
Abstract
Diabetic nephropathy (DN) is a leading cause of end stage renal failure and there is an urgent need to identify new clinical biomarkers and targets for treatment to effectively prevent and slow the progression of the complication. Many lines of evidence show that inflammation is a cardinal pathogenetic mechanism in DN. Studies in animal models of experimental diabetes have demonstrated that there is a low-grade inflammation in the diabetic kidney. Both pharmacological and genetic strategies targeting inflammatory molecules have been shown to be beneficial in experimental DN. In vitro studies have cast light on the cellular mechanisms whereby diabetes triggers inflammation and in turn inflammation magnifies the kidney injury. Translation of this basic science knowledge into potential practical clinical applications is matter of great interest for researchers today. This review focuses on key pro-inflammatory systems implicated in the development of DN: the tumor necrosis factor(TNF)-α/TNF-α receptor system, the monocyte chemoattractant protein-1/CC-chemokine receptor-2 system, and the Endocannabinoid system that have been selected as they appear particularly promising for future clinical applications.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Diabetic nephropathy (DN) affects around 30 % of diabetic patients and is a leading cause of end stage renal failure (ESRD) in the Western World. Furthermore, the cardiovascular risk of diabetic patients raises progressively as DN develops and most of patients with DN die for cardiovascular events [1].
Increased glomerular permeability to proteins is a characteristic feature of the complication and albuminuria is a well-established clinical biomarker of DN. However, recent studies have shown that albuminuria is a less precise predictor of overt nephropathy risk than originally thought and the clinical relevance of albuminuria as a surrogate outcome in chronic kidney disease (CKD) has not been confirmed [2–4]. In addition, a substantial percentage of diabetic patients develops CKD, while remaining normoalbuminuric, and reliable biomarkers are lacking in this subset of patients [5, 6]. There is thus increasing quest to find novel clinical biomarkers, other than albuminuria, to identify individuals at risk of DN both onset and progression.
Both hyperglycemia and glomerular hypertension are key determinants in the pathogenesis of functional and structural abnormalities of DN [7–10] that comprise excessive mesangial matrix deposition, resulting in glomerulosclerosis and renal function decline, and podocyte abnormalities, leading to albuminuria. Current strategies for DN treatment focus on achieving optimal glycaemic control, blood pressure lowering, and blockade of the renin angiotensin system (RAS) [1]. The Steno-2 trial has shown that intensive multifactorial intervention, using all the currently available therapeutic strategies, was effective in reducing the progression from microalbuminuria to overt DN. However, 20 % of intensively treated patients still developed clinical nephropathy over the 7.8-year follow-up period, despite optimal treatment [11]. Therefore, it would be important to identify new mediators and thus novel potential targets for treatment to tackle this high residual risk.
Major obstacles in identifying novel biomarkers and therapeutic tools in DN are the limited availability of human histological data as kidney biopsies are rarely performed for clinical purposes, the lack of suitable experimental animal models of DN, and the existence of distinct forms of the complication (classical albuminuric/non-albuminuric DN) that likely differ in biomarkers and targets for treatment.
In recent years, growing evidence has emphasized the critical role of inflammation in both the pathogenesis and the progression of DN. As detailed below, renal monocyte infiltration occurs in both human and experimental DN. Expression of cell adhesion molecules, chemokines, and pro-inflammatory cytokines is increased in the renal tissues of diabetic patients. Strategies targeted to reduce inflammatory processes either attenuate or prevent the development of kidney injury in animal models of DN, establishing a causal relationship between inflammation and DN. Whether this line of research will lead to the identification of novel clinical biomarkers and/or therapeutic applications in humans is unknown, but there is increasing interest on its potential future clinical developments.
Micro-inflammation in DN
Inflammation is characterized by inflammatory cell accrual, increased expression of adhesion molecules, chemokines, and inflammatory cytokines. These features are seen in DN, though they are quite mild compared with classic inflammatory diseases. Therefore, the low-grade inflammation that occurs in DN is termed “microinflammation” to distinguish it from classic inflammation.
In human DN, there is a glomerular infiltration of monocytes/macrophages, which is not secondary to fibrosis as it occurs in mild and moderate glomerulosclerosis and recedes as sclerosis progresses [12]. In addition, tubulo-interstitial damage triggers an inflammatory response with mononuclear cell infiltration that is strongly related to disease progression [13]. Studies in experimental diabetes have clarified that macrophage infiltration occurs at an early stage of the disease and correlates with renal injury [14–16]. Deletion of genes encoding proteins crucial in driving renal monocyte recruitment such as the chemokine monocyte chemoattractant protein-1 and the adhesion molecule intercellular adhesion molecule-1 (ICAM-1) prevents the development of kidney injury in experimental diabetes [17, 18]. Moreover, a causal link between macrophages and renal damage has been recently established by demonstrating that macrophage depletion significantly reduces albuminuria, kidney macrophage recruitment, and glomerular histological changes and preserves expression of podocyte proteins, such as nephrin and podocin, important in maintaining glomerular permselectivity [19].
The mechanism whereby macrophages contributes to the renal damage in DN remains elusive; however, activated macrophages are capable of secreting a wide range of potentially cytotoxic products, including proteolytic enzymes and reactive oxygen species (ROS), as well as both pro-inflammatory [tumor necrosis factor-α (TNF-α), interferon-γ (IFN-γ), interleukin-1 (IL-1)], and prosclerotic [transforming growth factor-β1 (TGF-β1)] cytokines that can contribute to renal cell dysfunction/injury. Consistent with this notion, in vitro studies have shown that mesangial cell exposure to macrophage-conditioned medium increases expression of extracellular matrix components [20], such as fibronectin, collagen type IV, laminin, and tissue inhibitor of metalloproteinases, which inhibits extracellular matrix degradation. In addition, coculture of macrophages and mesangial cells leads to a synergistic increase in fibronectin production by mesangial cells [20]. Studies on cultured podocytes have demonstrated that podocyte exposure to conditioned medium from activated macrophages causes cell shrinkage, disorganization of F-actin microfilaments, loss of cell processes, and downregulation of both nephrin and podocin [21]. The specific factor secreted by macrophages, which is responsible for these alterations is often undetermined, but the pro-inflammatory cytokines TNF-α and IL-1 appear to play a predominant role. Finally, the direct physical interaction between monocytes/macrophages and glomerular cells via the adhesion molecule ICAM-1 [22] can amplify the inflammatory process by favoring glomerular monocyte infiltration through chemokine secretion and by enhancing the release of inflammatory cytokines by both cell types.
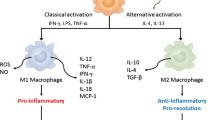
Besides enhancing renal monocyte recruitment, diabetes also affects the phenotype of macrophages. Infiltrating macrophages can polarize toward either pro-inflammatory M1 macrophages that play a key role in tissue injury or anti-inflammatory M2 macrophages that are important in tissue repair. In experimental diabetes, both subsets of macrophages are increased in the diabetic kidney, though M1 macrophages appear predominant [19, 23, 24]. In addition, a recent in vitro study has demonstrated that M1, but not M2, macrophages impair integrity of podocyte exposed to high glucose, leading to enhanced permeability to albumin [19]. Strategies that increase M2 macrophages relative to M1 at sites of kidney injury have been proposed for renal protection [25]. Notably adoptive transfer of M2 macrophages to mice with streptozotocin (STZ)-induced diabetes has been shown to decrease both renal macrophage accumulation and kidney damage [26].
Given the important role of micro-inflammation in the pathogenesis of DN, a large number of inflammatory mediators have been investigated to assess their potential relevance as clinical biomarkers and/or molecular targets in DN. These studies have clarified that some pro-inflammatory systems not only control infiltration and activation of inflammatory cells and thus indirectly contribute to the kidney damage, but they also have direct deleterious effects on resident kidney cells that occur independently of local monocyte accrual, opening an entirely new scenario in our understanding of the role of inflammatory processes in DN and in our possibility to exploit it for clinical purposes (Fig. 1). In this review, we will specifically focus on three of these pro-inflammatory systems: the tumor necrosis factor(TNF)-α/TNF-α receptor system, the monocyte chemoattractant protein-1/CC-chemokine receptor-2 system, and the endocannabinoid system (ECS) as recent studies have highlighted their potential clinical relevance in DN. TNF-α has long been implicated in the pathogenesis of DN, but recent epidemiological studies suggest that TNF-α receptors may also serve as novel biomarkers of renal function decline. Convincing preclinical data have demonstrated that blockade of the MCP-1/CCR2 system is beneficial in experimental diabetes, but phase I and II trials have only recently provided preliminary evidence of effectiveness and safety in humans. The ECS has been extensively studied in both obese and type 2 diabetic patients because of its important metabolic effects, but a previously unsuspected role of the ECS in DN has been only recently elucidated.

Central role of TNF-α and MCP-1 in the inflammatory cascade leading to diabetes-associated renal injury. Exposure of kidney cells to diabetes-related insults, such as hyperglycemia, advanced glycation end products (AGEs), glomerular hypertension, and angiotensin II (Ang-II), induces activation of the pro-inflammatory transcription factor NF-κB that enhances TNF-α and MCP-1 expression. The inflammatory cytokine TNF-α and the chemokine MCP-1 interact at multiple levels and can contribute to renal cell injury both directly by binding to their receptors exposed by renal cells (dotted red line) and indirectly through local recruitment and activation of inflammatory cells (continuous red line)
The TNF-α/TNF-α receptor system
Tumor necrosis factor α (TNF-α) is a type II transmembrane protein of 26 kDa that can be cleaved by the metalloprotease TNF-α-converting enzyme (TACE) to a 17 kDa TNF-α soluble form. TNF-α binds to TNF-α receptor 1 and 2 (TNFR1, TNFR2). TNFR1 is expressed on almost all cell types, is activated via both membrane-bound and soluble TNF-α, and is a potent inducer of apoptosis and activation of the transcription factor NF-κB. TNFR2 is expressed only in specific cell types, is predominantly activated by membrane-bound TNF-α, and induces a long-lasting NF-κB activation. Both TNFRs are shed from the cell surface and released into circulation as functional soluble forms that may represent a buffer system that prolong TNF-α biological actions or function as decoys for TNF-α [27]. Potentially relevant effects of TNF-α in DN include expression of adhesion molecules and chemokines [28], citotoxicity and apoptosis/necroptosis of susceptible cells [29, 30], alterations of intraglomerular blood flow and GFR, increased endothelial permeability [31], and induction of oxidative stress [32].
Infiltrating monocytes/macrophages are a major source of TNF-α in the diabetic kidney [33]. Furthermore, mesangial cells [34], podocytes [35], and tubular epithelial cells [36] can also release TNF-α upon stimulation and both hyperglycemia and AGE are potent TNF-α inducers in resident renal cells [37, 38]. TNFR1 is expressed by all resident kidney cells, while TNFR2 expression, which is almost undetectable in normal kidneys, raises in pathological conditions, including DN [39]. Studies in experimental diabetes have shown that TNF-α expression is increased in diabetic kidneys [40] in both the glomerulus and the tubulo-interstitium. Furthermore, administration of infliximab, a chimeric monoclonal antibody directed against TNF-α, markedly reduces albuminuria in STZ-induced diabetic rat [41]. Finally, TNF-α, inhibition with a soluble TNFR2 fusion protein (etanercept) improves the early stage of DN in the type 2 diabetic model of the KK-Ay mouse [39]. Taken together, these data provide evidence for a role of TNF-α in the pathogenesis of experimental DN.
In the kidney TNF-α can trigger and magnify the inflammatory processes by increasing the expression of adhesion molecules [42] and by inducing the release of both chemokines [43–46], and macrophage colony-stimulating factor [47]. Therefore, TNF-α is a major inducer and driver of renal micro-inflammation. Overexpression of both adhesion molecules and chemokines has also been observed in isolated glomeruli, indicating that TNF-α induces these pro-inflammatory effects by binding to TNFR exposed by resident cells [48].
TNF-α, released by either infiltrating or resident cells, can also directly contribute to the renal damage in DN and in vitro studies on glomerular cells have partially clarified the underlying cellular mechanisms. In cultured mesangial cells, TNF-α enhances oxidative stress by inducing ROS production [32], increases cytotoxicity through nitric oxide production [49], and acts synergistically with the prosclerotic cytokine TGF-β1 in promoting deposition of extracellular matrix components by increasing expression of both fibronectin and TIMP-1 [50]. The role of TNF-α in promoting oxidative stress has also been highlighted by a recent study showing that TNF-α activates NADPH oxidase in isolated glomeruli and prompts the local ROS generation via a phosphodiesterase-dependent mechanism [51]. In cultured podocytes, TNF-α lowers nephrin promoter activity leading to reduced nephrin gene expression [52] via activation of the PI3K/Akt pathway [53] and induces a rapid and reversible redistribution and loss of nephrin from the podocyte cell surface [54], probably through a reorganization of actin cytoskeleton and focal adhesions [55]. Of interest, TNF-α also compromises cell viability of podocytes through a decrease in Akt activity and this occurs specifically in podocytes from diabetic db/db mice [56]. These preclinical studies, which demonstrate the importance of TNF-α in experimental DN, have prompted studies in humans to assess the potential relevance of TNF-α as target for therapy and/or clinical biomarker.
TNF-α/TNF-α receptor system as therapeutic target
Over the last decade, a number of new ways to inhibit the actions of TNF-α have been developed including monoclonal antibodies (e.g., infliximab), circulating receptor fusion proteins (e.g., etanercept), and small molecule inhibitors (e.g., pentoxyphyline, bupropion, and 5-HT2a agonists). Among them pentoxifylline (PTF) is a methyl xanthine derivative that functions in vivo as a phosphodiesterase inhibitor with anti-TNF-α properties. Specifically, PTF inhibits both TNF-α gene transcription and TNF-α mRNA accumulation [57, 58]. Small intervention studies have shown that PTF significantly decreases proteinuria in both type 1 and type 2 diabetic patients [58, 59], and this anti-proteinuric effect was associated with a reduction in circulating TNF-α levels. A recent meta-analysis reviewing all randomized controlled trials has shown that PTF is an efficacious anti-proteinuric agent in patients with DN [60]. Finally, an open-label, randomized, 2-year-intervention trial (PREDIAN) has recently demonstrated that addition of PTF to RAS blockade reduces eGFR decline and residual albuminuria in patients with type 2 diabetes and stages 3–4 CKD [61]. However, these are small studies and additional research is needed to determine whether PTF could be a pharmacologic option for delaying or preventing the development of DN. Given that the primary role of TNF-α is the regulation of immune cells, any therapy targeting this axis would need to be extensively tested to define any side effect.
TNF-α/TNF-α receptor system as clinical biomarker
Recently, epidemiological and intervention studies in humans have shown that TNF-α and its receptors are also valuable biomarkers in DN. Both TNF-α and TNFRs are present in the circulation as soluble forms [62]. In patients with type 2 diabetes, serum TNF-α levels correlate with albuminuria [63] and urinary TNF-α levels with clinical markers of DN and disease progression [64]. In the Eurodiab study, TNF-α levels were associated with diabetic complications, including DN, and the association between NT-proBNP and diabetic complications was TNF-α-dependent [65]. Circulating TNFR2 levels were inversely and significantly correlated with eGFR in a cross-sectional study in type 2 diabetic patients [66]. A study examining serum inflammatory markers for association with GFR in type 1 diabetic patients without proteinuria has shown that both TNFRs were cross-sectionally associated with renal function decline even after adjustment for urinary albumin excretion [67].
Recently, longitudinal studies have confirmed that TNFR1 or TNFR2 are excellent predictors of progressive kidney disease in patients with a wide variety of stages and both types of diabetes [68–72] (Table 1). Type 1 diabetic patients with normo/microalbuminuria and TNFR2 levels in the highest quartile had a 55 % cumulative incidence of reaching stage 3 CKD compared with less than a 15 % incidence for patients with TNFR2 levels in the lower 3 quartiles after 12 years of follow-up [68]. In the Diabetes Control and Complications Trial (DCCT), both TNFR1 and TNFR2 were associated with an increased risk for the development of overt nephropathy [70]. Type 2 diabetic patients with proteinuria and TNFR1 levels in the highest quartile had a nearly 80 % cumulative incidence of progressing to ESKD after 12 years of follow-up, while the rate was less than 20 % in those with TNFR1 levels in the lowest 3 quartiles [69]. Collectively these data indicate that TNFRs hold great promise as biomarkers for renal function decline in diabetic patients. Consistent with this, a recent prospective study performed on the FinnDiane cohort has shown that TNRF1 is independently associated with the cumulative incidence of ESRD and has an added value as a biomarker of ESRD risk in patients with type 1 diabetes with macroalbuminuria [71]. However, prolonged longitudinal studies are needed to validate these biomarkers in a broad range of populations prior to implementation in routine diabetes management.
The MCP-1/CCR2 system
MCP-1 (also known as CCL2), the most thoroughly characterized CC chemokine, is secreted by mononuclear cells and by a variety of mesenchymal cells, including renal resident cells, and regulates the recruitment and activation of monocytes by binding to CC-chemokine receptor-2 (CCR2). In experimental settings, enhanced expression of MCP-1 has been demonstrated within the diabetic glomeruli. This occurs in an early phase of the disease, but persists during disease progression and correlates with macrophage infiltration [14, 18, 23, 73]. Upregulation of MCP-1 was also observed in tubular cells in both experimental diabetes and human DN [18, 74]. Furthermore, glomerular expression of CCR2 is strongly upregulated in renal biopsies from type 2 diabetic patients with overt nephropathy and closely correlates with the extent of proteinuria [75], indicating that in DN there is an increase in both expression of MCP-1 and responsiveness to MCP-1.
In vitro studies have clarified that diabetes-related insults, such as high glucose [76–81], advanced glycation end products (AGEs) [82–84], angiotensin II [85], mechanical stretch [85], which mimics glomerular capillary hypertension, and TGF-β1 [86, 87] are potent MCP-1 inducers in glomerular cells, providing a cellular mechanism for MCP-1 upregulation. Inflammatory cytokines also contribute as both IL-1 and TNF-α can enhance MCP-1 expression [44, 47, 88]. Furthermore, in established DN both excess mesangial matrix deposition and plasma protein leaking from injured glomeruli may further increase MCP-1 expression [89] as mesangial cell adhesion to extracellular matrix components promotes MCP-1 expression and protein overload up-regulates MCP-1 expression in tubular cells [90, 91]. The transcription factor NF-κB is a convergence point for insults inducing MCP-1 overexpression. Consistently, we have shown that a ligand of peroxisome proliferator-activated receptor-γ prevents MCP-1 secretion in response to both stretch and high glucose by inhibiting NF-κB activity [85].
Local recruitment/activation of monocytes/macrophages is considered the predominant way by which MCP-1 contributes to the pathogenesis and the progression of DN through the mechanisms described above. However, we have demonstrated that direct effects of MCP-1 on glomerular cells also play an important role [92]. The MCP-1 receptor CCR2 is exposed by resident glomerular cells both in vitro [75, 93, 94] and in vivo [75, 94, 95]. In mesangial cells MCP-1 binding to CCR2 induces ICAM-1 upregulation and has thus a direct pro-inflammatory activity [93]. Furthermore, it enhances fibronectin production through a NF-κB-TGF-β1-dependent mechanism, resulting in prosclerotic effects [96]. Finally, MCP-1 mediates at least in part high glucose-induced TGF-β1, fibronectin, and collagen type IV production [97]. There is also evidence that MCP-1 has direct deleterious effects in podocytes as activation of the CCR2 receptor by MCP-1 downregulates nephrin expression via a CCR2-Rho-kinase-dependent mechanism [75], enhances apoptosis via TGF-β1 [98], and is the mediator of high glucose-induced podocyte apoptosis [98]. MCP-1 also promotes podocyte migration [94] that is of relevance in the setting of diabetes as podocyte foot process effacement is considered a migratory event. Consistently, TGF-β has been shown to induce expression of MCP-1 in cultured podocytes and MCP-1, in turn, causes rearrangement of the actin cytoskeleton, cellular motility, and increased podocyte permeability to albumin [81]. Taken together these data indicate that MCP-1 can directly induce pleiotropic effects on resident glomerular cells that may contribute to the development of the phenotypic abnormalities characteristic of DN.
Studies in experimental diabetes have convincingly demonstrated the causative role of MCP-1 in the pathogenesis of DN. We and others have shown that deletion of the MCP-1 gene prevents the development of albuminuria and the rise in serum creatinine in STZ-induced diabetic mice [18, 75] and significantly reduces albumin leakage in obese ob/ob diabetic animals [24]. Furthermore, we found that nephrin downregulation was completely prevented [75] and overexpression of both fibronectin and collagen type IV significantly reduced in STZ-induced diabetic mice lacking MCP-1 [96], providing a mechanism for the anti-proteinuric and renoprotective effect of MCP-1 deprivation. Consistently, gene transfer of the 7ND gene, a N-terminal deletion mutant of human MCP-1, ameliorates glomerulosclerosis in iNOS transgenic diabetic mice [99] and attenuates diabetes-induced glomerular hypertrophy, and glomerulosclerosis in STZ-induced diabetic rats [100]. An alternative approach used to lower MCP-1 signalling is to block the MCP-1 receptor CCR2. In keeping with studies directly targeting MCP-1, diabetic CCR2 knockout mice show less albuminuria and reduced expression of both fibronectin and inflammatory cytokines [101]. More recently, oral CCR2 antagonists, such as RS504393, RS102895, RO5234444, TLK-19705, have been shown to ameliorate DN functional/structural alterations in both Ins2Akita mice and db/db mice by reducing macrophage infiltration, inflammation, oxidative stress, and fibrosis [102–104]. Finally, treatment with CCX140-B that, at variance with other CCR2 antagonists, does not rise MCP-1 circulating levels has been shown to ameliorate glomerular hypertrophy, podocyte loss, and renal function in experimental DN [105]. Blood glucose control is, however, improved by CCR2 antagonists and likely contributes to the clinical benefit [106]. Collectively these preclinical data strongly support the hypothesis of a key role of the MCP-1/CCR2 in the pathogenesis of DN and have open the way to studies testing potential clinical applications in humans. However, agents modulating the MCP-1/CCR2 may behave differently in two species because murine MCP-1 is similar to human MCP-1, but not functionally analogous [107].
MCP-1/CCR2 system as therapeutic target
Whether the MCP-1/CCR2 system is a potential new target for treatment in humans remains to be proven, but there are several clinical trials currently testing this hypothesis. A Phase 2 multicenter study is currently recruiting type 2 diabetic patients with overt nephropathy to evaluate both the efficacy and safety of the CCR2/5 antagonist PF-04634817 that has been previously shown to be safe in healthy subjects. CCX140-B, a specific CCR2 antagonist, has been shown to be safe in a Phase 2 clinical trial in type 2 diabetic patients with normal renal function [108] and is currently tested in two European clinical trials for safety and efficacy in type 2 diabetic patients with DN. A number of Phase 1 and Phase 2 clinical trials are ongoing to investigate the potential therapeutic benefit of anti-MCP-1 Spiegelmer NOX-E36 (an anti-MCP-1-enantiomeric RNA aptamer) [109] in DN. A randomized, double blind, and placebo-controlled Phase 2a study has been recently completed and findings reported at the Late Breaking Clinical Trials Symposium at the 2014 ERA-EDTA Conference. These unpublished results show that NOX-36 (emapticap pegol) added to standard therapy, including RAS blockade, is well tolerated and reduces both albumin/creatinine ratio and HbA1c in type 2 diabetics with albuminuria. There is thus increasing interest in this area of research and targeting the MCP-1/CCR2 system appears a promising novel therapeutic approach.
MCP-1/CCR2 system as clinical biomarker
Studies assessing the role of MCP-1 as clinical biomarker of DN have been so far disappointing. Despite increased expression of MCP-1 in renal tissue, serum MCP-1 values were comparable in diabetic patients with and without DN in most of the studies and there was no correlation between serum MCP-1 levels and either renal structural abnormalities or monocyte infiltration [76, 110, 111]. Binding to glycosaminoglycan chains on the endothelium is crucial for MCP-1 actions in vivo as it ensures high local MCP-1 concentrations and local MCP-1 immobilization in the kidney may explain the lack of elevations of circulating MCP-1 and the limited value of serum MCP-1 levels as a marker of DN [112]. On the contrary, urinary MCP-1 concentration is increased in patients with DN and strongly correlates with levels of albuminuria [76, 113, 114]. However, urinary MCP-1 raises in an advanced stage of the complication and is not predictive of either onset or worsening of albuminuria [114]. In a small study performed in macroalbuminuric patients, urinary MCP-1 levels were correlated with the rate of GFR decline [114]. However, further studies are required to assess the potential clinical relevance of MCP-1 as biomarker of renal function.
The endocannabinoid system
The two endogenous cannabinoids (ECs), anandamide and 2-arachidonoylglycerol (2-AG), bind to the endocannabinoid receptor of type 1 (CB1) and type 2 (CB2) that are coupled to G proteins. The CB1 receptor is expressed predominantly in the central nervous system [115], but is also exposed by several other cell types in peripheral tissues where it displays potent oxidative, inflammatory, and profibrotic activity [116–118]. By contrast, the CB2 receptor is mainly expressed by immune cells and has strong anti-inflammatory properties [116–119]. We have recently reported that a full EC system is present within the kidney, comprising ECs, EC receptors, and enzymes involved in EC synthesis and degradation [120, 121]. In the normal glomeruli, constitutive EC receptor expression is low for CB1 and high for CB2 and localises predominantly to podocytes [120, 121]. In addition, both receptors are also expressed by monocytes/macrophages, implying relevance to inflammatory processes, and a recent study in monocytes has shown that CB1 activates an intracellular cascade leading to inflammatory cytokine production through induction of oxidative stress that is inhibited by CB2 activation [122].
Recently, we have reported that in STZ-induced diabetes, the CB1 receptor is overexpressed within the glomeruli predominantly by podocytes [121]. On the contrary, podocyte CB2 expression was strongly downregulated in human biopsies from patients with advanced DN and the only CB2 positive cells within the glomeruli were infiltrating monocytes [120]. In early experimental diabetes, CB2 expression was still unaltered, but there was a relative deficiency of 2-AG, the major CB2 ligand, in the renal cortex [120]. Collectively, these data suggest that in DN signalling through the deleterious CB1 receptor is enhanced, while the protective CB2 signalling is reduced. The underlying cellular mechanisms are unknown; however, in vitro high glucose increased CB1 expression in podocytes [123] and mesangial cells [124], whereas mechanical stretch downregulates CB2 in cultured podocytes [125], suggesting that the major insults involved in the pathogenesis of DN can modulate the response of glomerular cells to EC. Furthermore, exposure of cultured proximal tubular epithelial cells to albumin reduces CB2 expression, suggesting that proteinuria may diminish the constitutive anti-inflammatory activity of the tubulo-interstitium in advanced DN [126].
Recently, we have provided evidence of the important role of the EC system in the pathogenesis of DN. We have shown that blocking of CB1 receptors with AM251, a selective CB1 receptor antagonist, ameliorates albuminuria by preventing the downregulation of both nephrin and podocin in STZ-induced diabetic mice [121]. Other studies have shown that treatment with selective CB1 antagonists has also renoprotective and anti-proteinuric effects in obesity-induced nephropathy [127] and db/db mice [123], though beneficial effects may be partially ascribed to amelioration of metabolic control in these models. We have also demonstrated that activation of the CB2 receptor, using the selective CB2 agonist AM1241, reduces albuminuria, glomerular monocyte infiltration, and nephrin loss in STZ-induced diabetic mice [120], indicating that a strategy compensating for the relative deficiency of CB2 signalling can be beneficial. Recently, we have further confirmed the protective role of CB2 by showing that in diabetic mice deletion of the CB2 gene worsens proteinuria, mesangial matrix expansion, renal function loss, monocytes infiltration, downregulation of slit diaphragm proteins, and overexpression of extracellular matrix components [125]. Taken together these results demonstrated that both CB1 overexpression and CB2 downregulation play an important role in the pathogenesis of experimental DN and may thus represent novel targets for treatment.
The anti-proteinuric and renoprotective effect of CB2 is likely due to inhibition of inflammatory processes and we have shown the existence of an interaction between the CB2 receptor and the MCP-1/CCR2 system. Although pharmacological/genetic modulation of CB2 does not alter MCP-1 expression, CB2 activation reduces [120], whereas CB2 deletion strongly enhances CCR2 expression [125] in the renal cortex of both diabetic and non-diabetic mice. Consistently, in both cultured podocyte [120] and monocytes [128], CCR2 expression is downregulated by CB2 agonists and upregulated by CB2 antagonists. Therefore, CB2 appears an endogenous modulator of the MCP-1/CCR2 system and may represent a physiological target of therapies aiming to lower MCP-1 signalling (Fig. 2).

Relationship between the CB2 receptor and the MCP-1/CCR2 system in diabetic nephropathy. In diabetic nephropathy, there is overexpression of the chemokine MCP-1 and downregulation of the cannabinoid receptor of type 2 (CB2) within the glomeruli. CB2 downregulation induces overexpression of the MCP-1 receptor CCR2. Both increased expression and responsiveness to MCP-1 contribute to enhance the deleterious MCP-1 signalling in the glomeruli resulting in monocyte recruitment and glomerular injury
Of interest, we have recently shown, using adaptive transfer techniques, that worsening of DN in CB2 knockout mice is due to CB2 deficiency on podocytes rather than on monocytes [125], suggesting that the predominant mechanism of kidney damage in response to MCP-1 is not monocyte recruitment, but the direct MCP-1 effect on podocytes. Lowering of inflammatory processes may also contribute to the beneficial effects of CB1 blockade observed in animal model of DN. Consistently, CB1 antagonists reduce monocyte infiltration in STZ-diabetic mice (unpublished data), as well as MCP-1 expression in db/db mice [123]. The underlying mechanism remains elusive; however, CB1 is a potent inducer of oxidative stress pathways that are strictly interconnected with inflammatory cascades. Furthermore, CB1 promotes pro-inflammatory responses of macrophages through ROS production [122] and favors macrophage polarization toward a M1 phenotype in various tissues.
The ECS as therapeutic target
As far as potential clinical applications are concerned, CB1 antagonists cross the blood brain barrier and rimonabant, the best known CB1 antagonist, was first approved for the treatment of obesity in Europe, but then withdrawn from the market because of serious central side effects, such as depression and anxiety. However, a new class of peripheral restricted CB1 antagonists has been recently developed that do not cross the blood–brain barrier and are thus devoid of psychoactive effects, while retaining their peripheral beneficial effects [129]. There is thus increasing interest on the potential use of these compounds for the treatment of diabetes and diabetes-related complications, including DN.
Selective CB2 agonists are free from central side effects and promising tools for treatment of chronic diseases associated with a low-grade inflammation. A number of pharmaceutical companies have reported entering development with CB2 agonists, such as KHK6188b, ABT-521b, APD371, and S-777469, mostly for the treatment of pain. Insofar there are no ongoing trials testing the efficacy of these compounds in DN; however, this area of research is appealing as CB2 activation has also beneficial effects in animal models of atherosclerosis [130], cardiac injury [131], and diabetic neuropathy [132], raising the possibility that CB2 agonism may have positive effects on multiple vascular bed of diabetic complications. However, in many injury models, CB2 agonists appear to be most effective when given before the initiation of the insult, and may lose their efficacy or even promote inflammation when given at later time [133]. Furthermore, there is experimental evidence that CB2 activation may worsen obesity-driven inflammation in target organ of metabolism [134]. Thus, a better understanding of the underlying mechanisms is required for the development of meaningful therapeutic approaches. Finally, given the close relationship between the two EC receptors that share ligands and have also intertwined intracellular signalling pathways, a combined approach with CB1 antagonists and CB2 agonists may result in further benefit.
The ECS as clinical biomarker
There are no published data on circulating levels of EC in patients with DN. However, plasma EC is unlikely to be good biomarkers for clinical applications as their measurement requires sophisticated techniques that are very expensive and time-consuming.
Conclusions
Over the past decade, several studies have helped to elucidate the association between inflammation and DN. It is now clear that the inflammatory milieu in diabetes contributes significantly to the development of DN. The TNF-α/TNFR systems, the MCP-1/CCR2, and the ECS play a significant role in this scenario and research on these inflammatory mediators is rapidly moving toward their validation as clinical biomarkers and/or assessment of efficacy of targeting therapies in human clinical trials.
References
American Diabetes Association, Standards of medical care in diabetes. Diabetes Care 37(suppl.1), S42–S44 (2014)
L.A. Stevens, T. Greene, A.S. Levey, Surrogate end points for clinical trials of kidney disease progression. Clin. J. Am. Soc. Nephrol. 1, 874–884 (2006)
H.J. Lambers Heerspink, D. de Zeeuw, Debate: PRO position. Should microalbuminuria ever be considered as a renal endpoint in any clinical trial? Am. J. Nephrol. 31, 458–461 (2010)
R.J. Glassock, Debate: CON position. Should microalbuminuria ever be considered as a renal endpoint in any clinical trial? Am. J. Nephrol. 31, 462–465 (2010)
R.J. Macisaac, G. Jerums, Diabetic kidney disease with and without albuminuria. Curr. Opin. Nephrol. Hypertens. 20, 246–257 (2011)
G. Penno, A. Solini, E. Bonora, C. Fondelli, E. Orsi, G. Zerbini, R. Trevisan, M. Vedovato, G. Gruden, F. Cavalot, M. Cignarelli, L. Laviola, S. Morano, A. Nicolucci, G. Pugliese, Clinical significance of nonalbuminuric renal impairment in type 2 diabetes. J. Hypertens. 29, 1802–1809 (2011)
DCCT. The effect of intensive treatment of diabetes on the development and progression of long term complications in the insulin-dependent diabetes mellitus. N. Engl. J. Med. 329, 977–86 (1993)
UK Prospective Diabetes Study, UKPDS) Group. Intensive blood glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33. Lancet 12, 837–853 (1998)
E.J. Lewis, L.G. Hunsicker, R.P. Bain, R.D. Rohde, The effect of ACE inhibition on diabetic nephropathy. N. Engl. J. Med. 329, 1456–1462 (1993)
A.S. Krolewski, M. Canessa, J.H. Warram, L.M. Laffel, A.R. Christlieb, W.C. Knowler, L.I. Rand, Predisposition to hypertension and susceptibility to renal disease in insulin-dependent diabetes mellitus. N. Engl. J. Med. 318, 140 (1988)
P. Gaede, P. Vedel, H.H. Parving, O. Pedersen, Intensified multifactorial intervention in patients with type 2 diabetes mellitus and microalbuminuria: the Steno type 2 randomised study. Lancet 20(353), 617–622 (1999)
T. Furuta, T. Saito, T. Ootaka, J. Soma, K. Obara, K. Abe, K. Yoshinaga, The role of macrophages in diabetic glomerulosclerosis. Am. J. Kidney Dis. 21, 480–485 (1993)
C. Viedt, S.R. Orth, Monocyte chemoattractant protein-1 (MCP-1) in the kidney: does it more than simply attract monocytes? Nephrol. Dial. Transplant. 17, 2043–2047 (2002)
C. Sassy-Prigent, D. Heudes, C. Mandet, M.F. Bélair, O. Michel, B. Perdereau, J. Bariéty, P. Bruneval, Early glomerular macrophage recruitment in streptozotocin-induced diabetic rats. Diabetes 49, 466–475 (2000)
H. Sugimoto, K. Shikata, K. Hirata, K. Akiyama, M. Matsuda, M. Kushiro, Y. Shikata, N. Miyatake, M. Miyasaka, H. Makino, Increased expression of intercellular adhesion molecule-1 (ICAM-1) in diabetic rat glomeruli: glomerular hyperfiltration is a potential mechanism of ICAM-1 upregulation. Diabetes 46, 2075–2081 (1997)
S. Kato, V.A. Luyckx, M. Ots, K.W. Lee, F. Ziai, J.L. Troy, B.M. Brenner, H.S. MacKenzie, Renin-angiotensin blockade lowers MCP-1 expression in diabetic rats. Kidney Int. 56, 1037–1048 (1999)
S. Okada, K. Shikata, M. Matsuda, D. Ogawa, H. Usui, Y. Kido, R. Nagase, J. Wada, Y. Shikata, H. Makino, Intercellular adhesion molecule-1—deficient mice are resistant against renal injury after induction of diabetes. Diabetes 52, 2586–2593 (2003)
F.Y. Chow, D.J. Nikolic-Paterson, E. Ozols, R.C. Atkins, B.J. Rollin, G.H. Tesch, Monocyte chemoattractant protein-1 promotes the development of diabetic renal injury in streptozotocin-treated mice. Kidney Int. 69, 73–80 (2006)
H. You, T. Gao, T.K. Cooper, W. Brian Reeves, A.S. Awad, Macrophages directly mediate diabetic renal injury. Am. J. Physiol. Renal Physiol. 305, F1719–F1727 (2013)
I.Z. Pawluczyk, K.P. Harris, Macrophages promote prosclerotic responses in cultured rat mesangial cells: a mechanism for the initiation of glomerulosclerosis. J. Am. Soc. Nephrol. 8, 1525–1536 (1997)
Y. Ikezumi, T. Suzuki, T. Karasawa, H. Kawachi, D.J. Nikolic-Paterson, M. Uchiyama, Activated macrophages down-regulate podocyte nephrin and podocin expression via stress-activated protein kinases. Biochem. Biophys. Res. Commun. 376, 706–711 (2008)
C.W. Park, J.H. Kim, J.H. Lee, Y.S. Kim, H.J. Ahn, Y.S. Shin, S.Y. Kim, E.J. Choi, Y.S. Chang, B.K. Bang, High glucose-induced intercellular adhesion molecule-1 (ICAM-1) expression through an osmotic effect in rat mesangial cells is PKC-NF-kappa B-dependent. Diabetologia 43, 1544–1553 (2000)
F. Chow, E. Ozols, D.J. Nikolic-Paterson, R.C. Atkins, G.H. Tesch, Macrophage in mouse type 2 diabetic nephropathy: correlation with diabetic state and progressive renal injury. Kidney Int. 65, 116–128 (2004)
F.Y. Chow, D.J. Nikolic-Paterson, F.Y. Ma, E. Ozols, B.J. Rollins, G.H. Tesch, Monocyte chemoattractant protein 1-induced tissue inflammation is critical for the development of renal injury but not type 2 diabetes in obese db/db mice. Diabetologia 50, 471–480 (2007)
S.D. Ricardo, H. van Goor, A.A. Eddy, Macrophage diversity in renal injury and repair. J. Clin. Invest. 118, 3522–3530 (2008)
D. Zheng, Y. Wang, Q. Cao, V.W. Lee, G. Zheng, Y. Sun, T.K. Tan, Y. Wang, S.I. Alexander, D.C. Harris, Transfused macrophages ameliorate pancreatic and renal injury in murine diabetes mellitus. Nephron Exp. Nephrol. 118, e87–e99 (2011)
P.J. Naudé, J.A. den Boer, P.G. Luiten, U.L. Eisel, Tumor necrosis factor receptor cross-talk. FEBS J. 278, 888–898 (2011)
A. Ortiz, C. Bustos, J. Alonso, R. Alcázar, M.J. López-Armada, J.J. Plaza, E. González, J. Egido, Involvement of tumor necrosis factor-alpha in the pathogenesis of experimental, and human glomerulonephritis. Adv. Nephrol. Necker Hosp. 24, 53–77 (1995)
S.M. Laster, J.G. Wood, L.R. Gooding, Tumor necrosis factor can induce both apoptotic and necrotic forms of cell lysis. J. Immunol. 141, 2629–2634 (1988)
J.J. Boyle, P.L. Weissberg, M.R. Bennett, Tumor necrosis factor-alpha promotes macrophage-induced vascular smooth muscle cell apoptosis by direct and autocrine mechanisms. Arterioscler. Thromb. Vasc. Biol. 23, 1553–1558 (2003)
J.A. McKenzie, A.J. Ridley, Roles of Rho/ROCK and MLCK in TNF-alpha-induced changes in endothelial morphology and permeability. J. Cell Physiol. 213, 221–228 (2007)
H.H. Radeke, B. Meier, N. Topley, J. Flöge, G.G. Habermehl, K. Resch, Interleukin 1-alpha and tumor necrosis factor-alpha induce oxygen radical production mesangial cells. Kidney Int. 37, 767–775 (1990)
G. Hasegawa, K. Nakano, M. Sawada, K. Uno, Y. Shibayama, K. Ienaga, M. Kondo, Possible role of tumor necrosis factor and interleukin-1 in the development of diabetic nephropathy. Kidney Int. 40, 1007–1012 (1991)
L. Baud, J.P. Oudinet, M. Bens, L. Noe, M.N. Peraldi, E. Rondeau, J. Etienne, R. Ardaillou, Production of tumor necrosis factor by rat mesangial cells in response to bacterial lipopolysaccharide. Kidney Int. 35, 1111–1118 (1989)
A.K. Hughes, P.K. Stricklett, D.E. Kohan, Shiga toxin-1 regulation of cytokine production by human glomerular epithelial cells. Nephron. 88, 14–23 (2001)
A.M. Jevnikar, D.C. Brennan, G.G. Singer, J.E. Heng, W. Maslinski, R.P. Wuthrich, L.H. Glimcher, V.E. Kelley, Stimulated kidney tubular epithelial cells express membrane associated and secreted TNF-α. Kidney Int. 40, 203–211 (1991)
X.H. Wu, S.M. Huang, W.X. Fan, W.X. Tang, H.Y. Qiu, Influence of high glucose and mannose binding lectin complement pathway activation to IL-6 and TNF-alpha’s expression by human renal glomerular endothelial cells. Sichuan Da Xue Xue Bao Yi Xue Ban. 42, 90–94. Chinese. (2011)
H. Sugimoto, K. Shikata, J. Wada, S. Horiuchi, H. Makino, Advanced glycation end products-cytokine-nitric oxide sequence pathway in the development of diabetic nephropathy: aminoguanidine ameliorates the overexpression of tumour necrosis factor-alpha and inducible nitric oxide synthase in diabetic rat glomeruli. Diabetologia 42, 878–886 (1999)
K. Omote, T. Gohda, M. Murakoshi, Y. Sasaki, S. Kazuno, T. Fujimura, M. Ishizaka, Y. Sonoda, Y. Tomino, Role of the TNF pathway in the progression of diabetic nephropathy in KK-Ay mice. Am. J. Physiol. Renal Physiol. 306, F1335–F1347 (2014)
K. Kalantarinia, A.S. Awad, H.M. Siragy, Urinary and renal interstitial concentrations of TNF-α increase prior to the rise in albuminuria in diabetic rats. Kidney Int. 64, 1208–1213 (2003)
Y. Moriwaki, T. Inokuchi, A. Yamamoto, T. Ka, Z. Tsutsumi, S. Takahashi, T. Yamamoto, Effect of TNF-α inhibition on urinary albumin excretion in experimental diabetic rats. Acta Diabetol. 44, 215–218 (2007)
R. Pai, B. Bassa, M.A. Kirschenbaum, V.S. Kamanna, TNF-alpha stimulates monocyte adhesion to glomerular mesangial cells. The role of intercellular adhesion molecule-1 gene expression and protein kinases. J. Immunol. 156, 2571–2579 (1996)
S.K. Lee, J.Y. Park, S.J. Chung, W.S. Yang, S.B. Kim, S.K. Park, J.S. Park, Chemokines, osteopontin, ICAM-1 gene expression in cultured rat mesangial cells. J. Korean Med. Sci. 13, 165–170 (1998)
A. Marfaing-Koka, O. Devergne, G. Gorgone, A. Portier, T.J. Schall, P. Galanaud, D. Emilie, Regulation of the production of the RANTES chemokine by endothelial cells. Synergistic induction by IFN-gamma plus TNF-alpha and inhibition by IL-4 and IL-13. J. Immunol. 154, 1870–1878 (1995)
V.S. Kamanna, R. Pai, B. Bassa, M.A. Kirschenbaum, Activation of mesangial cells with TNF-alpha stimulates M-CSF gene expression and monocyte proliferation: evidence for involvement of protein kinase C and protein tyrosine kinase. Biochim. Biophys. Acta 1313, 161–172 (1996)
G. Wolf, S. Aberle, F. Thaiss, P.J. Nelson, A.M. Krensky, E.G. Neilson, R.A. Stahl, TNF alpha induces expression of the chemoattractant cytokine RANTES in cultured mouse mesangial cells. Kidney Int. 44, 795–804 (1993)
C. Zoja, J.M. Wang, S. Bettoni, M. Sironi, D. Renzi, F. Chiaffarino, H.E. Abboud, J. Van Damme, A. Mantovani, G. Remuzzi, Interleukin-1 beta and tumor necrosis factor-alpha induce gene expression and production of leukocyte chemotactic factors, colony-stimulating factors, and interleukin-6 in human mesangial cells. Am. J. Pathol. 138, 991–1003 (1991)
A. Taubitz, M. Schwarz, N. Eltrich, M.T. Lindenmeyer, V. Vielhaue, Distinct contributions of TNF receptor 1 and 2 to TNF-induced glomerular inflammation in mice. PLoS One 8, e68167 (2013)
Z. Hruby, K.F. Beck, Cytotoxic effect of autocrine and macrophage derived nitric oxide on cultured rat mesangial cells. Clin. Exp. Immunol. 107, 76–82 (1997)
I.Z. Pawluczyk, K.P. Harris, Cytokine interactions promote synergistic fibronectin accumulation by mesangial cells. Kidney Int. 54, 62–70 (1998)
N. Koike, T. Takamura, S. Kaneko, Induction of reactive oxygen species from isolated rat glomeruli by protein kinase C activation and TNF- stimulation, and effects of a phosphodiesterase inhibitor. Life Sci. 80, 1721–1728 (2007)
K. Yamauchi, Y. Takano, A. Kasai, K. Hayakawa, N. Hiramatsu, N. Enomoto, J. Yao, M. Kitamura, Screening and identification of substances that regulate nephrin gene expression using engineered reporter podocytes. Kidney Int. 70, 892–900 (2006)
Y. Takano, K. Yamauchi, K. Hayakawa, N. Hiramatsu, A. Kasai, M. Okamura, M. Yokouchi, A. Shitamura, J. Yao, M. Kitamura, Transcriptional suppression of nephrin in podocytes by macrophages: role of inflammatory cytokines and involvement of the PI3K/Akt pathway. FEBS Lett. 581, 421–426 (2007)
S. Doublier, V. Ruotsalainen, G. Salvidio, E. Lupia, L. Biancone, P.G. Conaldi, P. Reponen, K. Tryggvason, G. Camussi, Nephrin redistribution on podocytes is a potential mechanism for proteinuria in patients with primary acquired nephrotic syndrome. Am. J. Pathol. 158, 1723–1731 (2001)
S.B. Koukouritaki, E.A. Vardaki, E.A. Papakonstanti, E. Lianos, C. Stournaras, D.S. Emmanouel, TNF-alpha induces actin cytoskeleton reorganization in glomerular epithelial cells involving tyrosine phosphorylation of paxillin and focal adhesion kinase. Mol. Med. 5, 382–392 (1999)
T. Tejada, P. Catanuto, A. Ijaz, J.V. Santos, X. Xia, P. Sanchez, N. Sanabria, O. Lenz, S.J. Elliot, A. Fornoni, Failure to phosphorylate AKT in podocytes from mice with early diabetic nephropathy promotes cell death. Kidney Int. 73, 1385–1393 (2008)
J. Han, P. Thompson, B. Beutler, Dexamethasone and pentoxifylline inhibit endotoxin-induced cachectin/tumor necrosis factor synthesis at separate points in the signalling pathway. J. Exp. Med. 172, 393–394 (1990)
G.M. Doherty, J.C. Jensen, H.R. Alexander, C.M. Buresh, J.A. Norton, Pentoxifylline suppression of tumor necrosis factor gene transcription. Surgery. 110, 192–198 (1991)
M.C. Thomas, Emerging drugs for managing kidney disease in patients with diabetes. Expert Opin. Emerging Drugs. 18, 55–70 (2013)
B.B. McCormick, A. Sydor, A. Akbari, D. Fergusson, S. Doucette, G. Knoll, The effect of pentoxifylline on proteinuria in diabetic kidney disease: a meta-analysis. Am. J. Kidney Dis. 52, 454–463 (2008)
J.F. Navarro-González, C. Mora-Fernández, M. Muros de Fuentes, M.L. Méndez, E. Gallego, M. Macía, N. Del Castillo, A. Rivero, N. Del Castillo, M.A. Getino, P. García, A. Jarque, J. García, Effect of pentoxifylline on renal function and urinary albumin excretion in patients with diabetic kidney disease: the PREDIAN trial. J. Am. Soc. Nephrol. (2014). doi:10.1681/ASN.2014010012
M.H. Bemelmans, L.J. van Tits, W.A. Buurman, Tumor necrosis factor: function, release and clearance. Crit. Rev. Immunol. 16, 1–11 (1996)
Y. Moriwaki, T. Yamamoto, Y. Shibutani, E. Aoki, Z. Tsutsumi, S. Takahashi, H. Okamura, M. Koga, M. Fukuchi, T. Hada, Elevated levels of interleukin-18 and tumor necrosis factor-alpha in serum of patients with type 2 diabetes mellitus: relationship with diabetic nephropathy. Metabolism. 53, 605–608 (2003)
J.F. Navarro, C. Mora, M. Muros, J. García, Urinary tumour necrosis factor-α excretion independently correlates with clinical markers of glomerular and tubulointerstitial injury in type 2 diabetic patients. Nephrol. Dial. Transplant. 21(12), 3428–3434 (2006)
G. Gruden, F. Barutta, N. Chaturvedi, C. Schalkwijk, C.D. Stehouwer, S. Pinach, M. Manzo, M. Loiacono, M. Tricarico, G. Mengozzi, D.R. Witte, J.H. Fuller, P.C. Perin, G. Bruno, NH2-terminal probrain natriuretic peptide is associated with diabetes complications in the EURODIAB Prospective Complications Study: the role of tumor necrosis factor-α. Diabetes Care 35, 1931–1936 (2012)
J. Lin, F.B. Hu, E.B. Rimm, N. Rifai, G.C. Curhan, The association of serum lipids and inflammatory biomarkers with renal function in men with type II diabetes mellitus. Kidney Int. 69, 336–342 (2006)
M.A. Niewczas, L.H. Ficociello, A.C. Johnson, W. Walker, E.T. Rosolowsky, B. Roshan, J.H. Warram, A.S. Krolewski, Serum concentrations of markers of TNFα and Fas-mediated pathways and renal function in nonproteinuric patients with type 1 diabetes. Clin. J. Am. Soc. Nephrol. 4, 62–70 (2009)
T. Gohda, M.A. Niewczas, L.H. Ficociello, W.H. Walker, J. Skupien, F. Rosetti, X. Cullere, A.C. Johnson, G. Crabtree, A.M. Smiles, T.N. Mayadas, J.H. Warram, A.S. Krolewski, Circulating TNF receptors 1 and 2 predict stage 3 CKD in type 1 diabetes. J. Am. Soc. Nephrol. 23, 516–524 (2012)
M.A. Niewczas, T. Gohda, J. Skupien, A.M. Smiles, W.H. Walker, F. Rosetti, X. Cullere, J.H. Eckfeldt, A. Doria, T.N. Mayadas, J.H. Warram, A.S. Krolewski, Circulating TNF receptors 1 and 2 predict ESRD in type 2 diabetes. J. Am. Soc. Nephrol. 23, 507–515 (2012)
M.F. Lopes-Virella, N.L. Baker, K.J. Hunt, P.A. Cleary, R. Klein, G. Virella, DCCT/EDIC Research Group.: Baseline markers of inflammation are associated with progression to macroalbuminuria in type 1 diabetic subjects. Diabetes Care 36, 2317–2323 (2013)
C. Forsblom, J. Moran, V. Harjutsalo, T. Loughman, J. Wadén, N. Tolonen, L. Thorn, M. Saraheimo, D. Gordin, P.H. Groop, M.C. Thomas, on behalf of the FinnDiane Study Group, Added value of soluble Tumor Necrosis Factor Alpha Receptor-1 as a biomarker of ESRD risk. Diabetes Care (2014). doi:10.2337/dc14-0225
J. Lin, F.B. Hu, C. Mantzoros, G.C. Curhan, Lipid and inflammatory biomarkers and kidney function decline in type 2 diabetes. Diabetologia 53, 263–267 (2010)
F.T. Lee, Z. Cao, D.M. Long, S. Panagiotopoulos, G. Jerums, M.E. Cooper, J.M. Forbes, Interactions between angiotensin II and NF-kappaB-dependent pathways in modulating macrophage infiltration in experimental diabetic nephropathy. J. Am. Soc. Nephrol. 15, 2139–2151 (2004)
S. Mezzano, A. Droguett, M.E. Burgos, L.G. Ardiles, C.A. Flores, C.A. Aros, I. Caorsi, C.P. Vío, M. Ruiz-Ortega, J. Egido, Renin-angiotensin system activation and interstitial inflammation in human diabetic nephropathy. Kidney Int. Suppl. 86, S64–S70 (2003)
E. Tarabra, S. Giunti, F. Barutta, G. Salvidio, D. Burt, G. Deferrari, R. Gambino, D. Vergola, S. Pinach, P.C. Perin, G. Camussi, G. Gruden, Effect of the monocyte chemoattractant protein-1/CC chemokine receptor 2 system on nephrin expression in streptozotocin-treated mice and human cultured podocytes. Diabetes 58, 2109–2118 (2009)
N. Banba, T. Nakamura, M. Matsumura, H. Kuroda, Y. Hattori, K. Kasai, Possible relationship of monocyte chemoattractant protein-1 with diabetic nephropathy. Kidney Int. 58, 684–690 (2000)
H. Ha, M.R. Yu, Y.J. Choi, M. Kitamura, H.B. Lee, Role of high glucose–induced nuclear factor-B activation in monocyte chemoattractant protein-1 expression by mesangial cells. J. Am. Soc. Nephrol. 13, 894–902 (2002)
M. Naito, A. Shenoy, I. Aoyama, J.S. Koopmeiners, R. Komers, H.W. Schnaper, K. Bomsztyk, High ambient glucose augments angiotensin ii-induced proinflammatory gene mrNA expression in human mesangial cells: effects of valsartan and simvastatin. Am. J. Nephrol. 30, 99–111 (2009)
C.G. Ihm, J.K. Park, S.P. Hong, T.W. Lee, B.S. Cho, M.J. Kim, D.R. Cha, H. Ha, A high glucose concentration stimulates the expression of monocyte chemotactic peptide 1 in human mesangial cells. Nephron. 79, 33–37 (1998)
S.Y. Han, G.A. So, Y.H. Jee, K.H. Han, Y.S. Kang, H.K. Kim, S.W. Kang, D.S. Han, J.Y. Han, D.R. Cha, Effect of retinoic acid in experimental diabetic nephropathy. Immunol. Cell Biol. 82, 568–576 (2004)
E.Y. Lee, C.H. Chung, C.C. Khoury, T.K. Yeo, P.E. Pyagay, A. Wang, S. Chen, The monocyte chemoattractant protein-1/CCR2 loop, inducible by TGF-beta, increases podocyte motility and albumin permeability. Am. J. Physiol. Renal Physiol. 297, F85–F94 (2009)
T. Matsui, S. Yamagishi, M. Takeuchi, S. Ueda, K. Fukami, S. Okuda, Nifedipine, a calcium channel blocker, inhibits advanced glycation end product (AGE)-elicited mesangial cell damage by suppressing AGE receptor (RAGE) expression via peroxisome proliferatoractivated receptor-gamma activation. Biochem. Biophys. Res. Commun. 385, 269–272 (2009)
T. Matsui, S. Yamagishi, S. Ueda, K. Nakamura, T. Imaizumi, M. Takeuchi, H. Inoue, Telmisartan, an angiotensin II type 1 receptor blocker, inhibits advanced glycation end-product (AGE)-induced monocyte chemoattractant protein-1 expression in mesangial cells through downregulation of receptor for AGEs via peroxisome proliferator-activated receptor-gamma activation. J. Int. Med. Res. 35, 482–489 (2007)
L. Gu, S. Hagiwara, Q. Fan, M. Tanimoto, M. Kobata, M. Yamashita, T. Nishitani, T. Gohda, Z. Ni, J. Qian, S. Horikoshi, Y. Tomino, Role of receptor for advanced glycation end-products and signalling events in advanced glycation end-product-induced monocyte chemoattractant protein-1 expression in differentiated mouse podocytes. Nephrol. Dial. Transplant. 21, 299–313 (2006)
G. Gruden, G. Setti, A. Hayward, D. Sugden, S. Duggan, D. Burt, R.E. Buckingham, L. Gnudi, G. Viberti, Mechanical stretch induces monocyte chemoattractant activity via an NF-kappaB-dependent monocyte chemoattractant protein-1-mediated pathway in human mesangial cells: inhibition by rosiglitazone. J. Am. Soc. Nephrol. 16, 688–696 (2005)
J. Cheng, M.M. Diaz Encarnacion, G.M. Warner, C.E. Gray, K.A. Nath, J.P. Grande, TGF-beta1 stimulates monocyte chemoattractant protein-1 expression in mesangial cells through a phosphodiesterase isoenzyme 4-dependent process. Am. J. Physiol. Cell Physiol. 289, C959–C970 (2005)
W. Qi, X. Chen, T.S. Polhill, S. Sumual, S. Twigg, R.E. Gilbert, C.A. Pollock, TGF-β1 induces IL-8 and MCP-1 through a connective tissue growth factor-independent pathway. Am. J. Physiol. Renal Physiol. 290, F703–F709 (2006)
B.H. Rovin, T. Yoshiumura, L. Tan, Cytokine-induced production of monocyte chemoattractant protein-1 by cultured human mesangial cells. J. Immunol. 148, 2148–2153 (1992)
Y. Watanabe, M. Tamura, A. Osajima, H. Anai, N. Kabashima, R. Serino, Y. Nakashima, Integrins induce expression of monocyte chemoattractant protein-1 via focal adhesion kinase in mesangial cells. Kidney Int. 64, 431–440 (2003)
A.A. Eddy, C.M. Giachelli, Renal expression of genes that promote interstitial inflammation and fibrosis in rats with protein-overload proteinuria. Kidney Int. 47, 1546–1557 (1995)
Y. Wang, J. Chen, L. Chen, Y.C. Tay, G.K. Rangan, D.C. Harris, Induction of monocyte chemoattractant protein-1 in proximal tubule cells by urinary protein. J. Am. Soc. Nephrol. 8, 1537–1545 (1997)
S. Giunti, Targeting the MCP-1/CCR2 system in diabetic kidney disease. Curr. Vasc. Pharmacol. 8, 849–860 (2010)
S. Giunti, F. Barutta, P.C. Perin, G. Gruden, The MCP-1/CCR2 system has direct proinflammatory effects in human mesangial cells. Kidney Int. 69, 856–863 (2006)
D. Burt, G. Salvidio, E. Tarabra, F. Barutta, S. Pinach, P. Dentelli, G. Camussi, P.C. Perin, G. Gruden, The monocyte chemoattractant protein-1/cognate CC chemokine receptor 2 system affects cell motility in cultured human podocytes. Am. J. Pathol. 171, 1789–1799 (2007)
V.H. Rao, D.T. Meehan, D. Delimont, M. Nakajima, T. Wada, M.A. Gratton, D. Cosgrove, Role for macrophage metalloelastase in glomerular basement membrane damage associated with alport syndrome. Am. J. Pathol. 169, 32–46 (2006)
S. Giunti, G.H. Tesch, S. Pinach, D.J. Burt, M.E. Cooper, P. Cavallo-Perin, G. Camussi, G. Gruden, Monocyte chemoattractant protein-1 has prosclerotic effects both in a mouse model of experimental diabetes and in vitro in human mesangial cells. Diabetologia 51, 198–207 (2008)
J. Park, D.R. Ryu, J.J. Li, D.S. Jung, S.J. Kwak, S.H. Lee, T.H. Yoo, S.H. Han, J.E. Lee, D.K. Kim, S.J. Moon, K. Kim, D.S. Han, S.W. Kang, MCP-1/CCR2 system is involved in high glucose-induced fibronectin and type IV collagen expression in cultured mesangial cells. Am. J. Physiol. Renal Physiol. 295, F749–F757 (2008)
B.Y. Nam, J. Paeng, S.H. Kim, S.H. Lee, H. do Kim, H.Y. Kang, J.J. Li, S.J. Kwak, J.T. Park, T.H. Yoo, S.H. Han, D.K. Kim, S.W. Kang, The MCP-1/CCR2 axis in podocytes is involved in apoptosis induced by diabetic conditions. Apoptosis 17, 1–13 (2012)
H. Kanamori, T. Matsubara, A. Mima, E. Sumi, K. Nagai, T. Takahashi, H. Abe, N. Iehara, A. Fukatsu, H. Okamoto, T. Kita, T. Doi, H. Arai, Inhibition of MCP-1/CCR2 pathway ameliorates the development of diabetic nephropathy. Biochem. Biophys. Res. Commun. 360, 772–777 (2007)
P. Celec, J. Hodosy, R. Gardlík, M. Behuliak, R. Pálffy, M. Pribula, P. Jáni, J. Turňa, K. Sebeková, The effects of anti-Inflammatory and anti-angiogenic DNA vaccination on diabetic nephropathy in rats. Hum. Gene Ther. 23, 158–166 (2012)
A.S. Awad, G.R. Kinsey, K. Khutsishvili, T. Gao, W.K. Bolton, M.D. Okusa, Monocyte/macrophage chemokine receptor CCR2 mediates diabetic renal injury. Am. J. Physiol. Renal Physiol. 301, F1358–F1366 (2011)
S.J. Seok, E.S. Lee, G.T. Kim, M. Hyun, J.H. Lee, S. Chen, R. Choi, H.M. Kim, E.Y. Lee, C.H. Chung, Blockade of CCL2/CCR2 signalling ameliorates diabetic nephropathy in db/db mice. Nephrol. Dial. Transplant. 28, 1700–1710 (2013)
M. Okamoto, M. Fuchigami, T. Suzuki, N. Watanabe, A novel C-C chemokine receptor 2 antagonist prevents progression of albuminuria and atherosclerosis in mouse models. Biol. Pharm. Bull. 35, 2069–2074 (2012)
S.G. Sayyed, M. Ryu, O.P. Kulkarni, H. Schmid, J. Lichtnekert, S. Grüner, L. Green, P. Mattei, G. Hartmann, H.J. Anders, An orally active chemokine receptor CCR2 antagonist prevents glomerulosclerosis and renal failure in type 2 diabetes. Kidney Int. 80, 68–78 (2011)
T.J. Sullivan, Z. Miao, D.J. Dairaghi, A. Krasinski, Y. Wang, B.N. Zhao, T. Baumgart, L.S. Ertl, A. Pennell, L. Seitz, J. Powers, R. Zhao, S. Ungashe, Z. Wei, L. Boring, C.L. Tsou, I. Charo, R.D. Berahovich, T.J. Schall, J.C. Jaen, CCR2 antagonist CCX140-B provides renal and glycemic benefits in diabetic transgenic human CCR2 knockin mice. Am. J. Physiol. Renal Physiol. 305, F1288–F1297 (2013)
T.J. Sullivan, Z. Miao, B.N. Zhao, L.S. Ertl, Y. Wang, A. Krasinski, M.J. Walters, J.P. Powers, D.J. Dairaghi, T. Baumgart, L.C. Seitz, R.D. Berahovich, T.J. Schall, J.C. Jaen, Experimental evidence for the use of CCR2 antagonists in the treatment of type 2 diabetes. Metab. Clin. Exp. 62, 1623–1632 (2013)
J.F. Navarro-González, C. Mora-Fernández, M. de Muros Fuentes, J. García-Pérez, Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat. Rev. Nephrol. 7, 327–340 (2011)
M. Hanefeld, E. Schell, I. Gouni-Berthold, M. Melichar, I. Vesela, D. Johnson, S. Miao, T.J. Sullivan, J.C. Jaen, T.J. Schall, P. Bekker, the CCX140-B Diabetes Study Group, Orally-administered chemokine receptor CCR2 antagonist CCX140-B in type 2 diabetes: a pilot double-blind, randomized clinical trial. J. Diabetes Metabolism. 3, 9 (2012)
V. Ninichuk, S. Clauss, O. Kulkarni, H. Schmid, S. Segerer, E. Radomska, D. Eulberg, K. Buchner, N. Selve, S. Klussmann, H.J. Anders, Late onset of Ccl2 blockade with the Spiegelmer mNOX-E36-3’PEG prevents glomerulosclerosis and improves glomerular filtration rate in db/db mice. Am. J. Pathol. 172, 628–637 (2008)
S. Kiyici, E. Erturk, F. Budak, C. Ersoy, E. Tuncel, C. Duran, B. Oral, D. Sigirci, S. Imamoglu, Serum monocyte chemoattractant protein-1 and monocyte adhesion molecules in type 1 diabetic patients with nephropathy. Arch. Med. Res. 37, 998–1003 (2006)
T. Wada, K. Furuichi, N. Sakai, Y. Iwata, K. Yoshimoto, M. Shimizu, S.I. Takeda, K. Takasawa, M. Yoshimura, H. Kida, K.I. Kobayashi, N. Mukaida, T. Naito, K. Matsushima, H. Yokoyama, Up-regulation of monocyte chemoattractant protein-1 in tubulointerstitial lesions of human diabetic nephropathy. Kidney Int. 58, 1492–1499 (2000)
N.G. Frangogiannis, The prognostic value of monocyte chemoattractant protein-1/CCL2 in acute coronary syndromes. J. Am. Coll. Cardiol. 50, 2125–2127 (2007)
T. Morii, H. Fujita, T. Narita, T. Shimotomai, H. Fujishima, N. Yoshioka, H. Imai, M. Kakei, S. Ito, Association of monocyte chemoattractant protein-1 with renal tubular damage in diabetic nephropathy. J. Diabetes Complicat. 17(1), 11–15 (2003)
F.W. Tam, B.L. Riser, K. Meeran, J. Rambow, C.D. Pusey, A.H. Frankel, Urinary monocyte chemoattractant protein-1 (MCP-1) and connective tissue growth factor (CCN2) as prognostic markers for progression of diabetic nephropathy. Cytokine 47, 37–42 (2009)
M. Kano, T. Ohno-Shosaku, Y. Hashimotodani, M. Uchigashima, M. Watanabe, Endocannabinoid-mediated control of synaptic transmission. Physiol. Rev. 9, 309–380 (2009)
J. Tam, J. Liu, B. Mukhopadhyay, R. Cinar, G. Godlewski, G. Kunos, Endocannabinoids in liver disease. Hepatology 53, 346–355 (2011)
P. Pacher, P. Mukhopadhyay, R. Mohanraj, G. Godlewski, S. Bátkai, G. Kunos, Modulation of the endocannabinoid system in cardiovascular disease: therapeutic potential and limitations. Hypertension 52, 601–607 (2008)
C. Silvestri, V. Di Marzo, The endocannabinoid system in energy homeostasis and the etiopathology of metabolic disorders. Cell Metab. 17, 475–490 (2013)
N. Leleu-Chavain, M. Body-Malapel, J. Spencer, P. Chavatte, P. Desreumaux, R. Millet, Recent advances in the development of selective CB(2) agonists as promising anti-inflammatory agents. Curr. Med. Chem. 19, 3457–3474 (2012)
F. Barutta, F. Piscitelli, S. Pinach, G. Bruno, R. Gambino, M.P. Rastaldi, G. Salvidio, V. Di Marzo, P. Cavallo Perin, G. Gruden, Protective role of cannabinoid receptor type 2 in a mouse model of diabetic nephropathy. Diabetes 60, 2386–2396 (2011)
F. Barutta, A. Corbelli, R. Mastrocola, R. Gambino, V. Di Marzo, S. Pinach, M.P. Rastaldi, P.C. Perin, G. Gruden, Cannabinoid receptor 1 blockade ameliorates albuminuria in experimental diabetic nephropathy. Diabetes 59, 1046–1054 (2010)
K.H. Han, S. Lim, J. Ryu, C.W. Lee, Y. Kim, J.H. Kang, S.S. Kang, Y.K. Ahn, C.S. Park, J.J. Kim, CB1 and CB2 cannabinoid receptors differentially regulate the production of reactive oxygen species by macrophages. Cardiovasc. Res. 84, 378–386 (2009)
D.H. Nam, Blockade of cannabinoid receptor 1 improves insulin resistance, lipid metabolism, and diabetic nephropathy in db/db mice. Endocrinology 153, 1387–1396 (2012)
J.C. Lim, M.H. Lee, J.E. Kim, H.K. Song, Y.S. Kang, J.E. Lee, H.W. Kim, J.J. Cha, Y.Y. Hyun, S.H. Kim, S.Y. Han, K.H. Han, J.Y. Han, D.R. Cha, Cannabinoid receptor 1 mediates high glucose-induced apoptosis via endoplasmic reticulum stress in primary cultured rat mesangial cells. Am. J. Physiol. Renal Physiol. 301, F179–F188 (2011)
F. Barutta, S. Grimaldi, I. Franco, S. Bellini, R. Gambino, S. Pinach, A. Corbelli, G. Bruno, M.P. Rastaldi, T. Aveta, E. Hirsch, V. Di Marzo, G. Gruden, Deficiency of cannabinoid receptor of type 2 worsens renal functional and structural abnormalities in streptozotocin-induced diabetic mice. Kidney Int. (2014). doi:10.1038/ki.2014.165
K.A. Jenkin, A.J. McAinch, J.F. Briffa, Y. Zhang, D.J. Kelly, C.A. Pollock, P. Poronnik, D.H. Hryciw, Cannabinoid receptor 2 expression in human proximal tubule cells is regulated by albumin independent of ERK1/2 signaling. Cell. Physiol. Biochem. 32, 1309–1319 (2013)
P. Janiak, B. Poirier, J.P. Bidouard, C. Cadrouvele, F. Pierre, L. Gouraud, I. Barbosa, J. Dedio, J.P. Maffrand, G. Le Fur, S. O’Connor, J.M. Herbert, Blockade of cannabinoid CB1 receptor improves renal function, metabolic profile, and increased survival of obese Zucker rats. Kidney Int. 72, 1345–1357 (2007)
F. Montecucco, F. Burger, F. Mach, S. Steffens, CB2 cannabinoid receptor agonist JWH-015 modulates human monocyte migration through defined intracellular signaling pathways. Am. J. Physiol. Heart Circ. Physiol. 294, H1145–H1155 (2008)
J. Tam, V.K. Vemuri, J. Liu, S. Bátkai, B. Mukhopadhyay, G. Godlewski, D. Osei-Hyiaman, S. Ohnuma, S.V. Ambudkar, J. Pickel, A. Makriyannis, G. Kunos, Peripheral CB1 cannabinoid receptor blockade improves cardiometabolic risk in mouse models of obesity. J. Clin. Invest. 120, 2953–2966 (2010)
S. Steffens, N.R. Veillard, C. Arnaud, G. Pelli, F. Burger, C. Staub, M. Karsak, A. Zimmer, J.L. Frossard, F. Mach, Low dose oral cannabinoid therapy reduces progression of atherosclerosis in mice. Nature 434, 782–786 (2005)
G.D. Duerr, J.C. Heinemann, G. Suchan, E. Kolobara, D. Wenzel, C. Geisen, M. Matthey, K. Passe-Tietjen, W. Mahmud, A. Ghanem, K. Tiemann, J. Alferink, S. Burgdorf, R. Buchalla, A. Zimmer, B. Lutz, A. Welz, B.K. Fleischmann, O. Dewald, The endocannabinoid-CB2 receptor axis protects the ischemic heart at the early stage of cardiomyopathy. Basic Res. Cardiol. 109, 425 (2014)
E.J. Rahn, A.G. Hohmann, Cannabinoids as pharmacotherapies for neuropathic pain: from the bench to the bedside. Neurotherapeutics. 6, 713–737 (2009)
P. Pacher, R. Mechoulam, Is lipid signaling through cannabinoid 2 receptors part of a protective system? Prog. Lipid Res. 50, 193–211 (2011)
V. Deveaux, T. Cadoudal, Y. Ichigotani, F. Teixeira-Clerc, A. Louver, S. Manin, J. Tran-Van Nhieu, M.P. Belot, A. Zimmer, P. Even, P.D. Cani, C. Knauf, R. Burcelin, A. Bertola, Y. Le Marchand-Brustel, P. Gual, A. Mallat, S. Lotersztajn, Cannabinoid CB2 receptor potentiates obesity-associated inflammation, Insulin resistance and hepatic steatosis. PloS One. 4(6), e5844 (2009)
Acknowledgments
This work was supported by the European Federation for the Study of Diabetes, the Compagnia di San Paolo, and the University of Turin.
Conflict of interest
The authors declare that they do not have conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Barutta, F., Bruno, G., Grimaldi, S. et al. Inflammation in diabetic nephropathy: moving toward clinical biomarkers and targets for treatment. Endocrine 48, 730–742 (2015). https://doi.org/10.1007/s12020-014-0437-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-014-0437-1