receptors by 22- and 20-kilodalton human GH in IM-9 lymphocytes and the biological effects of the hormone in Nb2 lymphoma cells. Endocrinology. 1993;133:2809–17.
Asada N, Takahashi Y, Honjo M. Effects of 22K or 20K human growth hormone on lipolysis, leptin production in adipocytes in the presence and absence of human growth hormone binding protein. Horm Res. 2000;54:203–7.
Mullis PE, Eble A, Wagner JK, Holl RW, Silbergeld A, Laron Z. Effect of different serum concentrations of growth hormone-binding protein (GHBP) on the regulation of GH receptor/GHBP gene transcription in a human hepatoma cell line. Horm Res. 1997;47:73–80.
Amit T, Youdim MB, Hochberg Z. Clinical review 112: Does serum growth hormone (GH) binding protein reflect human GH receptor function? J Clin Endocrinol Metab. 2000;85:927–32.
Schilbach K, Bidlingmaier M. Growth hormone binding protein — physiological and analytical aspects. Best Pract Res Clin Endocrinol Metab. 2015;29:671–83.
Ezzat S, Forster MJ, Berchtold P, Redelmeier DA, Boerlin V, Harris AG. Acromegaly. Clinical and biochemical features in 500 patients. Medicine (Baltimore). 1994;73:233–40.
Clemmons DR. Roles of insulin-like growth factor-I and growth hormone in mediating insulin resistance in acromegaly. Pituitary. 2002;5:181–3.
Rizza RA, Mandarino LJ, Gerich JE. Effects of growth hormone on insulin action in man. Mechanisms of insulin resistance, impaired suppression of glucose production, and impaired stimulation of glucose utilization. Diabetes. 1982;31:663–9.
Higham CE, Rowles S, Russell-Jones D, Umpleby AM, Trainer PJ. Pegvisomant improves insulin sensitivity and reduces overnight free fatty acid concentrations in patients with acromegaly. J Clin Endocrinol Metab. 2009;94:2459–63.
Palmer AJ, Chung MY, List EO, Walker J, Okada S, Kopchick JJ, Berryman DE. Age-related changes in body composition of bovine growth hormone transgenic mice. Endocrinology. 2009;150:1353–60.
Kopchick JJ, Bellush LL, Coschigano KT. Transgenic models of growth hormone action. Annu Rev Nutr. 1999;19:437–61.
Holt RI, Simpson HL, Sonksen PH. The role of the growth hormone-insulin-like growth factor axis in glucose homeostasis. Diabet Med. 2003;20:3–15.
• Young J, Bell S, Qian Y, Hyman C, Berryman DE. Mouse models of growth hormone insensitivity. Rev Endocr Metab Disord. 2021;22:17–29. This is a comprehensive review of GH resistance and its effects on systemic metabolism.
Laron Z, Ginsberg S, Lilos P, Arbiv M, Vaisman N. Body composition in untreated adult patients with Laron syndrome (primary GH insensitivity). Clin Endocrinol (Oxf). 2006;65:114–7.
Junnila RK, Duran-Ortiz S, Suer O, Sustarsic EG, Berryman DE, List EO, Kopchick JJ. Disruption of the GH receptor gene in adult mice increases maximal lifespan in females. Endocrinology. 2016;157:4502–13.
Fan Y, Menon RK, Cohen P, Hwang D, Clemens T, DiGirolamo DJ, Kopchick JJ, Le Roith D, Trucco M, Sperling MA. Liver-specific deletion of the growth hormone receptor reveals essential role of growth hormone signaling in hepatic lipid metabolism. J Biol Chem. 2009;284:19937–44.
List EO, Berryman DE, Funk K, Jara A, Kelder B, Wang F, Stout MB, Zhi X, Sun L, White TA, LeBrasseur NK, Pirtskhalava T, Tchkonia T, Jensen EA, Zhang W, Masternak MM, Kirkland JL, Miller RA, Bartke A, Kopchick JJ. Liver-specific GH receptor gene-disrupted (LiGHRKO) mice have decreased endocrine IGF-I, increased local IGF-I, and altered body size, body composition, and adipokine profiles. Endocrinology. 2014;155:1793–805.
List EO, Berryman DE, Buchman M, Parker C, Funk K, Bell S, Duran-Ortiz S, Qian Y, Young JA, Wilson C, Slyby J, McKenna S, Jensen EA, Kopchick JJ. Adipocyte-specific GH receptor-null (AdGHRKO) mice have enhanced insulin sensitivity with reduced liver triglycerides. Endocrinology. 2019;160:68–80.
List EO, Berryman DE, Funk K, Gosney ES, Jara A, Kelder B, Wang X, Kutz L, Troike K, Lozier N, Mikula V, Lubbers ER, Zhang H, Vesel C, Junnila RK, Frank SJ, Masternak MM, Bartke A, Kopchick JJ. The role of GH in adipose tissue: lessons from adipose-specific GH receptor gene-disrupted mice. Mol Endocrinol. 2013;27:524–35.
Vijayakumar A, Wu Y, Sun H, Li X, Jeddy Z, Liu C, Schwartz GJ, Yakar S, LeRoith D. Targeted loss of GHR signaling in mouse skeletal muscle protects against high-fat diet-induced metabolic deterioration. Diabetes. 2012;61:94–103.
Wu Y, Liu C, Sun H, Vijayakumar A, Giglou PR, Qiao R, Oppenheimer J, Yakar S, LeRoith D. Growth hormone receptor regulates beta cell hyperplasia and glucose-stimulated insulin secretion in obese mice. J Clin Invest. 2011;121:2422–6.
Lu C, Kumar PA, Sun J, Aggarwal A, Fan Y, Sperling MA, Lumeng CN, Menon RK. Targeted deletion of growth hormone (GH) receptor in macrophage reveals novel osteopontin-mediated effects of GH on glucose homeostasis and insulin sensitivity in diet-induced obesity. J Biol Chem. 2013;288:15725–35.
Young JA, Jensen EA, Stevens A, Duran-Ortiz S, List EO, Berryman DE, Kopchick JJ. Characterization of an intestine-specific GH receptor knockout (IntGHRKO) mouse. Growth Horm IGF Res. 2019;46–47:5–15.
List EO, Berryman DE, Ikeno Y, Hubbard GB, Funk K, Comisford R, Young JA, Stout MB, Tchkonia T, Masternak MM, Bartke A, Kirkland JL, Miller RA, Kopchick JJ. Removal of growth hormone receptor (GHR) in muscle of male mice replicates some of the health benefits seen in global GHR−/− mice. Aging (Albany NY). 2015;7:500–12.
Jara A, Liu X, Sim D, Benner CM, Duran-Ortiz S, Qian Y, List EO, Berryman DE, Kim JK, Kopchick JJ. Cardiac-specific disruption of GH receptor alters glucose homeostasis while maintaining normal cardiac performance in adult male mice. Endocrinology. 2016;157:1929–41.
List EO, Duran-Ortiz S, Kopchick JJ. Effects of tissue-specific GH receptor knockouts in mice. Mol Cell Endocrinol. 2020;515:110919.
Furigo IC, Teixeira PDS, de Souza GO, Couto GCL, Romero GG, Perello M, Frazao R, Elias LL, Metzger M, List EO, Kopchick JJ, Donato J Jr. Growth hormone regulates neuroendocrine responses to weight loss via AgRP neurons. Nat Commun. 2019;10:662.
Cady G, Landeryou T, Garratt M, Kopchick JJ, Qi N, Garcia-Galiano D, Elias CF, Myers MG Jr, Miller RA, Sandoval DA, Sadagurski M. Hypothalamic growth hormone receptor (GHR) controls hepatic glucose production in nutrient-sensing leptin receptor (LepRb) expressing neurons. Mol Metab. 2017;6:393–405.
Lee KY, Russell SJ, Ussar S, Boucher J, Vernochet C, Mori MA, Smyth G, Rourk M, Cederquist C, Rosen ED, Kahn BB, Kahn CR. Lessons on conditional gene targeting in mouse adipose tissue. Diabetes. 2013;62:864–74.
Scacchi M, Pincelli AI, Cavagnini F. Growth hormone in obesity. Int J Obes Relat Metab Disord. 1999;23:260–71.
Iranmanesh A, Lizarralde G, Veldhuis JD. Age and relative adiposity are specific negative determinants of the frequency and amplitude of growth hormone (GH) secretory bursts and the half-life of endogenous GH in healthy men. J Clin Endocrinol Metab. 1991;73:1081–8.
Veldhuis JD, Liem AY, South S, Weltman A, Weltman J, Clemmons DA, Abbott R, Mulligan T, Johnson ML, Pincus S, et al. Differential impact of age, sex steroid hormones, and obesity on basal versus pulsatile growth hormone secretion in men as assessed in an ultrasensitive chemiluminescence assay. J Clin Endocrinol Metab. 1995;80:3209–22.
Garlaschi C, di Natale B, del Guercio MJ, Caccamo A, Gargantini L, Chiumello G. Effect of physical exercise on secretion of growth hormone, glucagon, and cortisol in obese and diabetic children. Diabetes. 1975;24:758–61.
Ferini-Strambi L, Franceschi M, Cattaneo AG, Smirne S, Calori G, Caviezel F. Sleep-related growth hormone secretion in human obesity: effect of dietary treatment. Neuroendocrinology. 1991;54:412–5.
Vahl N, Jorgensen JO, Skjaerbaek C, Veldhuis JD, Orskov H, Christiansen JS. Abdominal adiposity rather than age and sex predicts mass and regularity of GH secretion in healthy adults. Am J Physiol. 1997;272:E1108-1116.
Clasey JL, Weltman A, Patrie J, Weltman JY, Pezzoli S, Bouchard C, Thorner MO, Hartman ML. Abdominal visceral fat and fasting insulin are important predictors of 24-hour GH release independent of age, gender, and other physiological factors. J Clin Endocrinol Metab. 2001;86:3845–52.
Roelen CA, Koppeschaar HP, de Vries WR, Snel YE, Doerga ME, Zelissen PM, Thijssen JH, Blankenstein MA. Visceral adipose tissue is associated with circulating high affinity growth hormone-binding protein. J Clin Endocrinol Metab. 1997;82:760–4.
Hochberg Z, Hertz P, Colin V, Ish-Shalom S, Yeshurun D, Youdim MB, Amit T. The distal axis of growth hormone (GH) in nutritional disorders: GH-binding protein, insulin-like growth factor-I (IGF-I), and IGF-I receptors in obesity and anorexia nervosa. Metabolism. 1992;41:106–12.
Kratzsch J, Dehmel B, Pulzer F, Keller E, Englaro P, Blum WF, Wabitsch M. Increased serum GHBP levels in obese pubertal children and adolescents: relationship to body composition, leptin and indicators of metabolic disturbances. Int J Obes Relat Metab Disord. 1997;21:1130–6.
Rasmussen MH, Ho KK, Kjems L, Hilsted J. Serum growth hormone-binding protein in obesity: effect of a short-term, very low calorie diet and diet-induced weight loss. J Clin Endocrinol Metab. 1996;81:1519–24.
Fernandez-Real JM, Granada ML, Ruzafa A, Casamitjana R, Ricart W. Insulin sensitivity and secretion influence the relationship between growth hormone-binding-protein and leptin. Clin Endocrinol (Oxf). 2000;52:159–64.
Erman A, Veilleux A, Tchernof A, Goodyer CG. Human growth hormone receptor (GHR) expression in obesity: I. GHR mRNA expression in omental and subcutaneous adipose tissues of obese women. Int J Obes (Lond). 2011;35:1511–9.
Postel-Vinay MC, Saab C, Gourmelen M. Nutritional status and growth hormone-binding protein. Horm Res. 1995;44:177–81.
Nam SY, Lee EJ, Kim KR, Cha BS, Song YD, Lim SK, Lee HC, Huh KB. Effect of obesity on total and free insulin-like growth factor (IGF)-1, and their relationship to IGF-binding protein (BP)-1, IGFBP-2, IGFBP-3, insulin, and growth hormone. Int J Obes Relat Metab Disord. 1997;21:355–9.
Minuto F, Barreca A, Del Monte P, Fortini P, Resentini M, Morabito F, Giordano G. Spontaneous growth hormone and somatomedin-C/insulin-like growth factor-I secretion in obese subjects during puberty. J Endocrinol Invest. 1988;11:489–95.
Skaggs SR, Crist DM. Exogenous human growth hormone reduces body fat in obese women. Horm Res. 1991;35:19–24.
Van Vliet G, Bosson D, Rummens E, Robyn C, Wolter R. Evidence against growth hormone-releasing factor deficiency in children with idiopathic obesity. Acta Endocrinol Suppl (Copenh). 1986;279:403–10.
Loche S, Cappa M, Borrelli P, Faedda A, Crino A, Cella SG, Corda R, Muller EE, Pintor C. Reduced growth hormone response to growth hormone-releasing hormone in children with simple obesity: evidence for somatomedin-C mediated inhibition. Clin Endocrinol (Oxf). 1987;27:145–53.
Frystyk J, Vestbo E, Skjaerbaek C, Mogensen CE, Orskov H. Free insulin-like growth factors in human obesity. Metabolism. 1995;44:37–44.
Argente J, Caballo N, Barrios V, Pozo J, Munoz MT, Chowen JA, Hernandez M. Multiple endocrine abnormalities of the growth hormone and insulin-like growth factor axis in prepubertal children with exogenous obesity: effect of short- and long-term weight reduction. J Clin Endocrinol Metab. 1997;82:2076–83.
Maccario M, Tassone F, Gauna C, Oleandri SE, Aimaretti G, Procopio M, Grottoli S, Pflaum CD, Strasburger CJ, Ghigo E. Effects of short-term administration of low-dose rhGH on IGF-I levels in obesity and Cushing’s syndrome: indirect evaluation of sensitivity to GH. Eur J Endocrinol. 2001;144:251–6.
Jorgensen JO, Pedersen SB, Borglum J, Frystyk J, Ho KK, Christiansen JS, Orskov H, Blum WF, Richelsen B. Serum concentrations of insulin-like growth factors (IGFs), IGF binding proteins 1 and 3 and growth hormone binding protein in obese women and the effects of growth hormone administration: a double-blind, placebo-controlled study. Eur J Endocrinol. 1995;133:65–70.
Muller AF, Kopchick JJ, Flyvbjerg A, van der Lely AJ. Clinical review 166: growth hormone receptor antagonists. J Clin Endocrinol Metab. 2004;89:1503–11.
Lee AP, Mulligan K, Schambelan M, Murphy EJ, Weiss EJ. Growth hormone receptor antagonism with pegvisomant in insulin resistant non-diabetic men: a phase II pilot study. F1000Res. 2017;6:614.
Winter RJ, Phillips LS, Klein MN, Traisman HS, Green OC. Somatomedin activity and diabetic control in children with insulin-dependent diabetes. Diabetes. 1979;28:952–4.
Rogers DG, Sherman LD, Gabbay KH. Effect of puberty on insulinlike growth factor I and HbA1 in type I diabetes. Diabetes Care. 1991;14:1031–5.
Mercado M, Molitch ME, Baumann G. Low plasma growth hormone binding protein in IDDM. Diabetes. 1992;41:605–9.
Kratzsch J, Keliner K, Zilkens T, Schmidt-Gayk H, Selisko T, Scholz GH. Growth hormone-binding protein related immunoreactivity is regulated by the degree of insulinopenia in diabetes mellitus. Clin Endocrinol (Oxf). 1996;44:673–8.
Frystyk J, Skjaerbaek C, Vestbo E, Fisker S, Orskov H. Circulating levels of free insulin-like growth factors in obese subjects: the impact of type 2 diabetes. Diabetes Metab Res Rev. 1999;15:314–22.
Hanaire-Broutin H, Sallerin-Caute B, Poncet MF, Tauber M, Bastide R, Chale JJ, Rosenfeld R, Tauber JP. Effect of intraperitoneal insulin delivery on growth hormone binding protein, insulin-like growth factor (IGF)-I, and IGF-binding protein-3 in IDDM. Diabetologia. 1996;39:1498–504.
Thankamony A, Tossavainen PH, Sleigh A, Acerini C, Elleri D, Dalton RN, Jackson NC, Umpleby AM, Williams RM, Dunger DB. Short-term administration of pegvisomant improves hepatic insulin sensitivity and reduces soleus muscle intramyocellular lipid content in young adults with type 1 diabetes. J Clin Endocrinol Metab. 2014;99:639–47.
Storr HL, Chatterjee S, Metherell LA, Foley C, Rosenfeld RG, Backeljauw PF, Dauber A, Savage MO, Hwa V. Nonclassical GH insensitivity: characterization of mild abnormalities of GH action. Endocr Rev. 2019;40:476–505.
Soendergaard C, Young JA, Kopchick JJ. Growth hormone resistance—special focus on inflammatory bowel disease. Int J Mol Sci. 2017;18(5):1019. https://doi.org/10.3390/ijms18051019.
Yamamoto M, Iguchi G, Fukuoka H, Suda K, Bando H, Takahashi M, Nishizawa H, Seino S, Takahashi Y. SIRT1 regulates adaptive response of the growth hormone—insulin-like growth factor-I axis under fasting conditions in liver. Proc Natl Acad Sci U S A. 2013;110:14948–53.
Ben-Zvi D, Meoli L, Abidi WM, Nestoridi E, Panciotti C, Castillo E, Pizarro P, Shirley E, Gourash WF, Thompson CC, Munoz R, Clish CB, Anafi RC, Courcoulas AP, Stylopoulos N. Time-dependent molecular responses differ between gastric bypass and dieting but are conserved across species. Cell Metab. 2018;28:310-323 e316.
Davey HW, Xie T, McLachlan MJ, Wilkins RJ, Waxman DJ, Grattan DR. STAT5b is required for GH-induced liver IGF-I gene expression. Endocrinology. 2001;142:3836–41.
Udy GB, Towers RP, Snell RG, Wilkins RJ, Park SH, Ram PA, Waxman DJ, Davey HW. Requirement of STAT5b for sexual dimorphism of body growth rates and liver gene expression. Proc Natl Acad Sci U S A. 1997;94:7239–44.
Greenhalgh CJ, Metcalf D, Thaus AL, Corbin JE, Uren R, Morgan PO, Fabri LJ, Zhang JG, Martin HM, Willson TA, Billestrup N, Nicola NA, Baca M, Alexander WS, Hilton DJ. Biological evidence that SOCS-2 can act either as an enhancer or suppressor of growth hormone signaling. J Biol Chem. 2002;277:40181–4.
Heidt AB, Black BL. Transgenic mice that express Cre recombinase under control of a skeletal muscle-specific promoter from mef2c. Genesis. 2005;42:28–32.
Nagarajan A, Srivastava H, Jablonsky J, Sun LY. Tissue-specific GHR knockout mice: an updated review. Front Endocrinol (Lausanne). 2020;11:579909.
Seminara S, Filpo A, La Cauza F, Faedda A, Miola A, Pellizzone S, Casati M, Loche S. Growth hormone binding protein activity in obese children. J Endocrinol Invest. 1998;21:441–4.
Saitoh H, Kamoda T, Nakahara S, Hirano T, Nakamura N. Serum concentrations of insulin, insulin-like growth factor(IGF)-I, IGF binding protein (IGFBP)-1 and -3 and growth hormone binding protein in obese children: fasting IGFBP-1 is suppressed in normoinsulinaemic obese children. Clin Endocrinol (Oxf). 1998;48:487–92.
Nam SY, Kim KR, Song YD, Lim SK, Lee HC, Huh KB. GH-binding protein in obese men with varying glucose tolerance: relationship to body fat distribution, insulin secretion and the GH-IGF-I axis. Eur J Endocrinol. 1999;140:159–63.
Arslanian SA, Menon RK, Gierl AP, Heil BV, Foley TP Jr. Insulin therapy increases low plasma growth hormone binding protein in children with new-onset type 1 diabetes. Diabet Med. 1993;10:833–8.
Holl RW, Siegler B, Scherbaum WA, Heinze E. The serum growth hormone-binding protein is reduced in young patients with insulin-dependent diabetes mellitus. J Clin Endocrinol Metab. 1993;76:165–7.
Massa G, Dooms L, Bouillon R, Vanderschueren-Lodeweyckx M. Serum levels of growth hormone-binding protein and insulin-like growth factor I in children and adolescents with type 1 (insulin-dependent) diabetes mellitus. Diabetologia. 1993;36:239–43.
Menon RK, Arslanian S, May B, Cutfield WS, Sperling MA. Diminished growth hormone-binding protein in children with insulin-dependent diabetes mellitus. J Clin Endocrinol Metab. 1992;74:934–8.
Clayton KL, Holly JM, Carlsson LM, Jones J, Cheetham TD, Taylor AM, Dunger DB. Loss of the normal relationships between growth hormone, growth hormone-binding protein and insulin-like growth factor-I in adolescents with insulin-dependent diabetes mellitus. Clin Endocrinol (Oxf). 1994;41:517–24.
Munoz MT, Barrios V, Pozo J, Argente J. Insulin-like growth factor I, its binding proteins 1 and 3, and growth hormone-binding protein in children and adolescents with insulin-dependent diabetes mellitus: clinical implications. Pediatr Res. 1996;39:992–8.
Funding
X. D. received funding from the Zhejiang Provincial Natural Science Foundation of China (Grant No. LY21H070002). L. S. received funding from National Natural Science Foundation of China (Award No. 81701367). M. E. P. received grant support related to this project from National Institutes of Health R01 DK121995 and P30 DK036836 (DRC, Joslin Diabetes Center).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics Approval
This article does not contain any studies with human or animal subjects performed by any of the authors.
Conflict of Interest
X. D. and L. S. declare that they have no conflict of interest related to this manuscript. M. E. P. reports personal fees from Astra Zeneca, Fractyl, Hanmi Pharmaceutical, MBX Biosciences, Recordati, Poxel, Eiger Pharmaceuticals, and Xeris and grants from Chan-Zuckerberg Initiative, Dexcom, and Helmsley Trust, outside the submitted work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Pathogenesis of Type 2 Diabetes and Insulin Resistance
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Dong, X., Su, L. & Patti, ME. Growth Hormone and Counterregulation in the Pathogenesis of Diabetes. Curr Diab Rep 22, 511–524 (2022). https://doi.org/10.1007/s11892-022-01488-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11892-022-01488-7