
Abstract
Neuronal ceroid lipofuscinoses (NCL) are the most commonly inherited progressive encephalopathies of childhood. Pathologically, they are characterized by endolysosomal storage with different ultrastructural features and biochemical compositions. The molecular mechanisms causing progressive neurodegeneration and common molecular pathways linking expression of different NCL genes are largely unknown. We analyzed proteome alterations in the brains of a mouse model of human infantile CLN1 disease—palmitoyl-protein thioesterase 1 (Ppt1) gene knockout and its wild-type age-matched counterpart at different stages: pre-symptomatic, symptomatic and advanced. For this purpose, we utilized a combination of laser capture microdissection-based quantitative liquid chromatography tandem mass spectrometry (MS) and matrix-assisted laser desorption/ionization time-of-flight MS imaging to quantify/visualize the changes in protein expression in disease-affected brain thalamus and cerebral cortex tissue slices, respectively. Proteomic profiling of the pre-symptomatic stage thalamus revealed alterations mostly in metabolic processes and inhibition of various neuronal functions, i.e., neuritogenesis. Down-regulation in dynamics associated with growth of plasma projections and cellular protrusions was further corroborated by findings from RNA sequencing of CLN1 patients’ fibroblasts. Changes detected at the symptomatic stage included: mitochondrial functions, synaptic vesicle transport, myelin proteome and signaling cascades, such as RhoA signaling. Considerable dysregulation of processes related to mitochondrial cell death, RhoA/Huntington’s disease signaling and myelin sheath breakdown were observed at the advanced stage of the disease. The identified changes in protein levels were further substantiated by bioinformatics and network approaches, immunohistochemistry on brain tissues and literature knowledge, thus identifying various functional modules affected in the CLN1 childhood encephalopathy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The neuronal ceroid lipofuscinoses (NCL) constitute a group of inherited, progressive degenerative diseases. Thirteen genetically distinct NCL variants, categorized by age of onset and pathological features, have been identified. A majority of NCL types share some clinical features such as cognitive decline with dementia, motor deterioration, seizures and visual failure that eventually lead to blindness and early death (Palmer et al. 2013). NCL are categorized as lysosomal storage disorders (LSDs) due to the characteristic accumulation of autofluorescent, electron dense lipopigments (featuring both ceroid and lipofuscin) in the lysosomes of both neuronal and several non-neural cells (Boustany 2013). Ultrastructural features of the storage deposits characterize the different forms of NCL, either as a prominent signature of specific forms (e.g., curvilinear profiles [CVPs] in CLN2, finger print profile [FPPs] in CLN3 or granular osmiophilic deposits [GRODs], in CLN1, CLN4, CLN10) or as a combination of different features in the remaining NCL (Anderson et al. 2013). Interestingly, the ultrastructural pattern of the storage deposits seems to be related to its biochemical properties in three forms only (CLN1, CLN4 and CLN10), in which the common presence of GRODs correlates with the shared accumulation of sphingolipid activator proteins A and D. However, protein interaction studies (Lyly et al. 2008, 2009; Scifo et al. 2013; Vesa et al. 2002), and observations that similar functional modules can be affected in different forms of NCL (i.e., CLN1, CLN3, CLN5 and CLN10) (Koch et al. 2013; von Schantz et al. 2008), suggest that distinct NCL types are linked at the molecular level.
Prominent features of infantile-onset CLN1 disease include gradual accumulation of storage material, most notably in the nervous system (but also in some other tissues), progressive cortical atrophy (underlying neuronal degeneration), thinned corpus callosum and reduced volume of the hemispheric myelin (centrum semiovale) (Haltia et al. 1973a, b). Evidence of altered myelin signal derived from head-proton magnetic resonance spectroscopy (1H-MRS) and MRI studies, is suggestive for myelin impairment (Vanhanen et al. 2004). Inflammatory changes affecting myelinated regions of the brain were also described in a mouse model of the disease (Groh et al. 2013). Altogether, these findings might reflect various cellular mechanisms that underlie brain pathology in CLN1 disease, affecting the neuronal cell bodies and processes, associated with a related inflammatory response.
Classic infantile NCL (INCL/CLN1) are caused by mutations in the CLN1 gene (Vesa et al. 1995; Kousi et al. 2012). CLN1 encodes palmitoyl-protein thioesterase 1, PPT1 (Vesa et al. 1995), an enzyme that removes palmitate groups from S-acylated proteins (Camp and Hofmann 1993), in vitro. However, in vivo substrates of PPT1 are yet to be clarified.
In mammals, PPT1 is ubiquitously expressed with the highest levels in the brain, eye and spleen (Isosomppi et al. 1999; Suopanki et al. 1999a, b). It localizes to the lysosomal compartments of neurons but is also present at extralysosomal compartments (Ahtiainen et al. 2003; Heinonen et al. 2000; Lehtovirta et al. 2001). PPT1 has been demonstrated to associate with lipid rafts (Goswami et al. 2005) and vesicular assemblies in neurons (Ahtiainen et al. 2003; Heinonen et al. 2000; Isosomppi et al. 1999; Kim et al. 2008; Lehtovirta et al. 2001). It can also be secreted, which is suggestive of different functions and/or involvement with substrates in the extracellular space (Camp and Hofmann 1993; Heinonen et al. 2000).
The Ppt1 ∆x4 knockout mouse model (hereby referred to as Ppt1 −/−) replicates the features of CLN1 disease and is characterized by several neurological defects, including motor dysfunction and brain atrophy associated with dramatic death of cortical neurons (Jalanko et al. 2005). The neuropathological changes in the brain are linked to progressive accumulation of autofluorescent storage material and pronounced glial activation (mostly in the thalamocortical system), prior to neuronal loss (Jalanko et al. 2005). Gene expression profiling analyzes of cortical tissues indicated shared, affected molecular pathways in the Ppt1 −/−and Cln5 −/− mouse models that point to inflammation, cytoskeleton abnormalities and regulation of neuronal growth cone stability (Jalanko et al. 2005; von Schantz et al. 2008).
Integrative system-based proteomics approaches have been utilized for studies of neurodegenerative disorders, including NCL. Wang et al. (2011) profiled disease-related changes by combining two-dimensional electrophoresis with mass spectrometry in NCL-derived fibroblast cell lines. This work revealed changes related to multiple cellular functional categories, including: cell motility and development, apoptosis, membrane trafficking and calcium binding, but the identified changes were not validated in neuronal cells. Chan et al. (2012) profiled protein levels in blood plasma from CLN3 (Batten) disease patients using two-dimensional difference gel electrophoresis and multiplex suspension arrays combined with mass spectrometry. They identified statistically significant changes in the levels of proteins associated with immune response. Parallel metabolomic profiling experiments indicated changes in levels of metabolites linked to fatty acid and cholesterol metabolism, triglycerides as well as elevated levels of ganglioside GM2 (Chan et al. 2012).
Another way to profile disease-related changes is to isolate brain-specific cellular fractions (i.e., synaptosomes) and perform quantitative measurements, which can be further validated. We and others implemented this approach to study cathepsin D-deficiency (CLN10 disease) in the mouse brain and to investigate cysteine string protein alpha, CSPα (DNAJC5/CLN4, causative for autosomal dominant adult onset NCL)-interacting partners, respectively (Zhang et al. 2012; Koch et al. 2013). However, these quantitative approaches utilized material derived from whole brains and did not target specific, disease-affected brain regions.
Laser capture microdissection (LCM) enables targeted enrichment of specific cell types of interest, thereby minimizing the impact of heterogeneity (Braakman et al. 2012). Various molecular biology techniques have been combined with LCM to reveal molecular profiles specific for the cell type/disease state [reviewed in (Xu 2010)]. Such studies were used to, i.e., investigate the proteomes of Lewy bodies (Leverenz et al. 2007), composition of amyloid β plaques in various forms of Alzheimer’s disease (Miravalle et al. 2005; Philipson et al. 2012), or to search for potential biomarkers of breast cancer (Liu et al. 2012). Proteomic approaches utilizing targeted quantitative mass spectrometry reveal changes directly related to cellular dynamics and may well complement microarray-based profiling, which has previously been used extensively to study NCL-related changes (i.e., in the CLN1, CLN2, CLN3 and CLN5 mouse disease models) (von Schantz et al. 2008; Luiro et al. 2006; Blom et al. 2013; Cao et al. 2011). Quantitative label-free profiling in disease-affected brain areas facilitates recognition of disease-specific molecular features, including specific post-translational modifications associated with the disease.
Detection of protein expression directly on tissue slices from fresh-frozen and formalin-fixed paraffin-embedded tissue specimens can be achieved using MS tissue profiling and imaging [MALDI-MSI, reviewed in (Lalowski et al. 2013)]. Distribution of various peptides/proteins and numerous biological compounds in normal and diseased tissues including neurodegenerative disorders (Philipson et al. 2012; Wisztorski et al. 2008; Zimmerman et al. 2008), using MALDI-MSI, has been demonstrated.
The molecular mechanisms underlying CLN1 remain poorly understood. In this work, we combined targeted LCM-based proteomics in the thalamus (at different stages) with MALDI-MSI to characterize the progression and complexity of disease-associated changes in the Ppt1 −/− brain. Identified alterations in protein levels were further validated using bioinformatics/network approaches, immunohistochemistry on brain tissues and literature knowledge from other NCL models, thereby pinpointing various functional modules affected in the childhood encephalopathy.
Materials and Methods
Materials
ITO-coated glasses (indium tin oxide) were obtained from Bruker Daltonics (Bremen, Germany), PEN (polyethylene naphthalate) membrane (1 mm) covered glass slides, and adhesive caps were purchased from PALM laboratories (Carl Zeiss MicroImaging, GmbH, Munich, Germany), SuperFrost Ultra Plus glass slides were purchased from Thermo Scientific (Gerhard Menzel GmbH, Braunschweig, Germany), and sequencing grade-modified trypsin was obtained from Promega (Madison, WI, USA). RapiGest SF surfactant and LC glass vials were from Waters Corporation (Milford, MA, USA); HPLC-grade water and acetonitrile (ACN) were purchased from Fluka analytical (Sigma-Aldrich Corporation, St. Louis, MO, USA). Sinapinic acid (SA), dithiothreitol, iodoacetamide and trifluoroacetic acid (TFA) were purchased from Sigma-Aldrich (Missouri, USA).
Antibodies
For immunohistochemistry on fresh-frozen tissue section, we utilized the following antibodies: rabbit polyclonal anti-glial fibrillary acidic protein (GFAP, IgG fraction of antiserum, G9269, Sigma-Aldrich), rabbit polyclonal anti-oligodendrocyte-specific protein (OSP/claudin 11; ab7474, Abcam) and rat monoclonal anti-myelin basic protein (epitope aa82-87 of MBP, MCA409S, Serotec). Secondary horse anti-mouse and anti-rabbit antibodies were from Vectastain ABC Elite Kit (Vector Laboratories, Burlingame, CA, USA). Goat anti-rat antibody (21,543) was from Millipore Corp (Merck KGaA Darmstadt, Germany).
Brain Tissue Sampling and Sectioning
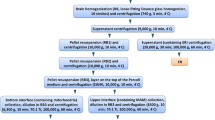
The biometric planning of the experiments was performed with the program G*Power 3.1.9.2 (Faul et al. 2007). For the power analysis, we assumed a first-class error with a probability of 0.05, a second-class error with a probability of 0.2 and a biologically relevant difference of 80 %. We therefore planned 4 animals per group for 3 time points, with effect strength of 2.63. Homozygous Ppt1 −/− (Ppt1 ∆ex4) (Jalanko et al. 2005) and wild-type littermate, age-matched control male mice of 1 month (pre-symptomatic), 3 months (symptomatic) and 5 months of age (advanced stage) from each genotype and disease stage were utilized in this study. Genotyping of the mice was done by PCR, using DNA from tail biopsies. Mice were euthanized by CO2 asphyxiation. After quick extraction from the skull, the brains were briefly washed with cold PBS to remove excess blood and gently dried with soft tissue. The brains were placed on an aluminum foil strip and transferred to an empty 50-ml Falcon tube, which was lowered into liquid nitrogen bath and kept there until the whole tissue was frozen. Brains were stored at −80 °C until sectioning. All animal procedures were performed according to protocols approved by the ethical boards for animal experimentation of the National Institute for Health and Welfare and University of Helsinki, as well as State Provincial offices of Finland (agreement number KEK12-015). All experiments were performed in accordance with good practice of handling laboratory animals and genetically modified organisms.
For MALDI-MSI, brain hemispheres from both genotypes and disease stages (n = 24) were coronally sectioned at 12 µm thickness (at the level of hippocampus and thalamic regions, between −2.46 and −3,00 mm relative to the bregma and interaural between 1.34 and 0.8 mm), using a Leica CM3050 cryostat (Leica Microsystems GmbH, Wetzlar, Germany), and thaw-mounted on conductive slides (indium tin oxide-coated slides, ITO from Bruker Daltonics, Bremen, Germany) using a minimum amount of TissueTek O.C.T. To avoid systematic experimental errors, the sections from different genotypes but the same time points were mounted on ITO slides. Sectioned tissues were fixed briefly in cold acetone and dried for 5 min in a speed-vac, prior to washing. In order to fix tissue and remove excessive salts, the cryosections were washed in 70 % EtOH for 45 s followed by two 30-s washes in 96 % EtOH, as described (Mainini et al. 2015). Sections were then dried in a speed-vac for 10 min. When possible, brain tissue sections were analyzed on the same day. For longer storage, the ITO slides were packed into boxes, flushed with argon and stored, sealed at −80 °C.
To perform thalamic laser capture microdissections (LCM), 12-μm tissue sections were utilized and mounted on PEN slides treated with 30-min UV light, prior to sectioning. Sections were stored at −80 °C until LCM and tissue slides allowed to defrost at room temperature for 5 min. The sections were fixed for 1 min in ice-cold 70 % ethanol. Slides were dehydrated using 100 % ethanol and allowed to dry at room temperature. Sections were then rehydrated in tap water for 15 s, stained in Mayer hematoxylin for 10 min, blued in tap water for 15 s and then dehydrated in increasing concentrations of ethanol/Milli-Q (v/v): 50 % (15 s), 70 % (15 s), 95 % (15 s) and 100 % (30 s, twice). Tissue sections were dried for 5 min at room temperature (RT) and further dried with vacuum for 2 min (Braakman et al. 2011). Laser capture microdissection and pressure catapulting (LCMPC) were performed shortly after staining. Cells were collected from ventral posteromedial nucleus (VPM), ventral posterolateral nucleus (VPL), ventral lateral nucleus (VL) and posterior nucleus (PO) areas of the thalamus (cut between −1.94 and −2.70 mm relative to the bregma and interaural between 1.86 and 1.10 mm), using a P.A.L.M. LMPC device (Carl Zeiss MicroBeam, GmbH, Munich, Germany). From each slide, an area of ~500,000 μm2 that corresponds to ~5000 cells [area × slide thickness/1000 μm3 cell volume (Umar et al. 2005)] was collected in P.A.L.M. adhesive caps. Collected tissues were stored at −80 °C until digestion. Digestions were performed in batches including all samples of the same time point (from both genotypes). Protein extraction and trypsin digestion were performed according to a modified protocol (Braakman et al. 2012). Collected brain tissue samples were defrosted on ice, dissolved in 40 μl of 0.2 % (w/v) RapiGest in 50 mM ammonium bicarbonate and lysed by repeated cycles of sonication in a water bath (6 cycles of 1-min sonication intermitted by 2-min incubation on ice). Proteins were denatured at 99 °C for 5 min, reduced with 5 mM dithiothreitol at 60 °C for 30 min and alkylated with 15 mM iodoacetamide in the dark, at RT for 60 min. Subsequently, 100 ng of trypsin was added (at approximately 1:4 ratio of enzyme to protein), and the samples were incubated at RT overnight with mixing. After digestion, RapiGest was degraded by acidifying to 6 % (v/v) TFA and incubating for 30 min at 37 °C. Samples were stored at −80 °C until LC-MSE analysis.
Liquid Chromatography-MSE
Samples were defrosted at room temperature, centrifuged at 14,000 rpm, and the supernatant was transferred to HPLC vials, which were stored at 4 °C prior to analyses. Nine microliters (out of total of 48 µl) of digested proteins/replicate (3 technical replicate runs per sample) was randomly injected for LC-MSE analysis. The peptides were separated by nanoAcquity UPLC system (Waters) equipped with a trapping column 5-μm Symmetry C18 180 μm × 20 mm C18 reverse phase (Waters), followed by an analytical 1.7 µm, 75 µm × 250 mm BEH-130 C18 reversed-phase column (Waters), in a single pump trapping mode. The injected sample analytes were trapped at a flow rate of 15 µl/min in 99.5 % of solution A (0.1 % formic acid). After trapping, the peptides were separated with a linear gradient of 3–35 % solution B (0.1 % formic acid/acetonitrile), for 90 min at a flow rate 0.3 µl/min and a stable column temperature of 35 °C. Each randomized sample run was succeeded by two empty runs to wash out any remaining peptides from previous runs. The samples were run in data independent analysis MSE mode (DIA-MSE), on a Synapt G2-S mass spectrometer (Waters), by alternating between low collision energy (6 V) and high collision energy ramp in the trap region (15–42 V) and using 0.5-s cycle time. The separated peptides were detected online with mass spectrometer, operated in positive, resolution mode in the range of m/z 50–2000 amu. 150 fmol/µl of human [Glu1]-fibrinopeptide B (Sigma) in 50 % acetonitrile/0.1 % formic acid solution at a flow rate of 0.3 µl/min was used for a lock mass correction and applied every 30 s.
Quantitation of LC-MSE Data
Relative quantification of randomized samples was performed with Progenesis QI™ for Proteomics software (Nonlinear Dynamics/Waters) and ProteinLynx Global Server (PLGS v3.0), using precursor ion intensities. MSE parameters were set as follows: low energy threshold of 135 counts, elevated energy threshold of 30 counts and intensity threshold of precursor/fragment ion cluster 750 counts. Chromatograms were automatically aligned by the Progenesis QI™ software, and those that had alignment score ≥ 85 % to the reference run, based on their retention and m/z values (2D map) were selected for further analysis. To compare the control(s) to other subjects, we utilized the between-subject design scheme of the software. The ANOVA calculation applied by this scheme assumes that the conditions are independent and applies a statistical test which presumes that means of the conditions are equal. Database searches were carried out against Mus musculus UniProtKB–SwissProt, reviewed, database (release 2014_03, 32,538 entries) with ion accounting algorithm and using the following parameters: peptide and fragment tolerance: automatic, maximum protein mass: 500 kDa, minimum fragment ions matches per protein ≥7, fragment ions matches per peptide ≥3, peptide matches per protein ≥1, primary digest reagent: trypsin, missed cleavages allowed: 2, fixed modification: carbamidomethylation C, variable modifications: phosphorylation of STY residues, oxidation of methionine (M) and false discovery rate (FDR) <4 %. All identifications were subsequently refined to Mus musculus only identifiers, and the protein lists simplified by protein grouping. Protein quantitation was performed entirely on non-conflicting proteins identifications, using precursor ion intensity data and standardized expression profiles. Additional filters in Progenesis QI™ applied to final data increased the stringency of accepted protein leads by limiting the ratio between treated as compared to controls [fold change, FC > 1.2, following the genomic findings described in (Blom et al. 2013; von Schantz et al. 2008)] in either direction of up- or down-regulation, computed from averaged, normalized protein intensities, and P < 0.05 by ANOVA for all comparisons. In subsequent steps, we curated the output lists, by screening for multiple UniProtKB–SwissProt identifiers and removal of known contaminants (i.e., skin keratins, hemoglobins). The lists of up-/down-regulated protein changes with their corresponding, unique UniProtKB–SwissProt identifiers served as inputs into ingenuity pathways (IPA, www.ingenuity.com), network and other functional analyses.
Functional and Statistical Analyses of Proteomic Data
Canonical pathway analysis, annotation of associated diseases and functions and linkage to upstream network regulators of thalamic datasets from different stages were performed with IPA. The IPA right-tailed Fisher’s exact test with post hoc Benjamini–Hochberg multiple testing corrections was used to determine a P value of significance in all functional analyses. The differentially expressed mouse gene products were connected to PPT1 utilizing GeneMania (www.GeneMania.org which indexes 2104 association networks containing 535,774,338 interactions mapped to 161,629 genes from 8 organisms, last update 10.07.2014) (Warde-Farley et al. 2010; Zuberi et al. 2013). Networks were calculated and drawn using GeneMania reference real- and binary-valued interaction networks consisting of physical, genetic, predicted and pathway interaction datasets. For weighting of the networks, we used gene ontology biological process (GO BP) algorithm from GeneMania, which assigns weights in order to maximize connectivity between all input genes in a given ontology class. Measured protein expression state was included as one of the parameters in network depiction. The connectivity to PPT1 interactome was realized by utilizing protein complex data from tandem affinity purification-MS experiments (Scifo et al. 2015a, b). Linkage to human/rat lipofuscin proteome was achieved by literature mining (Ng et al. 2008; Ottis et al. 2012; Warburton et al. 2005). Linking of network nodes to CLN3/CLN5 interactomes was performed by mining published literature data (Scifo et al. 2013) and utilizing the Hippie database [http://cbdm.mdc-berlin.de/tools/hippie/information.php (Schaefer et al. 2012)]. Comparisons of protein expression changes with gene expression profiles in the Ppt 1 −/− brain (from whole brain, cortex and thalamus) were performed using published datasets (Blom et al. 2013; Elshatory et al. 2003; Jalanko et al. 2005; von Schantz et al. 2008). Assignment of proteins with altered expression to myelin proteome was based on central nervous system (CNS) myelin datasets (de Monasterio-Schrader et al. 2012; Taylor et al. 2004; Vanrobaeys et al. 2005). Annotation of mitochondrial proteins in the datasets was performed with MitoMiner v3.1_2014-08 (http://mitominer.mrc-mbu.cam.ac.uk/). The MitoMiner database combines evidence for mitochondrial involvement derived from MitoMiner Reference Set (all species, 12,925 proteins), MitoCarta Inventory (human and mouse, 2682 proteins), IMPI—Integrated Mitochondrial Protein Index v.Q3_2014 (human, mouse, rat and cow, 6726 proteins) and annotations from GO cellular component (GO CC), as well as UniProtKB–SwissProt database knowledge combined with information from large-scale mass spectrometry studies and mitochondrial targeting sequence predictions. Proteins that were linked to the mitochondrial proteome in two or more databases were considered as positively identified.
MALDI-Mass Spectrometry Imaging
Deposition of matrix for mass spectrometry imaging (MSI) analyses was performed with the ImagePrep™ (Bruker Daltonics, Bremen, Germany) using a custom method to ensure matrix homogeneity and reproducibility across tissue sections. Matrix was sprayed over ITO-coated glass slides at 10 mg/ml concentration of SA in 50 % acetonitrile/0.2 % TFA. The sprayed tissue sections were analyzed using a MALDI-TOF/TOF mass spectrometer (UltrafleXtreme™, Bruker Daltonics) equipped with SmartBeam II Nd:YAG/355-nm laser operating in linear, positive mode at 2 kHz with a laser focus down to 70 µm diameter using the ‘large’ setting. Spectra were acquired in the range of m/z 2–30 kDa. Prior to image analysis, each MALDI plate was externally calibrated using a standard protein mix (Protein Calibration Standard I, calibration mass range ~4–20 kDa; Bruker Daltonics). For MSI data acquisition, 1000 shots were summed per array position with a spatial resolution of 150 µm × 150 µm. Preparation of sequence, acquisition and visualization of MSI data were accomplished with FlexControl™ (version 3.4) and FlexImaging™ software (version 4.0, Bruker Daltonics, Bremen, Germany). Data pre-processing by means of baseline reduction, normalization and data reduction was performed using MATLAB. Spectra were resampled uniformly in a range from m/z 3–25 kDa with a total number of 22,001 samples, finally resulting in one sample per Dalton. Subsequently, they were smoothed using a Savitzky–Golay filter employing a second-degree polynomial with a window span of 0.05 %, corresponding to a smoothing window of approximately 11 samples. Baseline was removed using a spline interpolation in a window encompassing 200 separation units. Spectra normalization was carried out by standardizing the area under curve (AUC) to the group median (i.e., one group per month and condition). Statistical comparisons of protein peaks between different stages and genotypes were subsequently performed in MATLAB executed using Kruskal–Wallis test [a nonparametric equivalent of ANOVA test since mass spectrometry data are not distributed normally and do not exhibit homoscedasticity (Caldwell and Caprioli 2005; Lehrer et al. 2003)]. Kruskal–Wallis statistic reports H values (in the same way as ANOVA’s F values), which approximate a Chi-squared distribution (Ikem 2010). No post hoc tests were further applied since only two groups were compared and the direction of the effect was determined by fold change values and the inspection of the spectra. The values from Kruskal–Wallis test and other statistical measures are summarized in supplemental Table S12, Online Resource 12. The Pearson product-moment correlation coefficient (r) was utilized to determine the strength of linear dependence between various detected m/z (Karlsson et al. 2014) and portrayed as a correlation matrix of dependencies.
After MSI was completed, the SA matrix was removed using 70 % EtOH and mild shaking. Tissues were then stained with hematoxylin and eosin (H&E) according to Stingl et al. (Stingl et al. 2011), to assess the histological accuracy of the MSI.
The comparison of differentially expressed m/z with other MALDI-MSI data was achieved using MaTisse (http://129.187.44.58:7171/MALDI/protein) (Maier et al. 2013) and MSiMass list databases (www.maldi-msi.org/mass) (McDonnell et al. 2014), and by screening available literature data.
Immunohistochemistry on Brain Slices
The observed protein expression changes were investigated using 1-, 3- and 5-month-old male Ppt1 −/− and wild-type control brains. Ten-micrometer-thick brain cortex tissue sections mounted on SuperFrost microscope slides were immunohistochemically stained for GFAP, MBP and CLD11. Briefly, sections were fixed by cold acetone for 7 min, quenched with 0.5 % hydrogen peroxide (H2O2) in methanol for 30 min, washed 3 times with cold PBS and blocked for 30 min with 2.5 % normal horse serum (NHS) in PBS, before overnight incubation at 4 °C with rabbit anti-CLD11 (1:100), rabbit anti-GFAP (1:200) or rat anti-MBP (1:500) antibodies diluted in PBS/10 % NHS. Sections were incubated for 30 min with secondary antisera (biotinylated horse anti-rabbit IgG; 1:200 in PBS/10 % NHS or biotinylated goat anti-rat IgG; pre-diluted) and, subsequently, for 30 min in an avidin-biotin-peroxidase complex in PBS (Vectastain ABC Elite Kit, Vector Laboratories). Immunoreactivity was visualized by 0.05 % 3.3′-diaminobenzidine tetrahydrochloride (DAB) and 0.001 % H2O2 in PBS for 5 min (DAB substrate kit, Vector Laboratories). The tissue sections were routinely counterstained with hematoxylin as described (Philipson et al. 2012). Negative controls included omission of primary and secondary antibodies. Sections were cover-slipped with DPX.
Whole Transcriptome Analysis in the CLN1 Patients’ Fibroblasts
Skin biopsies were performed for diagnostic purposes after obtaining informed consent by children’s parents, according to the rules of local ethics committees. All procedures complied with requirements of the Declaration of Helsinki. CLN1 fibroblasts were isolated from skin biopsies of two CLN1 patients carrying a homozygous missense mutation (c.665T > C/p.L222P; patient 1) and homozygous insertion mutation (c.169dupA/p.M57Asnfs*45; patient 2), respectively. Patient 1 was affected with the variant late infantile form associated with CLN1 disease (Simonati et al. 2009), whereas the classical, infantile-onset phenotype was observed in patient 2 (Santorelli et al. 1998). Three cell lines from healthy donors were used as controls to reduce the interindividual variability. Fibroblast cell lines were cultured in DMEM/high glucose supplemented with 10 % fetal bovine serum, penicillin/streptomycin and amphotericin B (Euroclone, Milan, Italy). CLN1 patients’ cells were collected in three independent experiments; control triplicate constituted of one specimen from each of the three control cell lines. Total RNA was isolated using TRI reagent (Life Technologies, USA). Indexed, directional cDNA libraries were prepared using TruSeq Stranded mRNA Sample Prep Kit (Illumina, San Diego, CA, USA) according to the manufacturer’s instructions. Reads with more than 10 % of undetermined bases or more than 50 bases with a quality score <7 were discarded. Alignment of reads to reference human genome (hg19) was performed using TopHat v2.0.9 (http://tophat.cbcb.umd.edu/) using default parameters. Transcript isoforms reconstruction, estimation of expression values, given as Fragments per Kilobase per Million Mapped Reads (FPKM) and differential expression analysis were performed with Cufflinks 2 package (http://cufflinks.cbcb.umd.edu/) utilizing RefSeq annotation as a reference with default parameters (Trapnell et al. 2012). To identify differentially expressed genes in CLN1 patients’ cells, the thresholds for module of log2 fold change of FPKM (log2FC) and for false discovery rate (FDR) were set at ≥1 and ≤0.05, respectively. The lists of differentially expressed transcripts with their corresponding log2FC values served as inputs into IPA pathway analyses.
Results
Extraction and trypsin digestion protocols were optimized to identify and quantify nearly 400 unique proteins in each LC-MSE run (Online Resource 1, Table S1A–C). Subtle differences in protein expression were assayed by label-free quantitation performed in three technical replicates per sample, which were used to determine a significant quantitative difference among Ppt1 −/− and wild-type age-matched control thalamic tissues. Based on these criteria, 36 proteins (7.8 % of all quantified proteins) were significantly differentially expressed at the pre-symptomatic stage, 38 (9.6 %) at the symptomatic and 80 (20.4 %) at the advanced stage of CLN1 disease (Online Resource 2, Table S2).
We linked the differentially expressed proteins in the Ppt1 −/− thalamus with literature knowledge using IPA algorithms at the pre-symptomatic stage and attained two distinct functional networks with the highest score, related to: (i) Cell Morphology, Cellular Assembly and Organization, Cellular Development (IPA score 46, formed by 35 molecules including 18 differentially expressed proteins) and (ii) Cellular Movement, Nervous System Development and Function, Auditory and Vestibular System Development and Function (IPA score 33 with 35 molecules encompassing 14 differentially expressed proteins; Fig. 1a and Online Resource 3, Table S3). Analysis of canonical pathways at the pre-symptomatic stage revealed a significant dysregulation of tricarboxylic acid cycle II (TCA, −log P value 5.136, with 3 differentially proteins involved) and mitochondrial dysfunction (−log P value 3.742, with 4 differentially expressed proteins involved; Fig. 1b and Online Resource 4, Table S4).

Analysis of the differentially expressed proteins in the Ppt1 −/− thalamus at the pre-symptomatic stage. a Proteins that were differentially expressed in the Ppt1 −/− thalamus were linked using ingenuity pathways (IPA). The networks with the highest IPA scores are portrayed. Network 1 linked 35 molecules including 18 differentially expressed proteins, while the second network linked 35 proteins including 14 differentially expressed ones. The network scores and corresponding names assigned by IPA are depicted. Open arrows point to differentially expressed proteins shown in (b). Canonical pathway analysis of differentially expressed proteins in the thalamus at the pre-symptomatic stage. Two pathways with the highest predicted P values, mitochondrial dysfunction and tricarboxylic acid cycle II (TCA) are depicted (rectangular). The up-regulated enzymes with their fold change values are indicated in the inset. Network numbering pinpoints to participation in the network (part A of the Figure and Online Resource 3, Table S3). Threshold for analyses: −log P value = 1.3
In an effort to obtain a more comprehensive picture of connectivity between differentially expressed proteins at this stage, we utilized GeneMANIA to extend the network analyses by bridging human orthologs of the differentially expressed mouse proteins with proteins in the same pathways/gene ontologies and PPT1. 86 proteins of the network were connected, by either physical interactions (85 links), genetic interactions (134 links), pathway sharing (75 links) or predicted interactions (104 links; Online Resource 5, Table S5A-C). The most enriched processes of the network encompassed involvement in TCA cycle (P = 1.1E−14, 10 proteins), participation in mitochondrial matrix (P = 6.58E−9; Fig. 2a), regulation of lipid metabolic process (1.76E−7) and protein targeting to ER (9.03E−7, Fig. 2b), supporting the findings of canonical pathway network analysis. Nodes of the network were further linked by screening for their connectivity to the PPT1 interactome or well-characterized lipofuscin and myelin proteomes. Interestingly, 5 proteins of the network, including three differentially expressed ones (Cmrp1/CRMP1, Map1b/MAP1B and Pdha1/PDHA1) form a core of the high-confidence PPT1 interactome (Scifo et al. 2015a, b). Twenty proteins in the network (23 %) could be assigned to the CNS myelin assembly (de Monasterio-Schrader et al. 2012), while 8 proteins were constituents of the lipofuscin proteome (Basp1/BASP1, Cnp/CNP, Dpysl2/DPYSL2, Mdh2/MDH2, Ndufa8/NDUFA8, Ndufs3/NDUFS3, Ndufv2/NDUFV2, Pfn2/PFN2, Ywhag/YWHAG; for full names see Online Resource 2, Table S2).

Interaction network linking differentially expressed proteins at the pre-symptomatic stage. Differentially expressed proteins in the thalamus of the Ppt1 −/− mice were connected using multiple interaction data and filtered for gene ontology biological process participation. Using these criteria, 86 proteins could be connected into a network. a The most enriched category participation in TCA cycle (P = E−14, 10 nodes of the network) is depicted. Inset presents a network module linked by proteins found as PPT1-interacting partners (Scifo et al. 2015a, b), PDHA1, DLAT and PDHB. b Three other enriched categories implicated in PPT1 function, namely: mitochondrial matrix (P = E−9), regulation of lipid metabolic process and protein targeting to ER (both P = E−7), are presented. The functional attributes of the network nodes are portrayed (for details see Online Resource 2 and 5, Tables S2 and S5)
In order to gain more insight into associated functions, we further mined the differentially expressed set of proteins using IPA database. Among the most enriched categories, Cellular Assembly and Organization together with Cellular Function and Maintenance (P = 7.65E−06 to P = 4.18E−02, with 16 proteins assigned) was selected (Fig. 3a and Online Resource 6, Table S6). In-depth mining of annotated functions pinpointed, i.e., down-regulation of defined processes important for the proper function of neurons, including neuritogenesis, branching and microtubule dynamics (bias-corrected activation scores Z = −1.863, Z = −1.866 and Z = −1.631, respectively; Fig. 3b and Online Resource 6, Table S6). A similar picture was observed in human CLN1 patients-derived fibroblasts, where over 100 differentially expressed transcripts shared between patients were annotated in relation to the down-regulated category: Cellular Assembly and Organization (P values = 2.85E−15 to P = 2.3E−08 from patient with missense mutation p.(Leu222Pro) and P = 3.68E−11 to P = 9.48E−08, from a patient with single adenine insertion p.(Met57Asnfs*45); Fig. 3c) encompassing functional categories related to, i.e., outgrowth of plasma membrane projections or microtubule dynamics. These observations support the findings and direction of changes observed in Ppt1 −/− mouse thalamus at the pre-symptomatic stage.

Disease and functional attributes analysis at the pre-symptomatic stage. Differentially expressed proteins in the thalamus of the Ppt1 −/− mice were analyzed for their involvement in the disease and participation in cellular processes. a Chart presenting the enriched associations sorted according to P values. Cellular Assembly and Organization and Cellular Function and Maintenance represented the most enriched categories (−log P values >5, marked by asterisks). b The specific categories pinned down with the broad terms shown in A, are depicted. The expression changes of several proteins at the pre-symptomatic stage are predicted to inhibit various neuronal functions, i.e., branching of neurons and neurites, neuritogenesis and microtubule dynamics. c Analysis of associated functions in the human CLN1 patients’ fibroblasts (n = 2). The associated diseases and functions within Cellular Assembly and Organization and Cellular Function and Maintenance, Cell Growth and Proliferation categories were compared using hierarchical agglomerative clustering utilizing average linkage and Euclidean distance metrics. The most affected, down-regulated cellular functions were linked to the dynamics of growth of plasma projections and cellular protrusions, similarly as in the Ppt1 −/− mouse thalamus. The respective Z scores are given in the accompanying columns. 1. CLN1 patient with single adenine insertion p.(Met57Asnfs*45), 2. CLN1 patient with missense mutation p.(Leu222Pro)
We studied the dynamics of CLN1 disease progression by performing similar calculations at the symptomatic stage (in 3-month-old thalamus). The distinct network with assigned functions ranging from: Cell-To-Cell Signaling and Interaction, Nervous System Development and Function, to Cellular Assembly and Organization was most significantly linked to 18 differentially regulated proteins at this stage (IPA score 45). The second network dealt with functions related to Cellular Compromise, Organismal injuries and Abnormalities and Skeletal and Muscle Disorders (IPA score 21, 10 nodes, Online Resource 15, Figure S1 and Online Resource 7, Table S7). Similarly to the pre-symptomatic stage, canonical pathway profiling identified 2-ketoglutarate dehydrogenase complex, TCA cycle II and mitochondrial dysfunction, as the most enriched modules linked to differentially expressed proteins (−log P values from 4.69 to 3.07, 6 proteins involved, Fig. 4a and Online Resource 8, Table S8). Interestingly, four of differentially expressed proteins (10.5 % of total) could be linked to signaling by Rho family GTPases (−log P value 3.03, including Rock1/ROCK1, Baiap2/BAIAP2, Vim/VIM, Gfap/GFAP; inset in Fig. 4a). The pathway connectivity analysis pinpointed two larger modules formed by mitochondrial dysfunction and actin nucleation by ARP-WASP complex bridged by PEDF signaling pathway node. The second subnetwork was formed by three nodes with high P values including glycolysis I and gluconeogenesis I bridged by NADH repair node (Fig. 4b).

Pathway analysis of differentially expressed proteins in the thalamus at the symptomatic stage. a Canonical pathway analysis of differentially expressed proteins at the symptomatic stage revealed significant association with events linked to mitochondrial dynamics and various signaling pathways including signaling by Rho family of GTPases (−log P values ≥3). The proteins with down-regulated expression linked to mitochondrial dysfunction and signaling by Rho family of GTPases are shown in the inset. Strong up-regulation of GFAP, associated with the last category, has been observed. b Network of associated functional modules at the symptomatic stage. The mitochondrial dysfunction is linked to Rho family of GTPases through PEDF signaling module. The number of common genes shared between the modules and respective P values is portrayed
The differentially expressed proteins were subsequently linked with associated functional categories, diseases and cellular functions. Cell-to-Cell Signaling and Interaction/ Nervous System Development and Function, as well as Psychological Disorders (−log P values above 5.0; Fig. 5a and Online Resource 9, Table S9), constituted the most enriched functional categories. Dissection of these functions pinpointed reactivation of astrocytes (P = 3.43E−6), dystrophy of neurites and neuroinflammation of the brain, highlighted by the strongly up-regulated protein Gfap/GFAP (Fig. 5b). Neurodegeneration of thalamus (P = 3.42E−5) could be interrelated with broad terms of neurological, psychological/movement disorders and more specifically with organismal injury and abnormalities, thereby validating the rationale of proteomic profiling in Ppt1 −/− thalamus.

Analysis of functional attributes at the symptomatic stage. a Chart depicting the enriched associations at the symptomatic stage ranked according to P values (from most enriched to less enriched). Cell-To-Cell Signaling and Interaction, Nervous System Development and Function and involvement in Psychological Disorders (−log P values >5, marked by asterisks) signified the most enriched classes. b Network of associations at the symptomatic stage. Twelve differentially down-regulated proteins and three up-regulated ones, including strongly differentially up-regulated glial fibrillary acid protein, GFAP, were linked to various neurodegenerative processes, including neurodegeneration of thalamus (P = 3.42E−05), reactivation of astrocytes, neuroinflammation of brain, dystrophy of neurites, mood disorders and dementia. For details see Online Resource 6, Table S6
Results from the protein interactome analysis encompassing the differentially expressed proteins in the symptomatic thalamus (88 proteins linked to PPT1), strongly supported the findings of IPA pathway analyses. Two of the most enriched processes: regulation of vesicle-mediated transport (14 proteins in the network, including PPT1; P = 6.58E−9, Online Resource 16, Figure S2a and Online Resource 10, Table S10a–c) and synaptic vesicle transport (9 proteins, P = 6.03E−7, Online Resource 16, Figure S2b), could be visualized. Down-regulation of Vamp2/VAMP2 and Stx1a/STX1A (vesicle-associated membrane protein 2/syntaxin 1A) axis is supported by literature data (Kim et al. 2008), while down-regulation of myelin oligodendrocyte glycoprotein (Mog/MOG) and up-regulation of Gfap/GFAP are consistent with Ppt1 −/−microarray data (Elshatory et al. 2003; von Schantz et al. 2008). We further verified the up-regulation of GFAP protein at the symptomatic and advanced-stage Ppt1 −/− thalamus by immunohistochemistry (Online Resource 17, Figure S3), confirming earlier findings (Bible et al. 2004; Blom et al. 2013; Macauley et al. 2011; Miller et al. 2015). Components of ATP synthase complex (Atp5b/ATP5B and Atp5a1/ATP5A1) are part of the PPT1 interactome (Lyly et al. 2008; Scifo et al. 2015a), while Calr/CALR, Slc25a5/SLC25A5 and Uqcrc2/UQCRC2 form a core of the combined CLN3/CLN5 interaction network (Scifo et al. 2013). Eight proteins of the network, including ATP5B, GFAP and VAMP2, could be assigned to the lipofuscin interactome, whereas over 30 % of network nodes are constituents of the myelin proteome.
Proteins that were differentially expressed at the advanced stage of CLN1 disease were grouped to similar functional categories as the symptomatic stage (Fig. 6a and Online Resource 11, Table S11). Functional grouping allocated eight proteins to an enriched, novel category, Huntington’s disease (HD) signaling (Capns1/CAPNS1, Dlg4/DLG4, Dnm3/DNM3, Gnai1/GNAI1, Hspa1 l/HSPA1L, Pacsin1/PACSIN1, Sdha/SDHA, Stx1a/STX1A, −log P value = 5.363; Fig. 6a). Dysregulation of some components of this pathway is supported by microarray data from the cortex of Ppt1 −/− mice [DLG4, STX1A and CTSD (von Schantz et al. 2008)]. Interestingly, up-regulation of several constituents of perinuclear inclusions in HD: HSPA1L (HSP70), DNM3 (DNM) and SDHA (SDH) was also observed (Fig. 6b). Five of the differentially expressed proteins were linked to signaling by Rho family GTPases (−log P value = 3.417, including Arpc3/ARPC3, Gfap/GFAP, Gna11/GNA11, Gnai1/GNAI1, Sept7/SEPT7). Strong down-regulation of this pathway in the Ppt1 −/− thalamus, which is supported by literature data (von Schantz et al. 2008) and results of transcriptomic analysis in CLN1 patients’ fibroblasts, is depicted in Online Resource 18, Figure S4.

Representation of signaling pathways affected at the advanced stage. a Canonical pathway analysis of affected modules at the symptomatic stage pinpointed to Huntington’s disease signaling (−log P value = 5.36) and signaling by Rho family of GTPases (−log P value = 3.42). b Huntington’s disease signaling module is depicted in detail. Differentially up-regulated proteins are shown in magenta, while down-regulated are depicted in light green. Insets magnify in detail a down-regulated module at the membrane including proteins Stx1A/STX1A-syntaxin 1A and post-synaptic density protein 95- PSD-95, associated with glutamate receptor, ionotropic, N-methyl-d-aspartate 2B GRIN2B, as well as several up-regulated components of perinuclear inclusions in Huntington’s disease
Mining of disease association and functions at the advanced stage, linked 15 differentially expressed proteins to a subnetwork of terms including nervous system development and function, hereditary, neurological and psychological disorders (P = 1.52E−7). Strikingly, 13 out of 15 (87 % of total) proteins of this subnetwork were associated with CNS myelin (Fig. 7a and Online resource 2, Table S2). Broad functional terms encompassed, i.e., ensheathment of axons (P = 1.23E−06), with a functional module formed by three down-regulated, CNS myelin proteins: myelin basic protein/MBP, proteolipid 1/PLP1 and claudin 11/CLDN11 and function of oligodendrocytes (P = 3.16E−07, Fig. 7a). Network analysis strongly linked CLDN11 to tight junction signaling (P = 5.11E−28, Online Resource 19, Figure S5). Using qualitative immunohistochemistry on fresh-frozen mouse brain slices, we have further verified the down-regulation of this protein at the symptomatic and advanced stage in Ppt1 −/− thalamus. CLDN11 antibody recognized myelin structures and various cell types including oligodendrocytes in the wild-type brain. The myelin structures were clearly less immunoreactive in the Ppt1 −/− thalamus (Fig. 7b).

Functional associations network at the advanced stage. a Network of associations at the symptomatic stage. Fourteen differentially down-regulated proteins were linked to various neuroinflammatory processes in the brain. Thirteen proteins were linked as components of myelin (Online resource 2, Table S2). Three strongly down-regulated proteins, including myelin basic protein (Mbp/MBP), proteolipid 1 (Plp1/PLP1) and claudin 11 (Cldn11/CLDN11), were associated with ensheathment of axons (P = 1.23E−06, indicated as boxed term). b Immunohistochemical analysis of claudin 11 immunoreactivity in thalamus. The immunoreactivity with claudin 11-specific antibodies was studied at symptomatic and advanced stages, respectively. A strong down-regulation of immunoreactivity specifically in myelin structures of Ppt1 −/− thalamus can be observed. Scale bars 200 and 50 µm, respectively
In order to portray the dynamic changes in protein distributions of the Ppt1 −/− cortex at different disease stages, we utilized proteomic profiling directly on tissues using MALDI-MSI. Several dysregulated m/z in the range of 2–25 kDa were detected (Online Resource 20, Figure S6 and Online resource 12, Table S12). For example, we detected down-regulation in the peak intensity of MBP isoform 8 in comparison with an age-matched control group. The down-regulation was most pronounced at the advanced stage in the Ppt1 −/− brain and resembled the pattern of a previously described MS imaging-based localization of MBP in the brain (observed as both singly and doubly charged ions; FC = 2.17, Fig. 8a, b) (Karlsson et al. 2014; Eberlin et al. 2011). The Pearson correlation matrix analysis of all detected m/z in the average spectrum revealed a strong positive linkage of m/z 14,144 Da and m/z 18,422 Da, which corresponded to the predicted mass of MBP isoform 4 (Fig. 8c). A similar trend of peak intensity down-regulation was observed for this MBP isoform (FC = 2.71, Fig. 8d). We verified the identity of MBP peaks by direct LC-MSE sequencing of trypsin-digested peptides from selected tissue regions, which were LCM dissected from consecutive slides (Online Resource 14, Supplemental Methods, Online Resource 21, Figure S7a and Online Resource 13, Table S13). The pattern of distribution of selected MBP peptides in MALDI-MSI experiments on trypsin-digested, adjacent wild-type brain tissue sections followed the intact proteins distribution and is presented in Online Resource 21, Figure S7b. We subsequently verified the down-regulation of MBP in the Ppt1 −/− brain with well-characterized antibodies (Fig. 8e), confirming earlier observations (Blom et al. 2013). We also observed a significant down-regulation of m/z corresponding to cytochrome c (m/z 12,135 Da; FC = 1.61) and its functional complex components: ATP synthase-coupling factor 6, mitochondrial (Atp5j/ATP5 J, m/z 8945 Da; FC = 1.958) and cytochrome c oxidase subunit 7C, mitochondrial (Cox7C/COX7C, m/z 5445 Da; FC = 2.056; Online Resource 12, Table S12 and Online Resource 22, Figure S8) (Maier et al. 2013; McDonnell et al. 2014), in our MS profiling experiments. A significant up-regulation of m/z 4749, likely corresponding to thymosin beta-4 fragment (FC = 4.37), was also observed. Increased thymosin beta-4 expression in glial cells, in concert with incremental severity of malignancy in brain gliomas, was recently described (Wirsching et al. 2014).

Down-regulation of myelin basic proteins in the Ppt1 −/− brain. a Ion density distributions of an average m/z 14,145 Da ([M + H]+) corresponding to myelin basic protein isoform 8. The increase in down-regulation of Mbp-8 isoform over time can be observed in the Ppt1 −/− brain. The zooming of averaged spectra focusing on m/z 14,145 peak region at 1 month (pre-symptomatic), 3 months (symptomatic) and 5 months (advanced stage), respectively. The mean of peak intensity is indicated. Relative intensity: dark blue 0 % intensity, red 100 % intensity. The maximum peak intensity in each image was set at 100 %. TIC—Total ion current normalization. Scale bar 5 mm. b Pearson’s correlation coefficient (r = 0.83) was calculated between detected [M + H]+ and [M + 2H]2+ of Mbp-8 isoform detected at m/z of 7072 Da. c Pearson’s correlation matrix analysis of m/z detected in MALDI-MSI experiments. A strong positive correlation between m/z 14,145 and m/z 18,422 is indicated (dotted circle). A down-regulation of this m/z, corresponding to myelin basic protein isoform 5 (Mbp-5), is provided in (d). e Immunohistochemical analysis of myelin basic protein immunoreactivity on consecutive slides. The area boxed in (a) and corresponding to barrel field 1 of somatosensory cortex, SB1F, which has projections to thalamus, is indicated. A strong down-regulation of Mbp immunoreactivity in myelin layer at the advanced stage is shown. Scale bars 200 and 50 µm, respectively
Discussion
Previous functional studies using different cell and disease models implicated PPT1 in various cellular functions including: lipid metabolism (Ahtiainen et al. 2007; Lyly et al. 2008), vesicular trafficking (Kim et al. 2008), endo- and exocytosis (Ahtiainen et al. 2006; Aby et al. 2013), synaptic function (Aby et al. 2013), synaptic vesicle recycling (Kim et al. 2008; Virmani et al. 2005), neuronal specification and axon guidance (Chu-LaGraff et al. 2010) and apoptosis (Cho and Dawson 2000; Z. Zhang et al. 2006). Microarray and lipid profiling (Kakela et al. 2003; Jalanko et al. 2005) suggested that PPT1 is linked to lipid alterations and neuroinflammation. Transcript profiling in the Ppt1 −/− mice at 10 weeks of age (Elshatory et al. 2003) pinpointed the changes in lipid metabolism, cell trafficking, glial activation and calcium homeostasis, previously linked to pathology of human CLN1 disease [reviewed in (Jalanko et al. 2006)]. It was reported earlier that protein expression changes in NCL might occur at the pre-/early symptomatic stages, in advance of morphologically detectable synaptic or axonal pathology, and display regional selectivity, occurring first within the thalamus and only later in the cortex (Gupta et al. 2001; Jalanko et al. 2005; Kielar et al. 2007, 2009; Kamate and Hattiholi 2012). Therefore, we decided to analyze the dynamics of protein expression changes in a well-characterized mouse model of the CLN1 disease (Jalanko et al. 2005) in relation to its wild-type age-matched controls, by utilizing LCM-based label-free proteomic profiling fortified with MALDI-MSI and bioinformatics. Most importantly, we focused our analyses on thalamic regions and studied the progression of changes over a period of time (from pre-symptomatic to advanced stages). The observed protein expression changes in the Ppt1 −/− mouse thalamus were fortified with results from global RNA sequencing analysis in CLN1 patients’ fibroblasts bearing severe disease-causing mutations in the CLN1 gene (the full characterization of changes observed in these cell lines is currently in progress, and only some results are being presented in this article; Pezzini et al. in preparation).
Despite varying clinical manifestations of neurodegenerative disorders, the fact that neurons rely heavily on oxidative energy metabolism suggests a unified mechanism that involves dysfunction in mitochondrial energy homeostasis (Schon and Manfredi 2003). We have observed dysregulation of several mitochondrial proteins at all stages of the CLN1 disease. At the early pre-symptomatic stage, up-regulation of tricarboxylic acid cycle II components and mitochondrial dysfunction (Fig. 1b and Online Resource 2, Table S2; 8 proteins) which is supported by network analyses (Fig. 2a) was evident. Up-regulation in expression of mitochondrial proteins might represent an early cellular response to correct the disturbed metabolism at this stage. A reversed trend was however noted at the symptomatic and advanced stages of CLN1 disease, with a down-regulation of various components of mitochondrial respiratory chain and mitochondrial proteome (Online Resource 2, Table S2). Anomalies at the level of mitochondrial ATP synthase, decreased activities of respiratory chain complexes II and IV and reduction in content of high-energy phosphate compounds in fibroblasts from children with various forms of NCL including CLN1, have previously been detected (Das et al. 1999, 2001). These findings are supported by studies in CLN1 patients’ fibroblasts demonstrating altered mitochondrial compartment and down-regulation of mitochondrial markers, VDAC1 and COX IV, respectively (Pezzini et al. 2011). Moreover, we have recently detected in MS-based, affinity pull-down experiments that several mitochondrial proteins, i.e., VDAC2, as well as constituents of the pyruvate dehydrogenase and ATP synthase complexes form a complex with PPT1 (Lyly et al. 2008; Scifo et al. 2015a), although the mechanism and the stoichiometry of these extravesicular interactions are currently not known. Two of these proteins were identified by us to be dysregulated in the Ppt1 −/− thalamus, namely ATP5B and PDHA1, at the pre-symptomatic stage, respectively.
Oxidative stress-mediated abnormalities in mitochondria activate the apoptotic caspase-9 pathway in CLN1 fibroblasts and in neurons (from another mouse model of CLN1 disease) (Kim et al. 2006). An abnormality in AMPK/SIRT1/PGC-1α signaling pathway of energy metabolism was also observed in CLN1 fibroblasts and mouse brains, which correlated with up-regulation of phosphorylated-ribosomal S6 protein kinase 1 levels and inversely affected the life span of the Ppt1 −/− mice (Wei et al. 2011). Taken together, our results from proteomic profiling in the thalamus of the Ppt1 −/− mice further support the findings of impaired mitochondria in CLN1 deficiency. An additional line of evidence in support of mitochondrial component involvement in the CLN1 disorder in this study is based on MALDI-MSI results from coronally cut brain tissue slices (Online Resource 12, Table S12 and Online Resource 22, Figure S8). We observed down-regulation of m/z 12,135 Da likely corresponding to cytochrome c (fold change 1.61; Kruskal–Wallis P value = 4.25 × 10−161, H test value = 731) in the Ppt1 −/− brain at the advanced stage of the disease. Interestingly, tumor necrosis factor alpha-initiated proteolytic processing of caspase 8 as well as the release of cytochrome c was impaired in PPT1-deficient fibroblasts as compared to control cells (Tardy et al. 2009). The other mitochondrial proteins that matched to previously identified m/z in the mouse brain [Online Resource 12, Table S12 and Online Resource 22, Figure S8 (Maier et al. 2013; McDonnell et al. 2014)] are parts of cytochrome c functional complex: Atp5j/ATP5 J and Cox7C/COX7C. ATP5 J is an ATP synthase, H + transporting, mitochondrial Fo complex, subunit F6 and part of complex V of oxidative phosphorylation (OXPHOS). Proteins of the ATP synthase complex, namely ATP5B and AT5A1, interact with PPT1 (Lyly et al. 2008; Scifo et al. 2015a; Kopra et al. 2004) and are dysregulated in the Ppt1 −/− brain. COX7C constitutes part of cytochrome c oxidase, which catalyzes electron transfer from a reduced form of cytochrome c to oxygen. Its down-regulation in conjunction with cytochrome c, which was observed at the advanced stage using MALDI-MSI is suggestive of severe disturbances in the mitochondrial respiration machinery of the CLN1 deficient brain.
The significant loss of axons is a common feature of many neurodegenerative disorders, i.e., Alzheimer’s, Parkinson’s and Huntington’s diseases, and it is consistent with the marked loss of neurons observed in these conditions. In NCL mouse models, severe axonal degeneration of long tracts in spinal cord and brain stem was observed in nclf mice, a spontaneous Cln6 mutation model of CLN6 disease (Bronson et al. 1998). Similarly, quantitative expression profiling of a subset of proteins implicated in regulation of axonal and synaptic vulnerability revealed changes in proteins involved in synaptic functions before the onset of axonal degeneration in both Ppt1 −/− and Cln6nclf mice (Kielar et al. 2009). MRI investigations disclosed focal or diffuse, low-grade fiber tract degeneration with loss of neurons in the brains of patients with neuronal ceroid lipofuscinosis (van der Knaap and Vaap 2005).
Wallerian degeneration of the nerve fibers or axonal tracts occurs rapidly following severe injury of the axons. In neurodegenerative disorders, axonal degeneration is a relatively slow process, probably due to neuronal cell body dysfunction; moreover, local cues along the axons may facilitate/enhance or even start axonal degeneration (Conforti et al. 2014). Modifier molecules such as pro-degenerative SARM1 (sterile alpha and TIR motif-containing 1) and ubiquitin ligase PHR1 which may alter axonal degeneration, have recently been identified (Gerdts et al. 2015). Conversely, overexpression of WLDS protein postpones axon degeneration in motor neuron and Parkinson’s disease models (reviewed in (Conforti et al. 2014). WLDS is a chimeric gene, which encodes full-length nicotinamide mononucleotide adenylyltransferase 1 (NMNAT1). Similarly to other NMNAT isoforms, NMNAT1 catalyzes NAD + synthesis from ATP and NMN, crucial for mitochondrial function. Disruption of axonal transport caused by genetic mutations or toxins, knockdown of Nmnat2 gene and mutations in Schwann cell genes cause WLDS-sensitive degeneration without physical injury. The key events in WLDS-sensitive degeneration can be portrayed in a conserved signaling pathway, consisting of initiation and execution phases, which are triggered by injury or other stresses (Conforti et al. 2014). Interestingly, the post-translational, covalent attachment of palmitic acid, usually to cysteine residues as in NMNAT2 is one of the elements of the initiation phase. Disturbing NMNAT2, pamitoylation-dependent localization to Golgi-derived vesicles by ubiquitylation and increased turnover strongly enhanced its protective capacity (Milde et al. 2013a, b). Since, in vivo substrates of PPT1 in the brain are not yet known, it is tempting to speculate whether this enzyme could play a role in palmitoylation/depalmitoylation dynamics important in the initiation phase of WLDS-sensitive degeneration.
Altogether, these findings demonstrate a number of down-regulated mitochondrial proteins to be affected along the disease course in Ppt1 −/− mice (summarized in Online Resource 2, Supplemental Table S2). Consequently, this may result in considerable hindrance of metabolic circuits in neurons, contributing to brain tissue damage. It would be of interest to target NAD+ and mitochondrial dynamics in future metabolomics profiling studies using PPT1-deficient brains.
Myelination in the brain is particularly important for upsurge in velocity at which impulses propagate along the myelinated fibers that facilitates the development of complex and compacted neural circuits. Defects in myelination perturb motor and sensory functions and are associated with cognitive impairment [reviewed in (Nave and Trapp 2008; Nave 2010)]. Myelination permits the saltatory conduction and is reasoned to save energy (Saab et al. 2013). The highest energy requirements occur along the internodes underneath the myelin sheath, and the majority of axonal mitochondria is located there (internodal stationary sites) (Ohno et al. 2011). Disturbances in myelination have been suggested for various NCL disorders, including: CLN5 (in Cln5 knockout mice), CLN8 (Cln8/mnd mutant), CLN10 (Cln10/Ctsd knockout mice) (Blom et al. 2013; Schmiedt et al. 2012; Kuronen et al. 2012; Koch et al. 2013; Mutka et al. 2010; Kopra et al. 2004) and CLN1 deficiency (Blom et al. 2013).
We have identified several dysregulated CNS myelin assembly proteins at various stages of disease progression. This down-regulation of various myelin components was the most pronounced at the advanced stage of the disease and included 9 structural myelin proteins and 15 myelin-associated ones (Fig. 7 and Online Resource 2, Table S2). Importantly, three myelin proteins: myelin basic protein/MBP, proteolipid 1/PLP1 and claudin 11/CLDN11 were found to be significantly down-regulated in their expression. Our previous proteomic studies on CLN10 deficiency using iTRAQ quantitation demonstrated that Plp1/PLP1 and Mbp/MBP were less abundant in Ctsd −/−mouse brains compared to wild type (Koch et al. 2013; Mutka et al. 2010). Down-regulation of all three myelin sheath components has previously been observed in microarray data from Cln1/Cn5 double knockout mouse 1-month brain cortex (Blom et al. 2013), which exhibits an exacerbated NCL phenotype earlier than either single knockout mouse. According to the calculated relative abundance of the CNS myelin-associated proteins by LC-MSE (Patzig et al. 2011; Jahn et al. 2009), PLP1, MBP and CLDN11 constitute 17, 8 and 1 % of the total myelin-associated proteins, respectively. Microarray analysis of porcine neural progenitor cells subjected to differentiation demonstrated strong up-regulation of transcripts for MBP, CLDN11 and GFAP together with notable increase in microtubule-associated protein 2 (MAP2) transcript (Yang et al. 2012). These results suggest that a differentiation cascade functionally links several genes/proteins observed to be down-regulated in the CLN1 deficiency. Down-regulation of MBP (a main protein constituent of the myelin sheath), over time was confirmed in our study by LC-MSE, MALDI-MSI and immunohistochemistry. MBP can induce chemotactic and inflammatory mediators, alter blood-brain barrier permeability (BBB) as well as tight junction expression, thus contributing to the BBB rupture characteristic of multiple sclerosis (D’Aversa et al. 2013). Interestingly, several MBP- and PLP1-derived peptides display encephalitogenic properties and can induce experimental autoimmune encephalomyelitis [EAE; reviewed in (McCarthy et al. 2012)]. Recently, Groh et al. (2013) investigated the role of inflammatory cells in the Ppt1 −/−mouse brain. In this study, an early infiltration of CD8+ T lymphocytes targeting Ppt1 −/− neurons and activation of microglia/macrophage-like cells were observed. The fact that damage was predominantly located at the juxtaparanodal domains of the node of Ranvier potentially defined this region as a new ‘hot spot’ for immune-mediated axonopathy.
Claudin 11 (Osp) and PLP1 are members of four-transmembrane (tetraspan) family of proteins abundant in CNS myelin (Jahn et al. 2009). Both proteins maintain compact myelin structure and exhibit overlapping functions. Mice in which the two genes were knocked out suffered from severe neurological deficits, particularly, abnormal myelin compaction and smaller axon diameters. However, when single genes were knocked out, the expression of the other gene was increased, pointing to compensatory effects (Chow et al. 2005). Claudin 11 is a member of claudin family, with 23 members in humans, and constitutes part of tight junctions (TJs) in CNS myelin (Findley and Koval 2009). Studies using Cldn11 −/−mice brains demonstrated a major function of claudin 11 TJs in preventing internodal depolarization at the myelin membrane, which has great impact on small diameter myelinated axons. Cldn11 −/− mice were characterized by preserved ultrastructure of myelinated axons, but displayed sheer decrease (~60 %) in conduction (Devaux and Gow 2008). Measured action potential was significantly increased in these mice, in addition to augmented juxtaparanodal potassium currents in small fibers lacking tight junctions. These data signified that TJs modulate the biophysical properties of myelin (Devaux and Gow 2008). Taken together, we hypothesize that low expression of claudin 11 in the CLN1-deficient brain may exert a similar negative effect on TJs, resulting in deficient salutatory conduction and in consequence leading to dysfunctional myelin.
Since the down-regulation in expression of various myelin proteins was very pronounced at the late stages in the Ppt1 −/− brain and preceded by changes in various mitochondrial proteins, we thus hypothesize that early mitochondrial dysfunction may contribute to both impaired axonal activities [leading eventually to axonal degeneration, a prominent feature of infantile CLN1 disease (Haltia 2006), and supported by our study] and hamper mechanisms for myelin renewal and maintenance, leading to decreased amounts of myelin proteins.
Electric signal transmission is one of the key properties of myelin, which is facilitated by tight wrapping of multilamellar membranes around axon fibers. The minor alterations in membrane lipid composition may result in aberrations in their distribution and domain size and have impact on adhesive properties (Lee et al. 2014). Myelin membranes constitute lipids enriched in cholesterol, galactolipids and glycosphingolipids (Chrast et al. 2011) and as such are sensitive to altered lipid metabolism. It has been hypothesized that NCL proteins are directly implicated in lipid metabolism/transport and myelination processes (Schmiedt et al. 2012; Kuronen et al. 2012). PPT1 regulates lipid raft properties such as glycosphingolipid content and transport (Goswami et al. 2005). Disturbances in lipid metabolism were postulated to affect the assembly of the myelin sheath, leading to defective myelination (Blom et al. 2013). Moreover, alterations in all major lipid classes, sphingolipids, phospholipids and cholesterol in the Ppt1-deficient brain have been reported (Lyly et al. 2008). Therefore, myelin disruption might also result from primary disturbances in lipidation, lipid metabolism and/or palmitoylation.
Taken together, the common origin of myelin dysfunction may be due to: (i) defects in lipidation, including palmitoylation which consequently leads to impaired myelin assembly, (ii) mitochondrial dysfunction and impaired energy supply which affect myelin maintenance and stabilization, (iii) myelin breakdown related to lowered expression of various myelin proteins, which may be secondary to axonal degeneration. Moreover, our study demonstrates the myelin alterations occur very early in the CLN1 disease process and may contribute to neuronal networks malfunctioning.
Dysregulation of various (mostly metabolic) pathways has been suggested in NCL disorders. Confirming these observations at the protein level in the Ppt1-deficient thalamus at different disease stages, we also linked dysregulated thalamic proteins to a variety of signaling pathways (Online Resource, Tables S4, S6 and S8). For example, we have observed a significant down-regulation of signaling by Rho family GTPases (Online Resource 18, Figure S4), which confirms previous observations of global gene expression microarray profiling in the brain of these mice (von Schantz et al. 2008), and is strongly supported by the results of RNA sequence analysis in CLN1 patients’ fibroblasts (respective Z scores −1.941 and −2.324) with 8 common transcripts shared between patients with different CLN1 mutations (Online Resource 18, Figure S4). It is known that Rho family GTPases, key regulators of actin dynamics and organization, play critical roles in orchestrating the development and remodeling of spines and synapses. Aberrant Rho GTPase signaling induces spine and synapse defects as well as cognitive impairments, leading to intellectual disabilities (Ba et al. 2013). Rho GTPase signaling is strongly affected in multiple neurodevelopmental, neuropsychiatric and neurodegenerative disorders (Ba et al. 2013; Newey et al. 2005; Fiala et al. 2002) and linked to abnormalities/loss of dendritic spines (van Spronsen and Hoogenraad 2010). It is therefore very likely that the same pathway(s) are affected in the CLN1 deficiency, where the intellectual disabilities occur at the very early stage of the disease. Interestingly, a dramatic reduction in density of dendritic spines has recently been reported in the mouse brain cortex of Cln6 nclf mice, which is suggestive of a reduction in synaptic strength following disruption in CLN6 (Morgan et al. 2013). Proteins linked to Rho GTPase signaling are also critical for neuronal growth cone—cytoskeletal dynamics and subsequently to axon guidance/growth processes. The aberration in the dynamics of the neuronal outgrowth, including neuritogenesis, branching and microtubule dynamics, was observed already at the pre-symptomatic stage in the Ppt1 −/− brain, with several differentially expressed proteins bioinformatically linked to these processes; these findings were further supported by RNA-seq analysis data in the CLN1 patients’ fibroblasts from an advanced stage of the disease and by studies with CLN1 patient-derived iPS cell lines (Uusi-Rauva et al., unpublished). Since CLN1 deficiency is also linked to axonal degeneration (Kielar et al. 2009; von Schantz et al. 2008), we speculate that down-regulation of Rho GTPase signaling in the brain may be linked to severe neurodegenerative phenotype observed in this disorder.
Another, novel pathway found to be affected at the advanced stage in the Ppt1 −/−thalamus, is Huntington’s disease (HD) signaling (8 proteins assigned, Fig. 6). Disturbances in HD signaling were, so far, not specifically associated with CLN1 deficiency and may underlie the general cognitive and behavioral disturbances, similarly as recently described for 22q11 deletion syndrome (22q11DS), linked to schizophrenia (van Beveren et al. 2012). Similarly to other NCL disorders, CLN1 shares some common features with HD (Margolis and Ross 2003), including: neuronal degeneration, caudate involvement and mental deterioration, with cognitive decline starting early in childhood (Hofmann et al. 1999). Interestingly, 3 of the differentially expressed proteins linked to Huntington’s disease signaling category are also components of the perinuclear inclusions in HD (Fig. 6), while Dlg4/DLG4 and Stx1a/STX1A contribute to lipofuscin proteome (Online Resource 2, Table S2). It is tempting to speculate that both HD and CLN1 may also be linked by the involvement of the same oligomerization-prone proteins in the process of amyloid/ lipofuscin aggregates formation, thus playing a role in disease pathologies.
Proteomic approaches utilizing targeted, label-free LCM-based quantitative mass spectrometry may reveal changes directly related to cellular dynamics in disease-affected brain regions. This approach allowed us to pinpoint several molecular processes, pathways and disease-associated functional modules related to progression of CLN1 deficiency in the mouse brain. Our transdisciplinary approaches (proteomic disease profiling and analysis of protein assemblies by network analysis, fortified by RNA sequence analysis in CLN1 patients’ fibroblasts) are well suited for studies of various neurodegenerative disorders.
Abbreviations
- FDR:
-
False discovery rate
- INCL/CLN1:
-
Classic infantile NCL
- NCL:
-
Neuronal ceroid lipofuscinoses
- LCM:
-
Laser capture microdissection
- LC-MSE :
-
Liquid chromatography tandem mass spectrometry
- LSDs:
-
Lysosomal storage disorders
- MALDI-MSI:
-
Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry imaging
- PPT1:
-
Palmitoyl-protein thioesterase 1
- Ppt1 −/− :
-
Ppt1 gene knockout
- RNA-seq:
-
RNA sequence analysis
References
Aby, E., Gumps, K., Roth, A., Sigmon, S., Jenkins, S. E., Kim, J. J., et al. (2013). Mutations in palmitoyl-protein thioesterase 1 alter exocytosis and endocytosis at synapses in Drosophila larvae. Fly (Austin), 7(4), 267–279.
Ahtiainen, L., Kolikova, J., Mutka, A. L., Luiro, K., Gentile, M., Ikonen, E., et al. (2007). Palmitoyl protein thioesterase 1 (Ppt1)-deficient mouse neurons show alterations in cholesterol metabolism and calcium homeostasis prior to synaptic dysfunction. Neurobiology of Diseases, 28(1), 52–64.
Ahtiainen, L., Luiro, K., Kauppi, M., Tyynela, J., Kopra, O., & Jalanko, A. (2006). Palmitoyl protein thioesterase 1 (PPT1) deficiency causes endocytic defects connected to abnormal saposin processing. Experimental Cell Research, 312(9), 1540–1553.
Ahtiainen, L., Van Diggelen, O. P., Jalanko, A., & Kopra, O. (2003). Palmitoyl protein thioesterase 1 is targeted to the axons in neurons. The Journal of Comparative Neurology, 455(3), 368–377.
Anderson, G. W., Goebel, H. H., & Simonati, A. (2013). Human pathology in NCL. Biochimica et Biophysica Acta, 1832(11), 1807–1826.
Ba, W., van der Raadt, J., & Nadif Kasri, N. (2013). Rho GTPase signaling at the synapse: Implications for intellectual disability. Experimental Cell Research, 319(15), 2368–2374.
Bible, E., Gupta, P., Hofmann, S. L., & Cooper, J. D. (2004). Regional and cellular neuropathology in the palmitoyl protein thioesterase-1 null mutant mouse model of infantile neuronal ceroid lipofuscinosis. Neurobiology of Diseases, 16(2), 346–359.
Blom, T., Schmiedt, M. L., Wong, A. M., Kyttala, A., Soronen, J., Jauhiainen, M., et al. (2013). Exacerbated neuronal ceroid lipofuscinosis phenotype in Cln1/5 double-knockout mice. Disease Models & Mechanisms., 6(2), 342–357.
Boustany, R. M. (2013). Lysosomal storage diseases—The horizon expands. Nature Reviews. Neurology, 9(10), 583–598.
Braakman, R. B., Luider, T. M., Martens, J. W., Foekens, J. A., & Umar, A. (2011). Laser capture microdissection applications in breast cancer proteomics. Methods in Molecular Biology, 755, 143–154.
Braakman, R. B., Tilanus-Linthorst, M. M., Liu, N. Q., Stingl, C., Dekker, L. J., Luider, T. M., et al. (2012). Optimized nLC-MS workflow for laser capture microdissected breast cancer tissue. Journal of Proteomics, 75(10), 2844–2854.
Bronson, R. T., Donahue, L. R., Johnson, K. R., Tanner, A., Lane, P. W., & Faust, J. R. (1998). Neuronal ceroid lipofuscinosis (nclf), a new disorder of the mouse linked to chromosome 9. American Journal of Medical Genetics, 77(4), 289–297.
Caldwell, R. L., & Caprioli, R. M. (2005). Tissue profiling by mass spectrometry: A review of methodology and applications. Molecular and Cellular Proteomics, 4(4), 394–401.
Camp, L. A., & Hofmann, S. L. (1993). Purification and properties of a palmitoyl-protein thioesterase that cleaves palmitate from H-Ras. Journal of Biological Chemistry, 268(30), 22566–22574.
Cao, Y., Staropoli, J. F., Biswas, S., Espinola, J. A., MacDonald, M. E., Lee, J. M., et al. (2011). Distinct early molecular responses to mutations causing vLINCL and JNCL presage ATP synthase subunit C accumulation in cerebellar cells. PLoS ONE, 6(2), e17118.
Chan, C. H., Want, E. J., Geraets, R., Weber, K., Hersrud, S., & Pearce, D. A. (2012) Biomarker Discovery in Batten Disease. In NCL2012, Roayl Holloway College, London, UK, 2012.
Cho, S., & Dawson, G. (2000). Palmitoyl protein thioesterase 1 protects against apoptosis mediated by Ras-Akt-caspase pathway in neuroblastoma cells. Journal of Neurochemistry, 74(4), 1478–1488.
Chow, E., Mottahedeh, J., Prins, M., Ridder, W., Nusinowitz, S., & Bronstein, J. M. (2005). Disrupted compaction of CNS myelin in an OSP/Claudin-11 and PLP/DM20 double knockout mouse. Molecular and Cellular Neuroscience, 29(3), 405–413.
Chrast, R., Saher, G., Nave, K. A., & Verheijen, M. H. (2011). Lipid metabolism in myelinating glial cells: Lessons from human inherited disorders and mouse models. Journal of Lipid Research, 52(3), 419–434.
Chu-LaGraff, Q., Blanchette, C., O’Hern, P., & Denefrio, C. (2010). The batten disease palmitoyl protein thioesterase 1 gene regulates neural specification and axon connectivity during drosophila embryonic development. PLoS ONE, 5(12), e14402.
Conforti, L., Gilley, J., & Coleman, M. P. (2014). Wallerian degeneration: An emerging axon death pathway linking injury and disease. Nature Reviews Neuroscience, 15(6), 394–409.
Das, A. M., Jolly, R. D., & Kohlschutter, A. (1999). Anomalies of mitochondrial ATP synthase regulation in four different types of neuronal ceroid lipofuscinosis. Molecular Genetics and Metabolism, 66(4), 349–355.
Das, A. M., von Harlem, R., Feist, M., Lucke, T., & Kohlschutter, A. (2001). Altered levels of high-energy phosphate compounds in fibroblasts from different forms of neuronal ceroid lipofuscinoses: Further evidence for mitochondrial involvement. European Journal of Paediatric Neurology, 5(Suppl A), 143–146.
D’Aversa, T. G., Eugenin, E. A., Lopez, L., & Berman, J. W. (2013). Myelin basic protein induces inflammatory mediators from primary human endothelial cells and blood-brain barrier disruption: Implications for the pathogenesis of multiple sclerosis. Neuropathology and Applied Neurobiology, 39(3), 270–283.
de Monasterio-Schrader, P., Jahn, O., Tenzer, S., Wichert, S. P., Patzig, J., & Werner, H. B. (2012). Systematic approaches to central nervous system myelin. Cellular and Molecular Life Sciences, 69(17), 2879–2894.
Devaux, J., & Gow, A. (2008). Tight junctions potentiate the insulative properties of small CNS myelinated axons. Journal of Cell Biology, 183(5), 909–921.
Eberlin, L. S., Liu, X., Ferreira, C. R., Santagata, S., Agar, N. Y., & Cooks, R. G. (2011). Desorption electrospray ionization then MALDI mass spectrometry imaging of lipid and protein distributions in single tissue sections. Analytical Chemistry, 83(22), 8366–8371.
Elshatory, Y., Brooks, A. I., Chattopadhyay, S., Curran, T. M., Gupta, P., Ramalingam, V., et al. (2003). Early changes in gene expression in two models of Batten disease. FEBS Letters, 538(1–3), 207–212.
Faul, F., Erdfelder, E., Lang, A. G., & Buchner, A. (2007). G*Power 3: A flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behavior Research Methods, 39(2), 175–191.
Fiala, J. C., Spacek, J., & Harris, K. M. (2002). Dendritic spine pathology: Cause or consequence of neurological disorders? Brain Research. Brain Research Reviews, 39(1), 29–54.
Findley, M. K., & Koval, M. (2009). Regulation and roles for claudin-family tight junction proteins. IUBMB Life, 61(4), 431–437.
Gerdts, J., Brace, E. J., Sasaki, Y., DiAntonio, A., & Milbrandt, J. (2015). SARM1 activation triggers axon degeneration locally via NAD(+) destruction. Science, 348(6233), 453–457.
Goswami, R., Ahmed, M., Kilkus, J., Han, T., Dawson, S. A., & Dawson, G. (2005). Differential regulation of ceramide in lipid-rich microdomains (rafts): Antagonistic role of palmitoyl:protein thioesterase and neutral sphingomyelinase 2. Journal of Neuroscience Research, 81(2), 208–217.
Groh, J., Kuhl, T. G., Ip, C. W., Nelvagal, H. R., Sri, S., Duckett, S., et al. (2013). Immune cells perturb axons and impair neuronal survival in a mouse model of infantile neuronal ceroid lipofuscinosis. Brain, 136(Pt 4), 1083–1101.
Gupta, P., Soyombo, A. A., Atashband, A., Wisniewski, K. E., Shelton, J. M., Richardson, J. A., et al. (2001). Disruption of PPT1 or PPT2 causes neuronal ceroid lipofuscinosis in knockout mice. Proceedings of the National Academy of Sciences of the United States of America, 98(24), 13566–13571.
Haltia, M. (2006). The neuronal ceroid-lipofuscinoses: from past to present. Biochimica et Biophysica Acta, 1762(10), 850–856.
Haltia, M., Rapola, J., & Santavuori, P. (1973a). Infantile type of so-called neuronal ceroid-lipofuscinosis. Histological and electron microscopic studies. Acta Neuropathologica, 26(2), 157–170.
Haltia, M., Rapola, J., Santavuori, P., & Keranen, A. (1973b). Infantile type of so-called neuronal ceroid-lipofuscinosis. 2. Morphological and biochemical studies. Journal of the Neurological Sciences, 18(3), 269–285.
Heinonen, O., Kyttala, A., Lehmus, E., Paunio, T., Peltonen, L., & Jalanko, A. (2000). Expression of palmitoyl protein thioesterase in neurons. Molecular Genetics and Metabolism, 69(2), 123–129.
Hofmann, S. L., Das, A. K., Yi, W., Lu, J. Y., & Wisniewski, K. E. (1999). Genotype-phenotype correlations in neuronal ceroid lipofuscinosis due to palmitoyl-protein thioesterase deficiency. Molecular Genetics and Metabolism, 66(4), 234–239.
Ikem, A. (2010). Measurement of volatile organic compounds in bottled and tap waters by purge and trap GC–MS: Are drinking water types different? Journal of Food Composition and Analysis, 23(1), 70–77.
Isosomppi, J., Heinonen, O., Hiltunen, J. O., Greene, N. D., Vesa, J., Uusitalo, A., et al. (1999). Developmental expression of palmitoyl protein thioesterase in normal mice. Brain Research. Developmental Brain Research, 118(1–2), 1–11.
Jahn, O., Tenzer, S., & Werner, H. B. (2009). Myelin proteomics: Molecular anatomy of an insulating sheath. Molecular Neurobiology, 40(1), 55–72.
Jalanko, A., Tyynela, J., & Peltonen, L. (2006). From genes to systems: New global strategies for the characterization of NCL biology. Biochimica et Biophysica Acta, 1762(10), 934–944.
Jalanko, A., Vesa, J., Manninen, T., von Schantz, C., Minye, H., Fabritius, A. L., et al. (2005). Mice with Ppt1Deltaex4 mutation replicate the INCL phenotype and show an inflammation-associated loss of interneurons. Neurobiology of Diseases, 18(1), 226–241.
Kakela, R., Somerharju, P., & Tyynela, J. (2003). Analysis of phospholipid molecular species in brains from patients with infantile and juvenile neuronal-ceroid lipofuscinosis using liquid chromatography-electrospray ionization mass spectrometry. Journal of Neurochemistry, 84(5), 1051–1065.
Kamate, M., & Hattiholi, V. (2012). Novel neuroimaging finding in palmitoyl protein thioesterase-1-related neuronal ceroid lipofuscinosis. Pediatric Neurology, 46(5), 325–328.
Karlsson, O., Bergquist, J., & Andersson, M. (2014). Quality measures of imaging mass spectrometry aids in revealing long-term striatal protein changes induced by neonatal exposure to the cyanobacterial toxin beta-N-methylamino-l-alanine (BMAA). Molecular and Cellular Proteomics, 13(1), 93–104.
Kielar, C., Maddox, L., Bible, E., Pontikis, C. C., Macauley, S. L., Griffey, M. A., et al. (2007). Successive neuron loss in the thalamus and cortex in a mouse model of infantile neuronal ceroid lipofuscinosis. Neurobiology of Diseases, 25(1), 150–162.
Kielar, C., Wishart, T. M., Palmer, A., Dihanich, S., Wong, A. M., Macauley, S. L., et al. (2009). Molecular correlates of axonal and synaptic pathology in mouse models of batten disease. Human Molecular Genetics, 18(21), 4066–4080.
Kim, S. J., Zhang, Z., Lee, Y. C., & Mukherjee, A. B. (2006). Palmitoyl-protein thioesterase-1 deficiency leads to the activation of caspase-9 and contributes to rapid neurodegeneration in INCL. Human Molecular Genetics, 15(10), 1580–1586.
Kim, S. J., Zhang, Z., Sarkar, C., Tsai, P. C., Lee, Y. C., Dye, L., et al. (2008). Palmitoyl protein thioesterase-1 deficiency impairs synaptic vesicle recycling at nerve terminals, contributing to neuropathology in humans and mice. The Journal of Clinical Investigation, 118(9), 3075–3086.
Koch, S., Scifo, E., Rokka, A., Trippner, P., Lindfors, M., Korhonen, R., et al. (2013). Cathepsin D deficiency induces cytoskeletal changes and affects cell migration pathways in the brain. Neurobiology of Diseases, 50, 107–119.
Kopra, O., Vesa, J., von Schantz, C., Manninen, T., Minye, H., Fabritius, A. L., et al. (2004). A mouse model for Finnish variant late infantile neuronal ceroid lipofuscinosis, CLN5, reveals neuropathology associated with early aging. Human Molecular Genetics, 13(23), 2893–2906.
Kousi, M., Lehesjoki, A. E., & Mole, S. E. (2012). Update on the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses. Human Mutation, 33(1), 42–63.
Kuronen, M., Hermansson, M., Manninen, O., Zech, I., Talvitie, M., Laitinen, T., et al. (2012). Galactolipid deficiency in the early pathogenesis of neuronal ceroid lipofuscinosis model Cln8mnd: Implications to delayed myelination and oligodendrocyte maturation. Neuropathology and Applied Neurobiology, 38(5), 471–486.
Lalowski, M., Magni, F., Mainini, V., Monogioudi, E., Gotsopoulos, A., Soliymani, R., et al. (2013). Imaging mass spectrometry: A new tool for kidney disease investigations. Nephrology, Dialysis, Transplantation, 28(7), 1648–1656.
Lee, D. W., Banquy, X., Kristiansen, K., Kaufman, Y., Boggs, J. M., & Israelachvili, J. N. (2014). Lipid domains control myelin basic protein adsorption and membrane interactions between model myelin lipid bilayers. Proceedings of the National Academy of Sciences of the United States of America, 111(8), E768–E775.
Lehrer, S., Roboz, J., Ding, H., Zhao, S., Diamond, E. J., Holland, J. F., et al. (2003). Putative protein markers in the sera of men with prostatic neoplasms. BJU International, 92(3), 223–225.
Lehtovirta, M., Kyttala, A., Eskelinen, E. L., Hess, M., Heinonen, O., & Jalanko, A. (2001). Palmitoyl protein thioesterase (PPT) localizes into synaptosomes and synaptic vesicles in neurons: implications for infantile neuronal ceroid lipofuscinosis (INCL). Human Molecular Genetics, 10(1), 69–75.
Leverenz, J. B., Umar, I., Wang, Q., Montine, T. J., McMillan, P. J., Tsuang, D. W., et al. (2007). Proteomic identification of novel proteins in cortical Lewy bodies. Brain Pathology, 17(2), 139–145.
Liu, N. Q., Braakman, R. B., Stingl, C., Luider, T. M., Martens, J. W., Foekens, J. A., et al. (2012). Proteomics pipeline for biomarker discovery of laser capture microdissected breast cancer tissue. J Mammary Gland Biol Neoplasia, 17(2), 155–164.
Luiro, K., Kopra, O., Blom, T., Gentile, M., Mitchison, H. M., Hovatta, I., et al. (2006). Batten disease (JNCL) is linked to disturbances in mitochondrial, cytoskeletal, and synaptic compartments. Journal of Neuroscience Research, 84(5), 1124–1138.
Lyly, A., Marjavaara, S. K., Kyttala, A., Uusi-Rauva, K., Luiro, K., Kopra, O., et al. (2008). Deficiency of the INCL protein Ppt1 results in changes in ectopic F1-ATP synthase and altered cholesterol metabolism. Human Molecular Genetics, 17(10), 1406–1417.
Lyly, A., von Schantz, C., Heine, C., Schmiedt, M. L., Sipila, T., Jalanko, A., et al. (2009). Novel interactions of CLN5 support molecular networking between Neuronal Ceroid Lipofuscinosis proteins. BMC Cell Biology, 10, 83.
Macauley, S. L., Pekny, M., & Sands, M. S. (2011). The role of attenuated astrocyte activation in infantile neuronal ceroid lipofuscinosis. Journal of Neuroscience, 31(43), 15575–15585.
Maier, S. K., Hahne, H., Gholami, A. M., Balluff, B., Meding, S., Schoene, C., et al. (2013). Comprehensive identification of proteins from MALDI imaging. Molecular and Cellular Proteomics, 12(10), 2901–2910.
Mainini, V., Lalowski, M., Gotsopoulos, A., Bitsika, V., Baumann, M., & Magni, F. (2015). MALDI-imaging mass spectrometry on tissues. Methods in Molecular Biology, 1243, 139–164.
Margolis, R. L., & Ross, C. A. (2003). Diagnosis of Huntington disease. Clinical Chemistry, 49(10), 1726–1732.
McCarthy, D. P., Richards, M. H., & Miller, S. D. (2012). Mouse models of multiple sclerosis: Experimental autoimmune encephalomyelitis and Theiler’s virus-induced demyelinating disease. Methods in Molecular Biology, 900, 381–401.
McDonnell, L. A., Walch, A., Stoeckli, M., & Corthals, G. L. (2014). MSiMass list: A public database of identifications for protein MALDI MS imaging. Journal of Proteome Research, 13(2), 1138–1142.
Milde, S., Fox, A. N., Freeman, M. R., & Coleman, M. P. (2013a). Deletions within its subcellular targeting domain enhance the axon protective capacity of Nmnat2 in vivo. Scientific Reports, 3, 2567.
Milde, S., Gilley, J., & Coleman, M. P. (2013b). Subcellular localization determines the stability and axon protective capacity of axon survival factor Nmnat2. PLoS Biology, 11(4), e1001539.
Miller, J. N., Kovacs, A. D., & Pearce, D. A. (2015). The novel Cln1R151X mouse model of infantile neuronal ceroid lipofuscinosis (INCL) for testing nonsense suppression therapy. Human Molecular Genetics, 24(1), 185–196.
Miravalle, L., Calero, M., Takao, M., Roher, A. E., Ghetti, B., & Vidal, R. (2005). Amino-terminally truncated Abeta peptide species are the main component of cotton wool plaques. Biochemistry, 44(32), 10810–10821.
Morgan, J. P., Magee, H., Wong, A., Nelson, T., Koch, B., Cooper, J. D., et al. (2013). A murine model of variant late infantile ceroid lipofuscinosis recapitulates behavioral and pathological phenotypes of human disease. PLoS ONE, 8(11), e78694.
Mutka, A. L., Haapanen, A., Kakela, R., Lindfors, M., Wright, A. K., Inkinen, T., et al. (2010). Murine cathepsin D deficiency is associated with dysmyelination/myelin disruption and accumulation of cholesteryl esters in the brain. Journal of Neurochemistry, 112(1), 193–203.
Nave, K. A. (2010). Myelination and support of axonal integrity by glia. Nature, 468(7321), 244–252.
Nave, K. A., & Trapp, B. D. (2008). Axon-glial signaling and the glial support of axon function. Annual Review of Neuroscience, 31, 535–561.
Newey, S. E., Velamoor, V., Govek, E. E., & Van Aelst, L. (2005). Rho GTPases, dendritic structure, and mental retardation. Journal of Neurobiology, 64(1), 58–74.
Ng, K. P., Gugiu, B., Renganathan, K., Davies, M. W., Gu, X., Crabb, J. S., et al. (2008). Retinal pigment epithelium lipofuscin proteomics. Molecular and Cellular Proteomics, 7(7), 1397–1405.
Ohno, N., Kidd, G. J., Mahad, D., Kiryu-Seo, S., Avishai, A., Komuro, H., et al. (2011). Myelination and axonal electrical activity modulate the distribution and motility of mitochondria at CNS nodes of Ranvier. Journal of Neuroscience, 31(20), 7249–7258.
Ottis, P., Koppe, K., Onisko, B., Dynin, I., Arzberger, T., Kretzschmar, H., et al. (2012). Human and rat brain lipofuscin proteome. Proteomics, 12(15–16), 2445–2454.
Palmer, D. N., Barry, L. A., Tyynela, J., & Cooper, J. D. (2013). NCL disease mechanisms. Biochimica et Biophysica Acta, 1832(11), 1882–1893.
Patzig, J., Jahn, O., Tenzer, S., Wichert, S. P., de Monasterio-Schrader, P., Rosfa, S., et al. (2011). Quantitative and integrative proteome analysis of peripheral nerve myelin identifies novel myelin proteins and candidate neuropathy loci. Journal of Neuroscience, 31(45), 16369–16386.
Pezzini, F., Gismondi, F., Tessa, A., Tonin, P., Carrozzo, R., Mole, S. E., et al. (2011). Involvement of the mitochondrial compartment in human NCL fibroblasts. Biochemical and Biophysical Research Communications, 416(1–2), 159–164.
Philipson, O., Lord, A., Lalowski, M., Soliymani, R., Baumann, M., Thyberg, J., et al. (2012). The Arctic amyloid-beta precursor protein (AbetaPP) mutation results in distinct plaques and accumulation of N- and C-truncated Abeta. Neurobiol Aging, 33(5), 1010e1–1010e13.
Saab, A. S., Tzvetanova, I. D., & Nave, K. A. (2013). The role of myelin and oligodendrocytes in axonal energy metabolism. Current Opinion in Neurobiology, 23(6), 1065–1072.
Santorelli, F. M., Bertini, E., Petruzzella, V., Di Capua, M., Calvieri, S., Gasparini, P., et al. (1998). A novel insertion mutation (A169i) in the CLN1 gene is associated with infantile neuronal ceroid lipofuscinosis in an Italian patient. Biochemical and Biophysical Research Communications, 245(2), 519–522.
Schaefer, M. H., Fontaine, J. F., Vinayagam, A., Porras, P., Wanker, E. E., & Andrade-Navarro, M. A. (2012). HIPPIE: Integrating protein interaction networks with experiment based quality scores. PLoS ONE, 7(2), e31826.
Schmiedt, M. L., Blom, T., Kopra, O., Wong, A., von Schantz-Fant, C., Ikonen, E., et al. (2012). Cln5-deficiency in mice leads to microglial activation, defective myelination and changes in lipid metabolism. Neurobiology of Diseases, 46(1), 19–29.
Schon, E. A., & Manfredi, G. (2003). Neuronal degeneration and mitochondrial dysfunction. The Journal of Clinical Investigation, 111(3), 303–312.
Scifo, E., Szwajda, A., Debski, J., Uusi-Rauva, K., Kesti, T., Dadlez, M., et al. (2013). Drafting the CLN3 protein interactome in SH-SY5Y human neuroblastoma cells: A label-free quantitative proteomics approach. Journal of Proteome Research, 12(5), 2101–2115.
Scifo, E., Szwajda, A., Soliymani, R., Pezzini, F., Bianchi, M., Dapkunas, A., et al. (2015a). Quantitative analysis of PPT1 interactome in human neuroblastoma cells. Data in Brief, 4, 207–216.
Scifo, E., Szwajda, A., Soliymani, R., Pezzini, F., Bianchi, M., Dapkunas, A., et al. (2015b). Proteomic analysis of the palmitoyl protein thioesterase 1 interactome in SH-SY5Y human neuroblastoma cells. J Proteomics, 123, 42–53.
Simonati, A., Tessa, A., Bernardina, B. D., Biancheri, R., Veneselli, E., Tozzi, G., et al. (2009). Variant late infantile neuronal ceroid lipofuscinosis because of CLN1 mutations. Pediatric Neurology, 40(4), 271–276.
Stingl, C., van Vilsteren, F. G., Guzel, C., Ten Kate, F. J., Visser, M., Krishnadath, K. K., et al. (2011). Reproducibility of protein identification of selected cell types in Barrett’s esophagus analyzed by combining laser-capture microdissection and mass spectrometry. Journal of Proteome Research, 10(1), 288–298.
Suopanki, J., Tyynela, J., Baumann, M., & Haltia, M. (1999a). The expression of palmitoyl-protein thioesterase is developmentally regulated in neural tissues but not in nonneural tissues. Molecular Genetics and Metabolism, 66(4), 290–293.
Suopanki, J., Tyynela, J., Baumann, M., & Haltia, M. (1999b). Palmitoyl-protein thioesterase, an enzyme implicated in neurodegeneration, is localized in neurons and is developmentally regulated in rat brain. Neuroscience Letters, 265(1), 53–56.
Tardy, C., Sabourdy, F., Garcia, V., Jalanko, A., Therville, N., Levade, T., et al. (2009). Palmitoyl protein thioesterase 1 modulates tumor necrosis factor alpha-induced apoptosis. Biochimica et Biophysica Acta, 1793(7), 1250–1258.
Taylor, C. M., Marta, C. B., Claycomb, R. J., Han, D. K., Rasband, M. N., Coetzee, T., et al. (2004). Proteomic mapping provides powerful insights into functional myelin biology. Proceedings of the National Academy of Sciences of the United States of America, 101(13), 4643–4648.
Trapnell, C., Roberts, A., Goff, L., Pertea, G., Kim, D., Kelley, D. R., et al. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature Protocols, 7(3), 562–578.
Umar, A., Dalebout, J. C., Timmermans, A. M., Foekens, J. A., & Luider, T. M. (2005). Method optimisation for peptide profiling of microdissected breast carcinoma tissue by matrix-assisted laser desorption/ionisation-time of flight and matrix-assisted laser desorption/ionisation-time of flight/time of flight-mass spectrometry. Proteomics, 5(10), 2680–2688.
van Beveren, N. J., Krab, L. C., Swagemakers, S., Buitendijk, G. H., Boot, E., van der Spek, P., et al. (2012). Functional gene-expression analysis shows involvement of schizophrenia-relevant pathways in patients with 22q11 deletion syndrome. PLoS ONE, 7(3), e33473.
van der Knaap, M. S., & Vaap, J. (2005). Magnetic resonance of myelination and myelin disorders (3rd ed.). Berlin: Springer.
van Spronsen, M., & Hoogenraad, C. C. (2010). Synapse pathology in psychiatric and neurologic disease. Current Neurology and Neuroscience Reports, 10(3), 207–214.
Vanhanen, S. L., Puranen, J., Autti, T., Raininko, R., Liewendahl, K., Nikkinen, P., et al. (2004). Neuroradiological findings (MRS, MRI, SPECT) in infantile neuronal ceroid-lipofuscinosis (infantile CLN1) at different stages of the disease. Neuropediatrics, 35(1), 27–35.
Vanrobaeys, F., Van Coster, R., Dhondt, G., Devreese, B., & Van Beeumen, J. (2005). Profiling of myelin proteins by 2D-gel electrophoresis and multidimensional liquid chromatography coupled to MALDI TOF-TOF mass spectrometry. Journal of Proteome Research, 4(6), 2283–2293.
Vesa, J., Chin, M. H., Oelgeschlager, K., Isosomppi, J., DellAngelica, E. C., Jalanko, A., et al. (2002). Neuronal ceroid lipofuscinoses are connected at molecular level: interaction of CLN5 protein with CLN2 and CLN3. Molecular Biology of the Cell, 13(7), 2410–2420.
Vesa, J., Hellsten, E., Verkruyse, L. A., Camp, L. A., Rapola, J., Santavuori, P., et al. (1995). Mutations in the palmitoyl protein thioesterase gene causing infantile neuronal ceroid lipofuscinosis. Nature, 376(6541), 584–587.
Virmani, T., Gupta, P., Liu, X., Kavalali, E. T., & Hofmann, S. L. (2005). Progressively reduced synaptic vesicle pool size in cultured neurons derived from neuronal ceroid lipofuscinosis-1 knockout mice. Neurobiology of Diseases, 20(2), 314–323.
von Schantz, C., Saharinen, J., Kopra, O., Cooper, J. D., Gentile, M., Hovatta, I., et al. (2008). Brain gene expression profiles of Cln1 and Cln5 deficient mice unravels common molecular pathways underlying neuronal degeneration in NCL diseases. BMC Genomics, 9, 146.
Wang, P., Ju, W., Wu, D., Wang, L., Yan, M., Zou, J., et al. (2011). A two-dimensional protein fragmentation-proteomic study of neuronal ceroid lipofuscinoses: Identification and characterization of differentially expressed proteins. Journal of Chromatography. B, Analytical Technologies in the Biomedical and Life Sciences, 879(5–6), 304–316.
Warburton, S., Southwick, K., Hardman, R. M., Secrest, A. M., Grow, R. K., Xin, H., et al. (2005). Examining the proteins of functional retinal lipofuscin using proteomic analysis as a guide for understanding its origin. Molecular Visions, 11, 1122–1134.
Warde-Farley, D., Donaldson, S. L., Comes, O., Zuberi, K., Badrawi, R., & Chao, P., et al. (2010). The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Research, 38(Web Server issue), W214–W220.
Wei, H., Zhang, Z., Saha, A., Peng, S., Chandra, G., Quezado, Z., et al. (2011). Disruption of adaptive energy metabolism and elevated ribosomal p-S6K1 levels contribute to INCL pathogenesis: Partial rescue by resveratrol. Human Molecular Genetics, 20(6), 1111–1121.
Wirsching, H. G., Krishnan, S., Florea, A. M., Frei, K., Krayenbuhl, N., Hasenbach, K., et al. (2014). Thymosin beta 4 gene silencing decreases stemness and invasiveness in glioblastoma. Brain, 137(Pt 2), 433–448.
Wisztorski, M., Croix, D., Macagno, E., Fournier, I., & Salzet, M. (2008). Molecular MALDI imaging: An emerging technology for neuroscience studies. Developmental Neurobiology, 68(6), 845–858.
Xu, B. J. (2010). Combining laser capture microdissection and proteomics: Methodologies and clinical applications. Proteomics. Clinical Applications, 4(2), 116–123.
Yang, J., Gu, P., Menges, S., & Klassen, H. (2012). Quantitative changes in gene transcription during induction of differentiation in porcine neural progenitor cells. Molecular Visions, 18, 1484–1504.
Zhang, Y. Q., Henderson, M. X., Colangelo, C. M., Ginsberg, S. D., Bruce, C., Wu, T., et al. (2012). Identification of CSPalpha clients reveals a role in dynamin 1 regulation. Neuron, 74(1), 136–150.
Zhang, Z., Lee, Y. C., Kim, S. J., Choi, M. S., Tsai, P. C., Xu, Y., et al. (2006). Palmitoyl-protein thioesterase-1 deficiency mediates the activation of the unfolded protein response and neuronal apoptosis in INCL. Human Molecular Genetics, 15(2), 337–346.
Zimmerman, T. A., Monroe, E. B., Tucker, K. R., Rubakhin, S. S., & Sweedler, J. V. (2008). Chapter 13: imaging of cells and tissues with mass spectrometry: Adding chemical information to imaging. Methods in Cell Biology, 89, 361–390.
Zuberi, K., Franz, M., Rodriguez, H., Montojo, J., Lopes, C. T., Bader, G. D., et al. (2013). GeneMANIA prediction server 2013 update. Nucleic Acids Research, 41, W115–W122.
Acknowledgments
The authors wish to thank the colleagues of the Functional Genomic Centre of Verona University, Verona, Italy, for performing RNA-seq analysis. Patients’ fibroblasts were referred to AS within the collaborative framework of CLNet, the Italian Network of CLN investigators. The authors also thank Teija Inkinen and Suvi Saarnio for excellent technical assistance, Essi Kaiharju for helping with mice preparations and Adjunct Professor Ove Eriksson for valuable comments on the manuscript. Professor Anna-Elina Lehesjoki is thanked for management of the DEM-CHILD project and overall support.
Funding
This study was funded by the European Community’s Seventh Framework Program (FP7/2007–2013) under Grant Agreement No. 281234; DEM-CHILD and partially by the Academy of Finland (#128600) to ML. AG was funded by the University of Helsinki and Aalto University collaborative grant.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no conflict of interest.
Additional information
Saara Tikka and Evanthia Monogioudi have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Tikka, S., Monogioudi, E., Gotsopoulos, A. et al. Proteomic Profiling in the Brain of CLN1 Disease Model Reveals Affected Functional Modules. Neuromol Med 18, 109–133 (2016). https://doi.org/10.1007/s12017-015-8382-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12017-015-8382-6