
Abstract
Intracellular β-amyloid (Aβ) accumulation is an early event in Alzheimer’s disease (AD) progression. Recently, it has been uncovered that presenilins (PSs), the key components of the amyloid precursor protein (APP) processing and the β-amyloid producing γ-secretase complex, are highly enriched in a special sub-compartment of the endoplasmic reticulum (ER) functionally connected to mitochondria, called mitochondria-associated ER membrane (MAM). A current hypothesis of pathogenesis of Alzheimer’s diseases (AD) suggests that MAM is involved in the initial phase of AD. Since MAM supplies mitochondria with essential proteins, the increasing level of PSs and β-amyloid could lead to metabolic dysfunction because of the impairment of ER-mitochondrion crosstalk. To reveal the early molecular changes of this subcellular compartment in AD development MAM fraction was isolated from the cerebral cortex of 3 months old APP/PS1 mouse model of AD and age-matched C57BL/6 control mice, then mass spectrometry-based quantitative proteome analysis was performed. The enrichment and purity of MAM preparations were validated with EM, LC-MS/MS and protein enrichment analysis. Label-free LC-MS/MS was used to reveal the differences between the proteome of the transgenic and control mice. We obtained 77 increased and 49 decreased protein level changes in the range of − 6.365 to + 2.988, which have mitochondrial, ER or ribosomal localization according to Gene Ontology database. The highest degree of difference between the two groups was shown by the ATP-binding cassette G1 (Abcg1) which plays a crucial role in cholesterol metabolism and suppresses Aβ accumulation. Most of the other protein changes were associated with increased protein synthesis, endoplasmic-reticulum-associated protein degradation (ERAD), oxidative stress response, decreased mitochondrial protein transport and ATP production. The interaction network analysis revealed a strong relationship between the detected MAM protein changes and AD. Moreover, it explored several MAM proteins with hub position suggesting their importance in Aβ induced early MAM dysregulation. Our identified MAM protein changes precede the onset of dementia-like symptoms in the APP/PS1 model, suggesting their importance in the development of AD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) and the other protein conformational diseases are caused by misfolding and aggregation of proteins and by the accumulation of intracellular and extracellular deposits [1]. Formation of toxic β-amyloid (Aβ) oligomers plays a pivotal role in AD pathogenesis [2]. Amyloid aggregation is based on the accumulation of Aβ monomer close to the generation from amyloid precursor protein (APP) [3]. Intracellular accumulation of Aβ could be a result of improper extent of amyloid clearance from the cell or it can be initiated by overproduction of APP. The cellular hot-spots of Aβ accumulation are the intracellular membrane regions containing high levels of APP and amyloidogenic proteases [4]. One of these cell compartments functioning as a special interface for intense molecule exchange, located between endoplasmic reticulum (ER) and mitochondria, is called mitochondria-associated ER membrane (MAM) [5]. Since intracellular accumulation of soluble Aβ induces mitochondrial dysfunction, ER stress, changes in glucose and lipid metabolism and dysfunctional Ca2+ homeostasis and signalling [6, 7], the MAM attracts particular attention in searching for initial phase process of AD [8,9,10].
MAM is a special sub-compartment of ER functionally connected to mitochondria that is involved in a number of key metabolic functions of vital importance [11] including cholesterol metabolism [12], synthesis and transfer of phospholipids between the ER and mitochondria [13] and calcium homeostasis [14]. Both in familial and sporadic AD subjects, increased level of cholesterol and phosphatidylserine of MAM origin can be detected [9]. Moreover, several MAM proteins are known to be overexpressed in human post-mortem AD brain samples [15]. Since the APP processing Presenilin-1 (PS1) and Presenilin-2 (PS2), the APP protein level and the γ-secretase activity itself are highly enriched in MAM compared to other cell compartments (plasma membrane, mitochondria and ER) [16], MAM could have a pivotal role in β-amyloid production. According to the MAM hypothesis of AD, elevated level of γ-secretase activity increases the Aβ abundance resulting into molecular, morphological and functional alterations in MAM [8]. At a certain level of amyloid monomers, the collision induced oligomerization of amyloid begins forming the toxic and diffusible Aβ oligomers. The toxic and hydrophobic Aβ oligomers could modify the protein and lipid structure of MAM which may result into spreading of toxic oligomers to the mitochondria and other cell organelles. In turn, as an initial phase of AD pathogenesis, MAM deregulates mitochondrial processes [17, 18], lipid metabolism [19], glucose metabolism [20] and induces inflammation [21].
The experimental data obtained in animal models may be translated to the human AD. Increasing amount of evidence suggests that Aβ accumulation in MAMs is an important factor in the initial phase of human AD as well. Electron microscopic studies revealed that the size of MAM increased in AD patients [9]. Deficiency of PS1 or PS2 increases the size and functionally upregulates MAM [10]. Furthermore, PS2 enhances the Ca2+ shuttle between ER and mitochondria by increasing the number of MAM patches [22]. Apolipoprotein E (ApoE) a well-known genetic risk factor protein of AD is present in the MAM [23] suggesting a certain role of MAM in AD-related lipid metabolism dysfunction.
MAM has a crucial role in glucose, phospholipid and cholesterol metabolism, apoptosis, regulation of Ca2+ homeostasis and signalling, all of which are affected by AD [24]. Proteins involved in Ca2+ signalling (e.g., Inositol trisphosphate (IP3) receptor)), molecular chaperones (e.g., sigma-1 receptor chaperone, BiP, calreticulin), Bcl-2 family proteins (e.g., Bcl-2), ubiquitin ligases (e.g., Amfr/Gp78), membrane tethering/vesicular transport proteins (e.g., mitofusion-2, phosphofurin acidic cluster sorting protein 2 (Pacs2)), and lipid synthases (e.g., PtSer synthase, acetyl-CoA: cholesterol acyltransferase) were reported to be localized in MAM [25]. This suggests that MAM is a hot-spot of molecular mechanisms maintaining the cellular energy homeostasis via controlling the mitochondrial energy production and metabolism.
It is widely accepted that initial phase processes of AD begins years before the first symptoms appear. Such an early stage of a human disease having no symptoms can be investigated only in animal models. The APP/PS1 mouse is a double transgenic, amyloid overexpressing model of AD [26, 27]. At 3 months of age, APP/PS1 mice perform without detectable memory deficit in behavioral tests [28], however, we described changes in mitochondrial proteome with 2D-DIGE based proteomics in our previous study [29].
In this report, we used 3 months old APP/PS1 mice and measured MAM proteome changes comparing to age matched control mice. Our data provide the basis for a hypothesis on the initial phase of AD elucidating the importance of early molecular changes in MAM. We report herein for the first time proteome changes in MAM before the symptoms of AD as memory deficit occur.
Methods
Animals
Three months old B6C3-Tg(APPswe,PSEN1dE9)85Dbo/J (APP/PS1) and C57BL/6 (B6) control mice were selected for proteomics (n = 6–6) and electron microscopy experiments (n = 2).
Animals were kept under standard laboratory conditions with 12-h light and dark periods (lights were on from 08.00 a.m. to 08.00 p.m.). Food and water were supplied ad libitum. The care and experimentation of all animals conformed to the Hungarian Act of Animal Care and Experimentation (1998, XXVIII) and to the guidelines of the European Communities Council Directive, 86/609/EEC as well as with local regulations for the care and use of animals for research.
Dissection of Cerebral Cortical Samples for MAM Preparation
The APP/PS1 and B6 mice were decapitated and the brains rapidly removed. The brains were washed in artificial cerebral spinal fluid (ACSF), the cerebral cortices were dissected on dry ice and the tissue samples were immediately frozen and stored at − 80 °C until use.
Isolation of MAM
The cortical mitochondria-associated endoplasmic reticulum membranes were isolated according to a previously published protocol [30] with minor modifications (Fig. 1). In detail, cortical tissue samples were homogenized in 1 ml of ice-cold homogenization buffer (225 mM mannitol, 75 mM sucrose, 0.5% BSA, 0.5 mM EGTA, 30 mM Tris-HCl pH 7.4, supplemented with 5–5 μl protease and phosphatase inhibitor cocktails), using pre-chilled “loose fitting” Dounce glass homogenizer (Kontes Glass Co.; eight-stroke per sample). To avoid post-mortem degradation, all steps were performed at 4 °C, in cold room or on ice with ice-cold buffers and solutions. The homogenates were centrifuged at 740×g for 10 min and the supernatants were collected and centrifuged at 9000×g for 10 min. The supernatants were discarded and the pellets were resuspended in 1 ml resuspending buffer I (225 mM mannitol, 75 mM sucrose, 0.5% BSA, 30 mM Tris-HCl pH 7.4) and centrifuged at 10,000×g for 10 min. The supernatants were again discarded and the pellets were resuspended in 1 ml resuspending buffer II (225 mM mannitol, 75 mM sucrose, 30 mM Tris-HCl pH 7.4) and were centrifuged at 10,000×g for 10 min. The resulting pellets that contains the crude mitochondria fraction were further resuspended in 1 ml resuspending buffer III (250 mM mannitol, 5 mM HEPES, 0.5 mM EGTA, pH 7.4). This was overlaid on 8 ml Percoll medium (225 mM mannitol, 25 mM HEPES, 1 mM EGTA, 30% (v/v) Percoll) and was centrifuged at 95,000×g for 30 min. The MAM fractions were collected from the upper interface and were diluted 10 times with resuspending buffer III and centrifuged at 100,000×g for 60 min. The MAM fraction was collected from the upper interface and precipitated overnight with ice-cold acetone.

Schematic figure of MAM fraction isolation from mouse cerebral cortex
Electron Microscopy Validation of MAM Samples
For the electron microscopic validation whole brain homogenization (n = 3), mitochondria (n = 3) and MAM fractions (n = 3) from cerebral cortex of B6 mice were fixed with 2% formaldehyde (freshly depolymerized from paraformaldehyde) and 0.5% glutaraldehyde in 0.1 M Na-cacodylate for 30 min at room temperature. The samples were post-fixed in 0.5% osmium tetroxide and 0.75% potassium hexacyano-ferrate for 45 min, dehydrated and embedded in LR White resin according to the manufacturer’s instructions. Ultrathin sections (70 nm) were examined in JEOL JEM 1011 electron microscope operating at 60 kV. Images were taken with Olympus Morada 11 megapixel camera and iTEM software (Olympus).
Proteolytic Digestion
The sample preparation for LC-MS analyses were implemented as previously described with minor modifications [31]. Briefly, the membrane samples were reconstituted in urea buffer (7 M urea, 2 M thiourea, 4% CHAPS, 100 mM DTT and 50 mM TEAB) and sonicated. Protein amounts were determined with the Pierce 660 protein assay. Fifty micrograms of samples were digested with trypsin (1:100 w/w) using the filter-aided sample preparation (FASP) [32, 33]. Tryptic peptides were extracted by successive washings of 50 μL of 50 mM TEAB buffer and 50 μL of 0.5 M NaCl followed by centrifugation at 13,000×g. Tryptophan fluorescence was used for the peptide quantification in protein digests [34]. All digests for label free analyses were desalted and concentrated with reversed-phase C18 resins. Lyophilized peptides were reconstituted in 5% formic acid and analyzed by LC-MS/MS.
Liquid Chromatography Tandem Mass Spectrometry
Samples were injected onto a Dionex Ultimate 3000 system (Thermo Fisher Scientific) coupled to a Q-Exactive mass spectrometer (Thermo Fisher Scientific, Germany). Carry overs from previous samples were minimized by implementing the following: (1) prevention of sample overloading by normalizing to the peptide amount (1 μg per analyses), (2) flushing of sample lines with ACN during peptide separation as implemented by a preconcentration setup and (3) injection of two BSA runs between different sample groups to reduce carry overs. Samples were analyzed in the LCMS into alternating groups of three with wild type groups before transgenic animals (3 B6, 3 APP/PS1, 3 B6, 3 APP/PS1) with two technical replicates per sample.
Software versions used for the data acquisition and operation of the Q-Exactive were Tune 2.5.0.2042 and Xcalibur 3.0.63. HPLC solvents were as follows: solvent A consisted of 0.1% formic acid (FA) in water and solvent B consisted of 0.1% formic acid in 80% acetonitrile. From a thermostated autosampler, 10 μL sample containing about 1 μg of the peptide mixture was loaded onto a trap column (PM100-C18 3 μm, 75 μm × 20 mm, Thermo Fisher Scientific, Austria) with a binary pump at a flow rate of 5 μL/min using 0.1% FA for loading and washing the pre-column. After washing, the peptides were eluted by forward-flushing onto a 50-cm analytical column with an inner diameter of 75 μm packed with 2 μm-C18 reversed phase filling material (PepMap-C18 2 μm, 75 μm × 500 mm, Thermo Fisher Scientific, Austria). The peptides were eluted from the analytical column with 235 min gradient ranging from 8 to 40% solvent B, followed by a 5-min gradient from 40 to 90% solvent B and finally, to 90% solvent B for 5 min before re-equilibration to 5% solvent B at a constant flow rate of 300 nL/min.
The LTQ Velos ESI positive ion calibration solution (Pierce, IL, USA) was used to externally calibrate the instrument prior to sample analysis and an internal calibration was performed on the polysiloxane ion signal at m/z 445.120024 from ambient air [35]. MS1 scans were performed from m/z 380–1800 at a resolution of 70,000. Using a data-dependent acquisition mode, the 20 most intense precursor ions of all precursor ions with + 2 to + 6 charge were isolated (within a 1.6 m/z window) and fragmented to obtain the corresponding MS2 spectrum. The fragment ions were generated in a higher-energy collisional dissociation (HCD) cell at an NCE of 27% with a fixed first mass of 100 m/z and were detected in an Orbitrap mass analyzer at a resolution of 17,500. The dynamic exclusion for the selected ions was 60 s. Maximal ion accumulation time allowed in MS and MS2 mode was 100 and 50 ms, respectively. Automatic gain control was used to prevent overfilling of the ion trap and was set to 3 × 106 ions and 1 × 105 ions for a full Fourier transform mass spectrometry scan and MS2, respectively.
Protein Identification and Label Free Quantitation
The acquired raw MS data files were processed in Proteome Discoverer (v 2.1, Thermo Fisher Scientific, IL, USA with ProteoWizard (v3.0.5009) and were searched against the mouse UniProt reference proteome database version v2015.11.11 (24,774 sequences with isoforms and appended with chimeric mouse-human APP1 and human PSEN1 sequences) with MASCOT (v2.3.02, MatrixScience, London, UK) [36] search engine. The search parameters were as follows: two tryptic missed cleavage sites, mass tolerances of ± 10 ppm and ± 0.02 Da for the precursor and fragment ions, respectively. Oxidation of methionine was set as variable modification, while carbamidomethylation of cysteine residues were set as fixed modifications. The data was also matched against a decoy database and using the Percolator [37] algorithm, q-values were obtained. Peptide identifications with 1% FDR (q value < 0.01) and protein identifications requiring a minimum of two peptides sequences were reported. LFQProfiler v2.0.2 [38] was used for label-free peptide and protein quantification within the Proteome Discoverer platform.
Bioinformatic Analysis
The enrichment analysis of MS identified proteins was performed by Funrich software tool [39]. Using hypergeometric test, Bonferroni and BH for enrichment statistics Funrich analysis identified the cellular components, biological pathways, biological processes and molecular functions that are enriched in the top 5 of the ranked protein list.
Significantly altered proteins were clustered according to their most relevant cellular functions on the basis of the protein annotations in the UniProt (http://www.uniprot.org/) and Gene Ontology (http://geneontology.org/) databases. The proteins were clustered into groups according to their most relevant cellular functions. The bioinformatic pathway analysis to uncover the linkage between significantly altered MAM proteins and AD was performed by Pathway Studio 11.0 software (Elsevier Life Science Solutions) and Cytoscape software (www.cytoscape.org).
Validation of Abcg1 Significantly Altered Proteins Via Western Blot (WB)
Among the mass spectrometry identified MAM protein changes Abcg1 was selected for WB validation as it showed the highest degree of change (− 6.365 in APP/PS1 mice). For validation, the same samples were used which were utilized in the mass spectrometry analysis. For immunoblotting, samples were reconstituted in SDS lysis buffer (1.5% SDS, 100 mM sodium chloride, 20 mM Tris-HCl pH 7.5 and supplemented with protease inhibitors). Samples were sonicated and centrifuged at 13,000×g for 10 min. Protein quantities were determined with the Pierce BCA protein assay (Thermo Fisher Scientific, IL, USA). Protein samples (15 μg) were warmed at 37 °C for 20 min in Laemmli sample buffer (Bio-Rad Laboratories, Hercules, CA, USA). Protein samples were separated on 10% SDS-PAGE gels and transferred onto PVDF membranes. Membranes were blocked with 5% non-fat dry milk (in TBS for 1 h at RT), and incubated with primary antibodies against rabbit polyclonal Abcg1 primary antibody (Proteintech, 13578-1-AP, 1:1000 dilution,) and mouse monoclonal Vdac1/Porin (Abcam, ab14734, 1:5000) in 2.5% BSA in TBS with 0.2% Tween 20 overnight at 4 °C). After incubation with HRP-conjugated secondary antibodies (in 2.5% BSA in TBS with 0.2% Tween 20) and exposed to ECL (GE Healthcare, Buckinghamshire, UK), membranes were scanned on a ChemiDOC™ + Imaging system (Bio-Rad Laboratories, Hercules, CA, USA). The densitometry data of protein band intensities were analyzed with ImageJ software (NIH, Bethesda). Densitometric values of the Abcg1 protein bands were normalized to the densities of Vdac1 in the same sample. Differences between APP/PS1 and B6 samples were statistically analyzed using independent two-sample t test.
Results
Validation of the Purity of MAM Fractions
For the validation of the quality of MAM preparation process from mouse brain cerebral cortex, we used two independent methods.
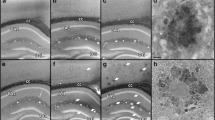
The different steps of MAM preparation from B6 control mice (n = 3) were quality controlled by using transmission electron microscopy. The initial whole brain homogenization contains large membrane fragments (black arrows), mitochondria (white arrowheads), synaptosomes (white arrows) and other cell fragments (Fig. 2a). Next, the separated mitochondria fraction was confirmed by the accumulation of round mitochondria with their cristae visible (white arrowheads) (Fig. 2b). The final purified sample contains closed electron-lucent membrane particles (black arrowheads) that most probably originate from mitochondrial outer membrane and the connecting ER (Fig. 2c, d). Essentially no intact organelles (like mitochondria in Fig. 2b), synaptosomes or large membranes (like in Fig. 2a) are present.

Electron microscopic images of the different steps of MAM preparation. The image of whole brain homogenization shows large membrane fragments (black arrows), mitochondria (white arrowheads), synaptosomes (white arrows) and other cell fragments (a). The image of separated mitochondria fraction shows round mitochondria with their cristae visible (white arrowheads) (b). The image of MAM fraction shows closed electron-lucent membrane particles (black arrowheads) that most probably originate from mitochondrial outer membrane and the attached ER (c). High magnification image of connecting membrane particles (black arrowheads) (d). Scale bar: 1 μm (A, B, C), 200 nm (d)
Results of LC-MS/MS protein identification of MAM preparation revealed that MAM fractions contained 11 known characteristic MAM proteins suggesting the MAM enrichment in the samples. Among them, Pacs2 main MAM regulator protein showed significant decreased. Moreover, MAM preparation contained 13 known AD-associated proteins confirming the literature results that MAM fraction is enriched in AD-related proteins [10]. The APP695 chimeric mouse human amyloid precursor protein (APP) showed significant increased protein abundance in APP/PS1 mice (Table 1).
According to the Funrich enrichment analysis of MAM samples, the mostly enriched cellular components are mitochondria, cytoplasm, ribosome, ER and Golgi apparatus, confirming the enrichment of MAM subcellular region in the sample. Moreover, most of the enriched biological pathways (like metabolism, citric acid cycle and respiratory electron transport), biological processes (like protein metabolism, energy pathways and transport) and molecular functions (like transporter-, catalytic- and oxidoreductase activity) are strongly related to mitochondrial-ER processes (Table 2).
Protein Identification and Label-Free Quantitation
A label-free LC-MS/MS proteomic approach was applied to analyze the differentially expressed proteins between the APP/PS1 mouse and the age-matching control groups. From each group, we collected 6 samples from the cortex of 6 animals and each sample was measured twice.
Reflecting the extreme complexity of the MAM region a total of 5957 proteins were identified. Among them, 3245 common proteins were successfully quantified from all individual samples, suggesting the reproducibility of MAM sample preparation. Of these, 335 proteins showed significant protein abundance changes (independent two sample t test, P ≤ 0.05 level of significance), while 317 proteins showed more than ± 1.2 protein fold changes. To graphically represent the significance and magnitude of protein changes, volcano plot—log10(P value) vs. log2(fold change of APP/PS1/B6 group)—was constructed. Points above the non-axial horizontal line represent proteins with significantly different abundances (P < 0.05). Points to the left of the left non-axial vertical line indicate protein fold changes of APP/PS1/B6 less than − 1.2, while points to the right of the right non-axial vertical line label protein fold changes of APP/PS1/B6 greater than + 1.2 (Fig. 3a). The LFQ intensities of the highest protein abundance changes represent the average protein abundances and the variance between the individual samples (Fig. 3b).

Volcano plot illustrates magnitude and significance of the protein comparisons between the APP/PS1 and B6 group. The −log10 (P value) is plotted against the log2 (fold change: APP/PS1/B6 group). The non-axial vertical lines denote ± 1.2-fold change while the non-axial horizontal line denotes P = 0.05 significance threshold (prior to logarithmic transformation) (a). The LFQ intensities of the highest protein abundance changes represent the average protein abundance and the variance between the individual samples (*p < 0.05, **p < 0.005, ***p < 0.001 using independent two-sample t-test; error bars indicate s.e.m.) (b)
Among the significant protein changes, 128 proteins have mitochondrial, ER or ribosomal localization according to Gene Ontology database. Of these, the abundance of 77 proteins significantly increased, while the abundance of 49 proteins significantly decreased. Protein abundance differences were in the range of − 6.365 to 2.988, and the number of proteins changed more than 2 fold was unusually high (n = 18, dark gray highlight in Table 3) suggesting a complex reorganization of the MAM in the Aβ overexpressing model. ATP-binding cassette sub-family G member 1 (Abcg1) showed the highest protein abundance change in APP/PS1 mice (− 6.365 fold change).
The significantly altered MAM protein changes participate in a variety of metabolic processes, including cytoplasmic protein synthesis (n = 32); mitochondrial protein synthesis (n = 16); lipid metabolism (n = 12); oxidative stress and apoptosis (n = 10); ER-associated protein degradation (n = 8); nucleotide metabolism (n = 5); electron transport chain (n = 5), mitochondrial protein transport (n = 5); signal transduction (n = 5); cytoplasmic protein transport and organelle trafficking (n = 4); cytoskeleton organization (n = 4); Ca2+ transport and homeostasis (n = 3); pyruvate metabolism (n = 3); glutamate metabolism (n = 3); neurotransmission (n = 3); ER-mitochondria morphology (n = 2); ER-mitochondria communication (n = 1); mitophagy (n = 1) and others (n = 2) based on Gene Ontology database. Based on these data, the cytoplasmic and mitochondrial protein synthesis changed in the greatest extent (Table 3).
To explore the relationships of the significantly altered MAM proteins, a localization network from a direct protein-protein interaction database was generated using Pathway Studio (Elsevier). The interaction network contained 55 connecting MAM proteins with 32 positive (green), 22 negative (red) and 80 other (gray) relationships suggesting strong connections between Aβ affected ER-mitochondrial processes. Among these proteins, there are important protein kinases (n = 2), receptors (n = 2), transcriptional factors (n = 3) and transporters (n = 6) (Fig. 4).

Direct interaction network of significantly altered MAM proteins according to their localization. Red/blue highlights indicate the significantly increased/decreased MAM proteins in APP/PS1 mice. Green/red/gray arrows indicate the positive/negative/other regulation between the proteins. Abbreviations: App—amyloid precursor protein; Psen1—Presenilin-1; Rpl3, 5, 10, 14, 17, 21, 27, 34, 35 - 60S ribosomal protein L3, 5, 10, 14, 17, 21, 27, 34, 35; RpsA, 2, 3, 10, 13, 15A 20, 21, 23, 24 - 40S ribosomal protein SA, 2, 3, 10, 13, 15A, 20, 21, 23, 24; Tmed2, 9 - Transmembrane emp24 domain-containing protein 2, 9; Nucb1—Nucleobindin-1; Cyp46a1—Cholesterol 24-hydroxylase; Syncrip—Heterogeneous nuclear ribonucleoprotein Q; Hnrnpd—Heterogeneous nuclear ribonucleoprotein D0; Csde1—Cold shock domain-containing protein E1; Ubqln1—Isoform 2 of Ubiquilin-1; Vps25—Vacuolar protein-sorting-associated protein 25; Synj2bp—Isoform 2 of Synaptojanin-2-binding protein; Fbxo6—F-box only protein 6; Grin1—Glutamate receptor ionotropic, NMDA 1; Abcg1—ATP-binding cassette sub-family G member 1; Mitch1—Mitochondrial carrier homolog 1, Pacs2—Phosphofurin acidic cluster sorting protein 2; Mapk8, 10—Mitogen-activated protein kinase 8, 10; Chchd2—Coiled-coil-helix-coiled-coil-helix domain-containing protein 2; Snd1—Staphylococcal nuclease domain-containing protein 1; Fkbp8—Isoform 2 of Peptidyl-prolyl cis-trans isomerase FKBP8; Rack1—Receptor of activated protein C kinase 1; Kras - GTPase Kras; Anp32a—Acidic leucine-rich nuclear phosphoprotein 32 family member; Sept4—Isoform 2 of Septin-4; Esyt1—Extended synaptotagmin-1; Rhoa—Transforming protein RhoA; Rab35, 1a—Ras-related protein Rab-35, 1A; Nefh—Neurofilament heavy polypeptide; Fdps—Farnesyl pyrophosphate synthase; Comt—Isoform Soluble of Catechol O-methyltransferase; Me1—NADP-dependent malic enzyme; Hmgb1—High mobility group protein B1; Cyp46a1—Cholesterol 24-hydroxylase
We also analyzed the interactions of MAM proteins with AD-related molecular alterations. We revealed that MAM protein changes have direct interactions with APP (22 proteins) and PS1 (7 proteins) AD-associated proteins, which show increased expression in APP/PS1 mice (Fig. 4).
Abcg1 Protein Changes Validation with Western Blot
WB analysis was performed for Abcg1 in the MAM fraction of cerebral cortex (Fig. 5). The protein expression of Abcg1 in the ratio of Vdac1 loading control protein (0.452 ± 0.169) showed significant decreasing level (p < 0.05) in the APP/PS1 MAM compared to the B6 control mice (0.856 ± 0.079). Thus, the WB results of Abcg1 protein confirmed the LC-MS/MS data.

Validation of the decreased expression of Abcg1 in MAM. Immunopositive bands are shown for Abcg1 and Vdac1 loading control. Densitometric analysis was performed for Abcg1 (n = 4). The expression of Abcg1 was significantly decreased in APP/PS1 mice compared to B6 controls (independent two sample t test, *p < 0.05). Error bars indicate SD
Bioinformatical Analysis
The Cytoscape interaction network analysis of protein synthesis related protein changes revealed several increased ribosomal protein with hub position suggesting their important role in Aβ affected cellular protein homeostasis (Fig. 6).

Interaction network of the protein synthesis related MAM protein changes. The color gradient of red and blue were used to visualize the increased or decreased abundances of APP/PS1 mouse proteins compared to the control group, respectively. Abbreviations: Hnrnpd—Heterogeneous nuclear ribonucleoprotein D0; Surf4—Surfeit locus protein 4; Rpsa, 3, 11, 13 and 24—40S ribosomal protein SA, S3, S11, S13 and S24; Eef1b—Elongation factor 1-beta; Sdcbp—Syntenin-1; Rpl5—60S ribosomal protein L5
We also analyzed the oxidative stress and signal transduction related protein changes and described several increased hub proteins (Fig. 7) suggesting their important protective role against Aβ induced MAM dysregulation.

Interaction network of the oxidative stress related MAM protein changes. The color gradient of red and blue were used to visualize the increased or decreased abundances of APP/PS1 mouse proteins compared to the control group, respectively. Abbreviations: App—Amyloid-beta A4 protein; Ryr1—Ryanodine receptor 1; Mapk8 and 10—Mitogen-activated protein kinase 8 and 10; Snd1—Staphylococcal nuclease domain-containing protein 1; Rack1—Receptor of activated protein C kinase 1; Ndusf3—NADH dehydrogenase [ubiquinone] iron-sulfur protein 3, mitochondrial; Anp32a—Acidic leucine-rich nuclear phosphoprotein 32 family member A; Chchd2—Coiled-coil-helix-coiled-coil-helix domain-containing protein 2; Nucb1—Nucleobindin-1; Hspa1b—Heat shock 70 kDa protein 1B; Hmbg1—High mobility group protein B1; Rhoa—Transforming protein RhoA
According to the interaction network analysis of lipid metabolism related protein changes, there is a strong interaction between Abcg1, ApoE and APP that may have an important role in Aβ affected MAM lipid metabolism (Fig. 8).

Interaction network of the lipid metabolism related MAM protein changes. The color gradient of red and blue were used to visualize the increased or decreased abundances of APP/PS1 mouse proteins compared to the control group, respectively. Abbreviations: App—Amyloid-beta A4 protein; Abcg1—ATP-binding cassette sub-family G member 1; Syncrip—Heterogeneous nuclear ribonucleoprotein Q; Coasy—Bifunctional coenzyme A synthase; Bckdha—2-oxoisovalerate dehydrogenase subunit alpha, mitochondrial; Amacr—Alpha-methylacyl-CoA racemase; Minpp1—Multiple inositol polyphosphate phosphatase 1; Fech—Ferrochelatase, mitochondrial
Discussion
The MAM Preparation
We report here an extensive proteome alteration pattern in MAM during in an experimental animal model of AD pathogenesis. The largest part of MAM protein fraction have mitochondrial, ER and ribosomal origin based on Gene Ontology database. Besides the exact mitochondria-ER connecting membrane region, mitochondrial matrix, ER membrane and ribosomal fragments can be present in low extent in enriched MAM samples.
For the validation of the quality of MAM preparation process from mouse brain cerebral cortex, we used two independent methods. Firstly, MAM samples were investigated by electron microscopy and no intact cell organelles (mitochondria, synaptosome, Golgi, etc) were found. Secondly, we analyzed the proteome of the MAM samples by mass spectrometry and identified a majority of known MAM marker proteins described in the literature and several AD associated proteins. We found plenty of ER and mitochondrial proteins although there was no intact mitochondrion observed in the electron microscopy images of the samples. This may suggest that we measured ER-associated mitochondrial proteins or mitochondrial proteins pooled in ER. It is interesting to note, that both mitochondrial and cytosolic ribosome proteins were found in our MAM samples. Although special ER-bound ribosomes exist [40], no intact ribosomes were observed from electron microscopic images taken from our samples. The detected ribosomal proteins are likely MAM-associated and not from intact ribosome contamination.
We emphasize that MAM showed an extraordinary change in a number of proteins in the early stage of amyloid accumulation. It supports the idea that MAM compartment could be of outmost importance for research of the pre-symptomatic early stage of AD.
Protein Synthesis and Folding as an Initial Process of AD
Most of the significantly altered protein levels in our study were involved in protein synthesis, folding and degradation. Among them, all of the cytoplasmic protein synthesis related, mainly ribosomal proteins showed increased abundance, while most of the mitochondrial protein synthesis associated proteins were downregulated (Table 3). Consistent with that, a number of mitochondrial protein transport-related protein changes were described. Both outer (Tom22 (mitochondrial import receptor subunit)) and inner mitochondrial membrane localized channel proteins (Tim17 and 16) showed decreased expression in the MAM region of APP/PS1 mice, which could perform a marked effect on transport of amyloid and other cytoplasmic proteins. The decreased levels of channel proteins can induce serious mitochondrial protein deficiency in citric acide cycle and oxidative phosphorylation processes, which leads to early metabolic dysfunctions in AD [29, 41].
β-Amyloid Induced ER-Associated Protein Degradation (ERAD)
Several ER-associated protein degradation (ERAD) related proteins showed increased abundance in APP/PS1 mice probably suggesting the increased amount of misfolded proteins. During ERAD, misfolded proteins of ER are targeted for ubiquitination and degradation by the proteasome. The ER-stress and misfolded protein aggregation induced ubiquitin proteasome system (UPS) activation is detectable already in the early phase of Alzheimer’s disease [42].
A main ERAD regulator protein, Sel1l (Protein Sel-1 homolog 1) showed significantly increased levels in APP/PS1 mice. The soluble form of Sel1l – Hrp1 (Ubiquitin C-terminal hidroxilase 6) complex supports the UPS degradation of APP. On the other hand, the increase of detergent resistant form of Sel1l and Hrp1 proteins correlates with Aβ accumulation, because the insoluble forms do not trigger APP degradation [43]. Based on these data, the increased Sel1l protein level in MAM region might have an important role in APP accumulation and Aβ production.
An important UPS and ERAD regulator protein, Ubqln1 (Isoform 2 of Ubiquilin-1) showed decreased protein level in APP/PS1 mice. There is a strong association between Ubqln1 genetic variants and familiar form of Alzheimer’s disease development [44, 45]. Ubqln1 shows also decreased protein level in AD, furthermore it has a role in APP biosynthesis, trafficking and Aβ secretion [8, 46]. Specifically, Ubqln1 is able to connect to Htra2 APP cleaving protein, that showed decreased level in mitochondria of 3 months old APP/PS1 mice in our previous study [29]. Moreover, the increased APP intracellular domain level in Ubqln1 overexpressing cell line suggests its role in non-amyloidogenic cleavage of APP [47]. According to these data, the decreased levels of Ubqln1 in the MAM fraction could have a significant role in Aβ accumulation.
Mitochondrial Apoptosis and Oxidative Stress Response
The protein network analysis of oxidative stress related proteins described several important hub proteins (Fig. 7). Among them, the Hspa1b chaperone protein (Heat shock 70 kDa protein 1B) showed increased expression in APP/PS1 mice. During protein synthesis Hsp40 binds to the newly synthesized proteins and carries it to Hsp70, which is a central player in protein folding and also effective against toxic misfolded protein aggregation in different disease models [48]. Hsp70 creates the conformation of the proteins by ATP binding and hydrolysis followed by the binding of a nucleotide-exchange factor catalyzing the ADP-ATP exchange, while further chaperonines connect to the synthetized protein [49]. In agreement with our findings, a previously published human study also described the increased Hsp70 protein level as a consequence of AD pathogenesis [50].
Hsp70 has also an important regulatory role in the apoptotic cascades, like JNK (Mitogen-activated protein kinase 8 (Mapk8)) related oxidative stress response [51]. Mapk8 has a role in Ca2+ transport from ER to mitochondria through IP3R, Ryr1 (Ryanodine receptor 1) and Vdac1 [52]. Although the misfolded protein aggregation related ER stress activates the Mapk8 pathway [53], the decreased abundance of Mapk8 and Ryr1 in APP/PS1 mice are suggesting their anti-apoptotic effect in APP/PS1 mice brain by the present study.
Moreover, we identified the decreased expression of the main MAM regulator protein Pacs2, which is a multifunctional vesicular sorting protein that controls ER–mitochondria tethering, membrane trafficking and Bid-mediated apoptosis [54]. The decreased abundance of Pacs2 in APP/PS1 mice proposes the inhibitory effect of amyloid on ER-mitochondria communication and mitochondrial apoptotic processes.
Several NADH dehydrogenase (ubiquinone) subunits showed a decrease in APP/PS1 mice. NADH dehydrogenase (also called electron transport chain complex I) catalyzes the transfer of electrons from NADH to ubiquinone, and furthermore it is the major source of mitochondrial superoxide anion production [55]. Consistent with Complex I decrease, Timmdc1 (Complex I assembly factor TIMMDC1) also showed decreased protein level in MAM. Based on these results, Aβ could have an early effect on MAM-associated electron leakage and reactive oxygen species (ROS) production.
Consistent with previous data, Fundc1 (FUN14 domain - containing protein 1) protein showed increased level in APP/PS1 mice. This protein exerts a main role in hypoxia-induced mitophagy, which represent an important mechanism in mitochondrial quality control [56]. The increased Fundc1 could have a protective role against increased oxidative stress and apoptotic processes [57].
Dysregulation of Cholesterol and Lipid Metabolism in MAM: the Outstanding Role of Abcg1 Protein in Alzheimer’s Disease
Among Alzheimer’s disease associated proteins, several proteins belong to the cholesterol and lipid metabolism functional group, as Abcg1, which showed the highest decrease in our study (− 6.365 fold change) (Fig. 8). Abcg1 is a cholesterol transporter, that has a main role in apolipoprotein mediated cholesterol transport, intraneuronal cholesterol distribution and neuronal cholesterol export. Therefore the decreased Abcg1 expression suggests cholesterol metabolism dysregulation and intracellular cholesterol accumulation in the APP/PS1 brain.
MAM has a pivotal role in lipid synthesis and contains large amounts of lipoproteins [12] and therefore MAM is a main pool of apolipoproteins (like ApoE). ApoE ε4 isoform is one of the main risk factors of AD [58] which has a smaller Aβ binding affinity [59]. Since the MAM region is one of the main Aβ producing compartment within the cells [60], it could have a significant role in ApoE - Aβ complex formation and soluble Aβ oligomers accumulation. A previous study described that Abca1 and Abcg1 play a significant role in the regulation of neuronal cholesterol efflux and in the suppression of APP processing to generate Aβ peptides [61]. Moreover, Abcg1 and Abcg4 protein supressing γ-secretase activity and Aβ production [62].
The intracellular cholesterol accumulation reaches the ER where it can be metabolized by Acat1 to a cholesteryl ester. Through another possible elimination pathway, accumulated cholesterol is metabolized to 24-hydroxycholesterol (24-HC) by Cyp46a1 (Cholesterol 24-hydroxylase) enzyme, that showed increased expression in APP/PS1 mice brain. The 24-HC can diffuse through the membranes and blood brain barrier and is therefore a potential serum marker candidate in AD [63].
Conclusion
According to our results, Aβ overexpression is associated with extensive MAM proteome changes in the cerebral cortex of APP/PS1 mice. Most of the MAM-localized proteins involved in protein synthesis, oxidative stress response and ERAD showed increased levels. However, the mitochondrial protein transport and energy metabolism related proteins were downregulated, suggesting the general inhibition of main mitochondrial metabolic processes. Applying bioinformatical analysis strategies, a strong relationship was identified between the detected MAM protein changes and the main pathological hallmarks of Alzheimer’s disease. The identified protein synthesis, oxidative stress and lipid metabolism related hub proteins is pointing their importance in Aβ affected MAM homeostasis, which could be a key mechanism in AD initiation. The MAM protein alterations reported in the current study appeared before development of any behavioral symptoms in APP/PS1 model, suggesting their early significant role in the development of AD.
We created a data-based model of MAM-related protein changes shown in Fig. 9. Our homeostatic-MAM hypothesis reports that the early stage of APP accumulation in the MAM the decreased mitochondrial protein transport and metabolism is leading to increased mitochondrial amyloid levels, but the dementia progression is compensated through increased oxidative stress response, ERAD, protein synthesis and other cell protecting mechanisms (green at Fig. 9). In late phase, the APP and the amyloid accumulation is so advanced, that the cell reparing mechanisms cannot reverse the dementia progression (yellow at Fig. 9).

The homeostatic-MAM hypothesis. The red and blue frames visualize the decreased or increased MAM processes of APP/PS1 mouse proteins compared to the control group, respectively. Red/blue gene names indicate the highest increasing/decreasing protein abundance changes in the cell processes. Abbreviations: App—Isoform APP695 chimeric mouse human amyloid precursor protein; Timm17b—Mitochondrial import inner membrane translocase subunit Tim17-B; Pam16—Mitochondrial import inner membrane translocase subunit TIM16; Ptcd3—Pentatricopeptide repeat domain-containing protein 3, mitochondrial; Abcg1—ATP-binding cassette sub-family G member 1; Mrpl 40 and 49—39S ribosomal protein L40 and L49, mitochondrial; Ndufb2—NADH dehydrogenase [ubiquinone] 1 beta subcomplex subunit 2, mitochondrial; Ndufs3 and 8—NADH dehydrogenase [ubiquinone] iron-sulfur protein 3 and 8, mitochondrial; Hmbg1—High mobility group protein B1; Sdf2—Stromal cell-derived factor 2; Emc10—Isoform 2 of ER membrane protein complex subunit 10; Rpl5—60S ribosomal protein L5; Rps21—40S ribosomal protein S21; Tmed2—Transmembrane emp24 domain-containing protein 2; Hnrnpd—Isoform 2 of Heterogeneous nuclear ribonucleoprotein D0
We propose studies on the ER-mitochondrion complex in the early stage of AD to disclose the molecular mechanisms and new molecular targets in that sensitive stage of the disease development.
References
Penke B, Bogar F, Fulop L (2016) Protein folding and misfolding, endoplasmic reticulum stress in neurodegenerative diseases: in trace of novel drug targets. Curr Protein Pept Sci 17:169–182
Hayden EY, Teplow DB (2013) Amyloid beta-protein oligomers and Alzheimer’s disease. Alzheimers Res Ther 5:60
Haass C, Selkoe DJ (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol 8:101–112
Choy RW, Cheng Z, Schekman R (2012) Amyloid precursor protein (APP) traffics from the cell surface via endosomes for amyloid beta (Abeta) production in the trans-Golgi network. Proc Natl Acad Sci U S A 109:E2077–E2082
Vance JE (1990) Phospholipid synthesis in a membrane fraction associated with mitochondria. J Biol Chem 265:7248–7256
LaFerla FM, Green KN, Oddo S (2007) Intracellular amyloid-beta in Alzheimer’s disease. Nat Rev Neurosci 8:499–509
Soejima N, Ohyagi Y, Nakamura N, Himeno E, Iinuma KM, Sakae N, Yamasaki R, Tabira T et al (2013) Intracellular accumulation of toxic turn amyloid-beta is associated with endoplasmic reticulum stress in Alzheimer’s disease. Curr Alzheimer Res 10:11–20
Area-Gomez E, Schon EA (2016) Mitochondria-associated ER membranes and Alzheimer disease. Curr Opin Genet Dev 38:90–96
Area-Gomez E, Del Carmen Lara Castillo M, Tambini MD, Guardia-Laguarta C, de Groof AJ, Madra M, Ikenouchi J, Umeda M et al (2012) Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J 31:4106–4123
Schon EA, Area-Gomez E (2013) Mitochondria-associated ER membranes in Alzheimer disease. Mol Cell Neurosci 55:26–36
Hayashi T, Rizzuto R, Hajnoczky G, Su TP (2009) MAM: more than just a housekeeper. Trends Cell Biol 19:81–88
Rusinol AE, Cui Z, Chen MH, Vance JE (1994) A unique mitochondria-associated membrane fraction from rat liver has a high capacity for lipid synthesis and contains pre-Golgi secretory proteins including nascent lipoproteins. J Biol Chem 269:27494–27502
Vance JE (2003) Molecular and cell biology of phosphatidylserine and phosphatidylethanolamine metabolism. Prog Nucleic Acid Res Mol Biol 75:69–111
Csordas G, Varnai P, Golenar T, Roy S, Purkins G, Schneider TG, Balla T, Hajnoczky G (2010) Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol Cell 39:121–132
Hedskog L, Pinho CM, Filadi R, Ronnback A, Hertwig L, Wiehager B, Larssen P, Gellhaar S et al (2013) Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer’s disease and related models. Proc Natl Acad Sci U S A 110:7916–7921
Area-Gomez E, de Groof AJ, Boldogh I, Bird TD, Gibson GE, Koehler CM, Yu WH, Duff KE et al (2009) Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am J Pathol 175:1810–1816
Castellani R, Hirai K, Aliev G, Drew KL, Nunomura A, Takeda A, Cash AD, Obrenovich ME et al (2002) Role of mitochondrial dysfunction in Alzheimer’s disease. J Neurosci Res 70:357–360
Szarka A (2015) The role of beta-amyloid and mitochondrial dysfunction in the pathogenesis of Alzheimer’s disease. Ideggyogy Sz 68:222–228
Wellington CL (2004) Cholesterol at the crossroads: Alzheimer’s disease and lipid metabolism. Clin Genet 66:1–16
Hoyer S (1991) Abnormalities of glucose metabolism in Alzheimer’s disease. Ann N Y Acad Sci 640:53–58
Akiyama H, Arai T, Kondo H, Tanno E, Haga C, Ikeda K (2000) Cell mediators of inflammation in the Alzheimer disease brain. Alzheimer Dis Assoc Disord 14(Suppl 1):S47–S53
Zampese E, Fasolato C, Kipanyula MJ, Bortolozzi M, Pozzan T, Pizzo P (2011) Presenilin 2 modulates endoplasmic reticulum (ER)-mitochondria interactions and Ca2+ cross-talk. Proc Natl Acad Sci U S A 108:2777–2782
Kim J, Basak JM, Holtzman DM (2009) The role of apolipoprotein E in Alzheimer’s disease. Neuron 63:287–303
Paillusson S, Stoica R, Gomez-Suaga P, Lau DH, Mueller S, Miller T, Miller CC (2016) There’s something wrong with my MAM; the ER-mitochondria axis and neurodegenerative diseases. Trends Neurosci 39:146–157
Fujimoto M, Hayashi T, Su TP (2012) The role of cholesterol in the association of endoplasmic reticulum membranes with mitochondria. Biochem Biophys Res Commun 417:635–639
Jankowsky JL, Slunt HH, Ratovitski T, Jenkins NA, Copeland NG, Borchelt DR (2001) Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol Eng 17:157–165
Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, Copeland NG, Lee MK et al (2004) Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet 13:159–170
Reiserer RS, Harrison FE, Syverud DC, McDonald MP (2007) Impaired spatial learning in the APPSwe + PSEN1DeltaE9 bigenic mouse model of Alzheimer’s disease. Genes Brain Behav 6:54–65
Volgyi K, Haden K, Kis V, Gulyassy P, Badics K, Gyorffy BA, Simor A, Szabo Z et al (2017) Mitochondrial proteome changes correlating with beta-amyloid accumulation. Mol Neurobiol 54:2060–2078
Wieckowski MR, Giorgi C, Lebiedzinska M, Duszynski J, Pinton P (2009) Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat Protoc 4:1582–1590
Sialana FJ, Gulyassy P, Majek P, Sjostedt E, Kis V, Muller AC, Rudashevskaya EL, Mulder J et al (2016) Mass spectrometric analysis of synaptosomal membrane preparations for the determination of brain receptors, transporters and channels. Proteomics 16:2911–2920
Manza LL, Stamer SL, Ham AJ, Codreanu SG, Liebler DC (2005) Sample preparation and digestion for proteomic analyses using spin filters. Proteomics 5:1742–1745
Wisniewski JR, Zougman A, Nagaraj N, Mann M (2009) Universal sample preparation method for proteome analysis. Nat Methods 6:359–362
Wisniewski JR, Gaugaz FZ (2015) Fast and sensitive total protein and peptide assays for proteomic analysis. Anal Chem 87:4110–4116
Olsen JV, de Godoy LM, Li G, Macek B, Mortensen P, Pesch R, Makarov A, Lange O et al (2005) Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C-trap. Mol Cell Proteomics 4:2010–2021
Perkins DN, Pappin DJ, Creasy DM, Cottrell JS (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20:3551–3567
Kall L, Canterbury JD, Weston J, Noble WS, MacCoss MJ (2007) Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat Methods 4:923–925
Veit J, Sachsenberg T, Chernev A, Aicheler F, Urlaub H, Kohlbacher O (2016) LFQProfiler and RNP(xl): open-source tools for label-free quantification and protein-RNA cross-linking integrated into proteome discoverer. J Proteome Res 15:3441–3448
Pathan M, Keerthikumar S, Ang CS, Gangoda L, Quek CY, Williamson NA, Mouradov D, Sieber OM et al (2015) FunRich: an open access standalone functional enrichment and interaction network analysis tool. Proteomics 15:2597–2601
Reid DW, Nicchitta CV (2012) Primary role for endoplasmic reticulum-bound ribosomes in cellular translation identified by ribosome profiling. J Biol Chem 287:5518–5527
Hauptmann S, Scherping I, Drose S, Brandt U, Schulz KL, Jendrach M, Leuner K, Eckert A et al (2009) Mitochondrial dysfunction: an early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol Aging 30:1574–1586
Hoozemans JJ, Scheper W (2012) Endoplasmic reticulum: the unfolded protein response is tangled in neurodegeneration. Int J Biochem Cell Biol 44:1295–1298
Kaneko M, Saito R, Okuma Y, Nomura Y (2012) Possible involvement of ubiquitin ligase HRD1 insolubilization in amyloid beta generation. Biol Pharm Bull 35:269–272
Hiltunen M, Lu A, Thomas AV, Romano DM, Kim M, Jones PB, Xie Z, Kounnas MZ et al (2006) Ubiquilin 1 modulates amyloid precursor protein trafficking and Abeta secretion. J Biol Chem 281:32240–32253
Stieren ES, El Ayadi A, Xiao Y, Siller E, Landsverk ML, Oberhauser AF, Barral JM, Boehning D (2011) Ubiquilin-1 is a molecular chaperone for the amyloid precursor protein. J Biol Chem 286:35689–35698
Mosser DD, Caron AW, Bourget L, Denis-Larose C, Massie B (1997) Role of the human heat shock protein hsp70 in protection against stress-induced apoptosis. Mol Cell Biol 17:5317–5327
Viswanathan J, Haapasalo A, Bottcher C, Miettinen R, Kurkinen KM, Lu A, Thomas A, Maynard CJ et al (2011) Alzheimer’s disease-associated ubiquilin-1 regulates presenilin-1 accumulation and aggresome formation. Traffic 12:330–348
Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM (2002) Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson’s disease. Science 295:865–868
Hartl FU, Hayer-Hartl M (2009) Converging concepts of protein folding in vitro and in vivo. Nat Struct Mol Biol 16:574–581
Yoo BC, Seidl R, Cairns N, Lubec G (1999) Heat-shock protein 70 levels in brain of patients with Down syndrome and Alzheimer's disease. J Neural Transm Suppl 57:315–22
Mosser DD, Caron AW, Bourget L, Denis-Larose C, Massie B (1997) Role of the human heat shock protein hsp70 in protection against stress-induced apoptosis. Mol Cell Biol 17(9):5317–5327
Verma G, Datta M (2012) The critical role of JNK in the ER-mitochondrial crosstalk during apoptotic cell death. J Cell Physiol 227:1791–1795
Rashid HO, Yadav RK, Kim HR, Chae HJ (2015) ER stress: autophagy induction, inhibition and selection. Autophagy 11:1956–1977
Simmen T, Aslan JE, Blagoveshchenskaya AD, Thomas L, Wan L, Xiang Y, Feliciangeli SF, Hung CH et al (2005) PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. EMBO J 24:717–729
Lenaz G, Fato R, Genova ML, Bergamini C, Bianchi C, Biondi A (2006) Mitochondrial complex I: structural and functional aspects. Biochim Biophys Acta 1757:1406–1420
Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C et al (2012) Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol 14:177–185
Kubli DA, Gustafsson AB (2012) Mitochondria and mitophagy: the yin and yang of cell death control. Circ Res 111:1208–1221
Liu CC, Liu CC, Kanekiyo T, Xu H, Bu G (2013) Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol 9:106–118
Tai LM, Koster KP, Luo J, Lee SH, Wang YT, Collins NC, Ben Aissa M, Thatcher GR et al (2014) Amyloid-beta pathology and APOE genotype modulate retinoid X receptor agonist activity in vivo. J Biol Chem 289:30538–30555
Schreiner B, Hedskog L, Wiehager B, Ankarcrona M (2015) Amyloid-beta peptides are generated in mitochondria-associated endoplasmic reticulum membranes. J Alzheimers Dis 43:369–374
Kim WS, Rahmanto AS, Kamili A, Rye KA, Guillemin GJ, Gelissen IC, Jessup W, Hill AF et al (2007) Role of ABCG1 and ABCA1 in regulation of neuronal cholesterol efflux to apolipoprotein E discs and suppression of amyloid-beta peptide generation. J Biol Chem 282:2851–2861
Sano O, Tsujita M, Shimizu Y, Kato R, Kobayashi A, Kioka N, Remaley AT, Michikawa M et al (2016) ABCG1 and ABCG4 suppress gamma-secretase activity and amyloid beta production. PLoS One 11:e0155400
Leoni V, Caccia C (2013) 24S-hydroxycholesterol in plasma: a marker of cholesterol turnover in neurodegenerative diseases. Biochimie 95:595–612
Acknowledgements
This work was supported by the KTIA NAP_13-2-2014-0004 (Katalin Völgyi and Edina Brigitta Udvari), KTIA_NAP_13-2-2015-0003 (Péter Gulyássy, László Drahos and Gábor Juhász) and FIEK_16-1-2016-0005 (Katalin Adrienna Kékesi and Gábor Juhász).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that there is no conflict of interest.
Rights and permissions
About this article

Cite this article
Völgyi, K., Badics, K., Sialana, F.J. et al. Early Presymptomatic Changes in the Proteome of Mitochondria-Associated Membrane in the APP/PS1 Mouse Model of Alzheimer’s Disease. Mol Neurobiol 55, 7839–7857 (2018). https://doi.org/10.1007/s12035-018-0955-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-018-0955-6