
Abstract
Psoriatic arthritis (PsA) is a chronic inflammatory condition characterized by psoriasis, synovitis, enthesitis, spondylitis, and the possible association with other extra-articular manifestations and comorbidities. It is a multifaceted and systemic disorder sustained by complex pathogenesis, combining aspects of autoinflammation and autoimmunity. Features of PsA autoinflammation include the role of biomechanical stress in the onset and/or exacerbation of the disease; the evidence of involvement of the innate immune response mediators in the skin, peripheral blood and synovial tissue; an equal gender distribution; the clinical course which may encounter periods of prolonged remission and overlapping features with autoinflammatory syndromes. Conversely, the role of autoimmunity is evoked by the association with class I major histocompatibility complex alleles, the polyarticular pattern of the disease which sometimes resembles rheumatoid arthritis and the presence of serum autoantibodies. Genetics also provide important insights into the pathogenesis of PsA, particularly related to class I HLA being associated with psoriasis and PsA. In this review, we provide a comprehensive review of the pathogenesis, genetics and clinical features of PsA that endorse the mixed nature of a disorder at the crossroads of autoinflammation and autoimmunity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
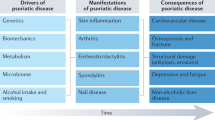
Psoriatic arthritis (PsA) is a chronic inflammatory condition sustained by genetic predisposition and environmental risk factors falling under the umbrella of spondyloarthropathies (SpA) [1]. It is characterized by psoriasis (PsO), synovitis, enthesitis, spondylitis, and the possible association with extra-articular manifestations and comorbidities. The chronic inflammation of the involved tissues can finally lead to structural damage and a reduction of function and quality of life [2]. It is such a complex and multifaceted condition that in 2006, Scarpa et al. proposed the concept of “psoriatic disease” to indicate a systemic disorder that begins in the skin, the bone marrow, or secondary lymph organs and then, in the same patient, can extend to several tissues and organ systems, such as the gut, eye, metabolic pathways, and cardiovascular system. The extraordinary effectiveness of tumor necrosis factor (TNF) inhibitors on the different clinical features of PsA provides indirect evidence of a shared pathogenetic mechanism, supporting the view of a unique condition, hence the term psoriatic disease [3]. Genetics investigations are challenging as PsA is a heterogenous disease: the identification of specific risk loci discriminating PsA from PsO is not trivial [4]. Yet, the debate on the appropriate definition still ranges from PsA to psoriatic disease and even psoriatic syndrome [5, 6], mirroring the variegated pathogenesis of a disorder that combines aspects of two opposite mechanisms, i.e., autoinflammation and autoimmunity [7]. Autoinflammatory disorders are generally characterized by unexplained and recurrent flares of inflammation triggered by local tissue factors at disease-susceptible sites, including trauma, necrosis, mechanical overload, and microbial agents or derivatives thereof, capable of activating the innate immune system, mainly macrophages and neutrophils, with resultant target tissue damage [7]. This concept is expressed by the theory of “danger signal” proposed by Matzinger, based on the assumption that it is not crucial for the immune system to discriminate between self and non-self-antigens, being the priority to respond to exogenous and endogenous danger signals [8]. Conversely, autoimmune diseases are systemic or organ-specific conditions determined by events occurring in primary and secondary lymphoid tissues, including bone marrow, thymus, lymph nodes, and spleen with consequent activation of the adaptive immune system due to aberrant self/non-self-discrimination and breakdown of immunological tolerance [7].
A “continuum model” of immunology has been suggested, with the two ends of autoinflammation and autoimmunity separated by several disorders in which a different relative contribution of both the innate and adaptive immune response could be recognized. In this model, PsA is located at the midpoint, joining autoinflammatory and autoimmune events that unravel the expression of a diversified clinical phenotype [7] (Fig. 1).

Autoinflammatory and autoimmune continuum in psoriatic arthritis
In this manuscript, we will review the current understanding of the pathogenetic and clinical features of PsA, providing insights about the complex nature of a disorder sharing autoinflammation and autoimmunity features.
Pathogenesis of Psoriatic Arthritis
Evidence for the Role of Autoinflammation
Growing evidence supports the role of innate immunity, strictly intertwined with the activation of the adaptive immune system, in the pathogenesis of PsA.
The Role of Trauma
A well-known risk factor for the onset of PsO and PsA is trauma, by means of skin breakdown, stress, medications, tattoos, wounds, and injuries. The onset of psoriatic lesions after a physical trauma is known as the Koebner phenomenon [9,10,11], while the term deep Koebner phenomenon refers to traumatic events causing enthesitis and arthritis [12]. Koebner phenomenon is deemed to be sustained by microdamage and activation of keratinocytes (KCs), subsequently leading to the production of inflammatory mediators, among which type I interferon (IFN), TNF, interleukin 1 (IL-1), and IL-6 are primarily involved and promote the activation of the adaptive immune system. Activated KCs further magnify the inflammatory response by releasing self-nucleic acids as well as antimicrobial peptides, such as LL-37 (the C-terminal part of the only human cathelicidin identified to date called human cationic antimicrobial protein) [13] and activate the Toll-like receptor (TLR) 7/8 pathway in plasmacytoid DCs (pDCs), favoring IFN and cytokine production [14, 15]. pDCs also release type I IFN in pre-psoriatic skin, triggering dermal DCs, also named Langerhans cells (LCs), to synthesize TNF and IL-23, which in turn activate the adaptive immune response [14]. The role of type I IFN in the pathogenesis of PsO is underlined by the evidence that up to 5% of patients treated with anti-TNF agents develop the so-called paradoxical psoriasis, which is explained as the consequence of an upregulation of type I IFN following TNF neutralization [14]. Again, in the deep Koebner phenomenon, an initiating environmental event in genetically susceptible individuals triggers the innate immune response. Due to genetic susceptibility, the abnormal innate response, interacting with the adaptive immune system, creates a self-perpetuating inflammation with joint damage [16]. In this view, the concept of “synovio-entheseal complex” (SEC) provides the anatomical basis for the role of trauma in PsA, because of the close contact between enthesis-associated fibrocartilage and other structures such as synovial membrane bursae, tendon sheaths, fat pads, and fasciae [17].
The Role of Enthesis
Enthesis microdamage with subsequent angiogenesis, inflammatory cell migration, and tissue repair may represent the trigger leading to the onset of PsA in genetically susceptible individuals [18, 19]. These events seem to be linked to an abnormal innate immune response rather than an adaptive immune pathway, at least in early PsA [20]. IL-23 is strictly related to enthesitis, which is observed in up to 50% of patients with PsA [21]. Also, several murine models of SpA have been studied, all of which show an enthesis-centered inflammatory disease linked to IL-23 [22, 23]. This cytokine directly affects enthesis-resident IL-23R + CD3 + CD4-CD8- lymphocytes, whose main population is composed of the γ/δ T cells [24]. Of interest, γ/δ T cells also accumulate at anatomic sites typically involved in SpA and subject to wear and tear, such as the aortic root and the ciliary body [24]. As with the Koebner phenomenon, microtrauma in all of these sites may trigger inflammation [25]. Another setting where microdamage affects the clinical phenotype of PsA is the distal interphalangeal joints, typically involved in the disease as a consequence of enthesitis of the extensor digitorum tendon, which trespasses on the fingernail and the bone of the distal phalanx [26].
The Role of Molecular and Cellular Pathways
Normally, in patients with PsO, pDCs are inhibited in the recognition of self-nucleic acids to hinder the activation of the immune cascade. However, the prolonged expression of self-peptides (LL-37, human β-defensin-2 and 3, lysozyme) together with DNA or RNA released by damaged KCs after the action of a trigger factor, may activate TLRs and lead to IFN-α production. LL-37 can also stimulate conventional DCs (cDCs), upregulating the release of IL-12 [27]. Finally, IL-12 synergizes with TNF and IL-23 to induce the maturation and differentiation of T helper 1 (Th1), Th22, Th17, and γ/δ T lymphocytes, with consequent production of IL-17, IL-22, IFN-γ, and more TNF, thus contributing to PsO appearance [27]. TNF is also responsible for a dramatic increase in the TNF-induced protein 3 (TNFAIP3 or A20) expression, which can inhibit the nuclear factor κB (NF-κB) signaling [9, 28], acting as a negative immune regulator during inflammatory states [29]. Genome-wide association studies have identified PsA-specific genetic variants at the TNFAIP3 locus, which are independent of those previously identified in PsO [30]. The expression of TNFAIP3 mRNA negatively correlated with the psoriatic area and severity index (PASI) as well as with the percentage of body surface area affected by the disease [31]. Consistently, a non-coding variant of the TNFAIP3 interacting protein 1 was associated with PsO, probably due to a reduced ability to suppress NFκB signaling [9].
Polymorphonuclear cells (PMNs) contribute to this wide inflammatory response by releasing DNA and RNA through their neutrophil extracellular traps (NETs) and involving LL-37 [32], which is stored in the secondary granules. When NETosis (the unique form of cell death that leads to the release of chromatin NETs [33]) is started, LL-37 can be extruded from the cells and, as above mentioned, initiates the inflammatory cascade by binding TLRs [32]. It was also demonstrated that LL-37 RNA is increased in the skin of psoriatic patients, moving a self-propagating pro-inflammatory cascade [32]. Activated neutrophils that produce NETs were found in significant quantities in psoriatic skin plaques and sera of patients with plaque-type and pustular PsO [34]. PMNs, especially neutrophils, may be involved in the pathogenesis of PsO also concerning the respiratory burst with the generation of reactive oxygen species (ROS) [34]. Besides, ROS participate in the expansion of anti-inflammatory type 2 macrophages in experimental mice, an effect associated with the protective role of macrophage mannose receptor (MR) toward PsO and PsA, as deficient MR mice develop a very severe form of both diseases [35]. In specific, it has been demonstrated that in a mouse model of mannan-induced PsA and PsO, Nos2-derived nitric oxide (NO) is pathogenic. The release of Nos2-dependent IL-1α from skin macrophages was essential for arthritis development by promoting IL-17 production of innate lymphoid cells [36].
While it seems clear that an exaggerated activation of T cells caused by the dysregulation of DCs can be considered part of the etiology of PsO, still no definite role has been outlined for LCs, a type of antigen-presenting cells [27]. However, their contribution to the inflammatory response is indisputable. Indeed, after a trauma, LCs produce nitric oxide (NO), causing the typical histological findings in the psoriatic skin, characterized by vasodilation, inhibition of platelet aggregation, and recruitment of T lymphocytes. LCs also activate KCs by producing endothelial growth factor that binds its receptor on these cells, stimulating their division [10]. Interestingly, an increased number of inducible nitric oxide synthase (iNOS) DCs were found in the skin of patients with PsO [37]. Genetic studies have identified an association for the inducible nitric oxide synthase (NOS2) gene to increase susceptibility for PsO and PsA, reported in 1352 PsA case and 2164 control DNA samples [38]. Furthermore, DCs express on their membrane the CD100 protein, also known as semaphorin 4D, which is implicated in the activation of T cells by binding the CD72 surface receptor [15]. Another protein, called phospholipase A2 lipid antigen, was found in psoriatic lesions. This enzyme is expressed by psoriatic KCs and mast cells, and its activation produces new lipid antigens that are afterward recognized by T cells, prompting the crosstalk between innate and adaptive immune systems [39].
TLRs, abundantly committed in the exacerbation of skin lesions, are also involved in the etiology of PsA, which develops in approximately 20–30% of PsO patients [40]. TLR2 and 4 expressions were increased on peripheral blood (PB) mononuclear cells and KCs from patients with PsA and PsO [41, 42], and a TLR2 polymorphism (rs5743708) is believed to produce a tenfold increased risk for the susceptibility to PsA [9, 43].
We previously investigated the biochemical characteristics of synovial fluid (SF) from patients with PsA to better define the local inflammatory response. In a cohort of 29 PsA patients, lower numbers (both in absolute numbers and in relative terms) of NK and NK-T cells were observed in the SF as compared to PB. In this study, the strong correlation found in SF between the absolute number of cell types that share an NK-like cytotoxic activity (γ/δ T cells and NK cells, as well as NK and NK T cells) suggested a downregulation of non-major histocompatibility complex (MHC) restricted cytotoxicity in the disease [44]. Furthermore, we analyzed the levels of some members of the IL-10 family in serum and SF from another cohort of 40 PsA patients. While IL-20 and IL-24 levels were increased in serum when compared with matched SF levels, reflecting their role in the systemic inflammatory responses, the ratio between IL-19 serum and SF levels showed that this cytokine was located mainly in the SF, supporting an intriguing role of this cytokine in joint inflammation [45]. Recently, two other cytokines have emerged as important mediators in the pathogenesis of PsO. Pustular PsO has been linked to mutations in the IL-36 receptor (IL-36R) gene. Because IL-36 enhances IL-1 production, not surprisingly, anakinra was recently reported to produce a good response in some clinical cases [14]. Another study focused on the possible role that IL-33 could have in the innate-adaptive immune cross-talk. It was hypothesized that IL-33 could be released by KCs after damage stimulating the production of other cytokines, via binding its ST2 receptor, expressed on DCs and Th2 cells [46]. Association studies have identified some risk alleles coding for cytokines or their receptors in patients with PsO and PsA, including IL-12A, IL-12B, and IL23R [30]. Conversely to what is typical for autoinflammatory diseases induced by inflammasomes, no IL-1 gene cluster mutations have been associated with PsA [47].
The Contribute of Genetics to Psoriatic Arthritis
It is well recognized that environmental and genetic factors contribute to PsA susceptibility. The concordance rate for psoriasis in MZ twins is between 20 and 64%, indicating that genetic factors account for roughly 70% of the population variance in the susceptibility to psoriasis [48].
Genome-wide association studies (GWAS) have been revolutionary and very informative in the investigation of genetic association with psoriatic disease. Over the last decade, GWAS have helped the identification of novel loci outside MHC region, resulting in 86 susceptibility regions associated with PsO [49]. Of interest, only a part of the susceptibility loci linked to PsO is associated with increased risk for PsA. This low number could be due to the lower power of statistical analysis due to very few GWAS investigating psoriatic disease sub-types, but also confirms that identifying PsA-specific variants (vs PsO vulgaris and/or cutaneous) is challenging. PsA-specific genetic variants independently associated from those found in PsO were found specifically near IL23R and TNFAIP3 (TNFα Induced Protein 3) [30].
In 2015, Barton’s group identified a specific PsA genetic variant at the IL23R locus, and a new PsA-specific association at chromosome 5q31, using an ImmunoChip array to fine-map immune-related and inflammatory susceptibility loci [38].
PsA is a very heterogeneous disease with skin, axial, and peripheral involvement. Cohort studies with detailed phenotyping are needed to identify specific genetic associations linked with specific PsA manifestations [4].
Evidence for the Role of Autoimmunity
Genetic Background
PsA and PsO have a polygenic background, which heavily affects the onset of both diseases. The recurrence risk ratio (i.e., the risk of disease manifestation in siblings vs. the risk in the general population) of PsA is greater than 27, which is substantially higher than the recurrence risk ratio for PsO or rheumatoid arthritis (RA) [50]. PsO and PsA are differently associated with class I MHC alleles. While HLA-C*06 is a major risk factor for PsO, PsA is associated with human leukocyte antigen (HLA)-B*08, B*27, B*38, and B*39, each supporting a different clinical subtype of the disease [51]. GWAS have identified HLA-C*0602 as the most probable PSORS1 (PsO susceptibility 1) gene. Nine regions of PsO susceptibility, called PSORS1-9, were identified of which only 3 were replicated (PSORS1, 2, and 4). PSORS 1 and 2 have also been linked with PsA; within the PSORS1 locus, two genes seem to determine a gender-dependent different risk for PsO [52]. More than 60% of patients with PsO carry HLA-Cw6 (that corresponds to HLA-C*0602), which could lead to a 9- up to 23-fold increased risk of developing the disease [53]. Interestingly, HLA-C*0602-negative patients with PsO respond significantly better to the anti-TNF agent adalimumab (ADA) than ustekinumab (a monoclonal antibody binding the p40 protein subunit shared by IL-12 and IL-23), but no benefit to ADA over ustekinumab has been recorded in HLA-C*0602-positive patients [54]. GWAS in PsA have shown an association with some polymorphisms in the gene encoding IL-23R, along with variants in NF-κB gene expression (TNFAIP3 interacting protein 1: TNIP1) and signaling (TNFAIP3), and TNF expression [54].
The Role of T Cells
The association of PsO and PsA with HLA genes underlines the pathogenetic role of T cells, further confirmed by the observation that these patients show an increased risk of developing human immunodeficiency virus (HIV) disease [55]. The onset of PsO during HIV is considered paradoxical since PsO is largely mediated by T cells, while HIV is characterized by T cell depletion [56]. PsO may appear in every phase of HIV infection, as an initial presentation, or as a feature of acquired immunodeficiency syndrome and HIV-related immune reconstitution inflammatory syndrome (IRIS). HIV could directly stimulate a proinflammatory reaction as a source of superantigens, since the negative regulatory factor, an HIV protein, could act as an antigen itself [57]. Also, bacterial and fungal infections, which may occur during the later phases of HIV infection, could favor the epitope spreading and the production of other antigens [57]. PsO can also be a manifestation of HIV-related IRIS, which can shortly appear after the initiation of antiretroviral therapy and may be the consequence of the shifting from a Th2 response (which predominates in the late phases of the HIV infection) to a Th1 or Th17 one [57]. Moreover, during an HIV infection, a Th2 environment tends to predominate, while PsO is caused by cytokines produced by Th1 and Th17 lymphocytes. It was hypothesized that CD8 + T cells play a pivotal role in the onset of HIV-associated PsO, with the enhanced production of IFN-γ [56]. It seems that the lesser the CD4 + T cell count, the more severe PsO. What is more startling is that therapies that decrease T-cell count seem to improve PsO [56]. Indeed, it was demonstrated that HIV-associated PsO is mediated by CD45RO + memory CD8 + T cells, which are expanded in HIV infection and produce IFN-γ [56]. It may be possible that IFN-γ stimulates KCs to express HLA-DR, facilitating the polyclonal activation of KCs by antigens that, as previously stated, can be present during HIV itself or other bacterial or fungal infections in the context of HIV infection [56, 57]. Moreover, HIV has been found in the skin cells of infected patients, supporting the hypothesis that this virus may have a direct role in the onset of PsO. The activated KCs can, therefore, produce TNF and favor the immune response [56]. Intriguingly, a single-nucleotide polymorphism in the HLA-B*5701 allele (rs3021366), associated with an increased risk of PsO, is protective in HIV-1 disease and is present in HIV long-term non-progressors phenotype, a group of patients that have a stable amount of CD4 + T cells for approximately 7–10 years [58]. Overall, HIV-infected patients with PsO have more severe disease, with prevailing guttate, inverse, and erythrodermic subtypes [59]. Likewise, patients with PsA generally develop a rapidly erosive arthropathy resistant to conventional treatment [60], and the number of their affected joints tends to increase over time [61].
The Role of Autoantigens
In addition to microbial antigens, LL-37 and the protein A disintegrin and metalloprotease domain-containing thrombospondin type 1 motif-like 5 (ADAMTSL5) can act as self-stimuli for autoimmunity [14, 39]. ADAMTSL5, normally expressed on epithelial cells, KCs, and connective tissues, is presented by HLA-C*0602 and activates Th17 cells [39]. IL-17 may, in turn, increase the expression of LL-37 and ADAMTSL5, perpetuating the autoimmune response [39, 62]. LL-37 promotes the production of C-X-C motif chemokine ligand 1 (CXCL1), which enhances the expansion of ADAMTSL5, causing additional expression of IL-17A and IFN-γ [62]. After the activation of DCs and KCs and the consequent production of cytokines, T cells migrate to the epidermis, where they recognize the above-mentioned autoantigens and produce IL-17 and IL-22 [14]. At this point, activated T cells induce the proliferation of epidermal cells by triggering KCs, which continue to release chemokines, upholding the autoimmune response.
It has been hypothesized that each clinical type of PsO has a different pathogenetic pathway [14]. Recently, an antigen array harboring putative autoantigens, followed by validation using individual ELISAs, allowed to identify potentially pathogenetic IgG autoantibodies targeting LL-37 and ADAMTS-L5 in the serum of patients with PsA. Because the serum levels were significantly higher compared to those found in patients with PsO without PsA, these molecules were deemed to be involved in the pathogenesis of PsA [63]. This is consistent with previous findings showing that autoantibodies may be found in patients with PsA [64]. Finally, a peculiar population of T cells expressing IL-17 and comprising mainly CD8 + T cells lacking traditional cytotoxic markers (IL-17 + CD4- T cells) was found at high levels in the SF from PsA patients, but not in RA SF. Interestingly, these cells correlated with clinical and laboratory parameters of disease activity, and with the presence of erosions in PsA patients, suggesting the participation in the pathogenesis of PsA and providing a prominent signal that PsA may be immunologically more similar to SpA than to RA [65].
Table 1 summarizes the pathogenetic features supporting a role for both autoinflammation and autoimmunity in PsA and PsO.
Clinical and Laboratory Features of Psoriatic Arthritis
Evidence for the Role of Autoinflammation
Several observations sustain the view of PsA as a predominantly autoinflammatory disease. These include gender distribution, the role of trauma in the onset or exacerbation of the disease, the clinical course of PsA which may encounter periods of prolonged remission, the emergence of enthesitis, and extra-articular manifestations as a consequence of inflammation in mechanically overloaded sites, the overlapping features with autoinflammatory conditions.
Gender Distribution
Differently from the classical autoimmune diseases which are more frequent in females, PsA occurs just as frequently in both sexes, and males are 2–3 times more susceptible to axial PsA [47]. In a recent systematic review and meta-analysis, the prevalence and incidence rates according to sex were not different in 12 studies, confirming that PsA affects equally men and women [66].
The Role of Trauma
As above mentioned, environmental factors, including trauma and obesity, have been implicated in the onset of psoriatic disease. Physical trauma is a known risk factor in PsO lesions and is a trigger for PsA development as well [47, 67]. There are three hypotheses linking trauma to PsA onset. First, trauma may activate the innate immune system in the SEC context as part of the deep Koebner phenomenon, resulting in the release of pro-inflammatory cytokines accessing the synovium to cause synovitis [18]. This hypothesis explains the relationship between psoriatic nail disease, the distal interphalangeal joints, and the entheses [12]. A second theory implies that trauma may cause the activation of nerve endings and the release of neuropeptides, such as substance P, with consequent activation of the inflammatory cascade and the development of PsA [12, 25, 68]. The third theory is based on the assumption that chronic injury and defective repair processes in psoriatic tissue may lead to prolonged Wnt signaling, innate immune activation, and the development of autoinflammatory lesions [69].
The first case of a patient with PsO who developed acro-osteolysis after trauma was described in 1959 [70]. Since then, several cases of onset or exacerbation of PsA following trauma were reported [12]. Among these, interesting reports are those of monozygotic twins developing PsA after trauma [71], the evidence of PsA following trauma in 24.6% of patients enrolled in a longitudinal study [72], and the observation that moving to a new house, injuries, and lifting heavy weights were associated with PsA [73, 74]. Besides, a high body mass index (BMI) in patients with PsO was associated with an increased risk of developing PsA, probably because obesity could trigger PsA as trauma does [75].
In a retrospective case series of patients with PsA, a higher prevalence of trauma was found before the onset of arthritis (8%) compared with patients with RA (1.7%) or ankylosing spondylitis (AS) (0.7%) [76]. In a similar study, a preceding acute event was documented in 9% of PsA patients compared with 1% of RA patients [77]. Furthermore, enthesophytes in mechanically exposed joints have been described in PsO patients, but not in healthy volunteers [78]. These findings have been recently confirmed in a PsO cohort study, where a previous joint or bone trauma was associated with PsA with multivariate hazard ratios of 1.50 and 1.46, respectively [25]. Interestingly, trauma may affect musculoskeletal sites other than joints, as an Achilles enthesitis was observed some weeks after an injury to calcaneus in a PsA patient [79], and a subject with PsO developing dactylitis 1 week after trauma was also described [80].
Clinical Presentation and Course
PsA is a chronic disease with variable periods of remission and flare. The clinical course of peripheral and axial PsA is usually less severe than RA and AS [81]. PsA has frequently a sudden onset, can be episodic, recurrent, or progressive and may show spontaneous remission. In a prospective, case–control study, the frequency of remission in peripheral PsA was higher compared with RA [82]. Besides, this study showed a persistent remission in PsA patients even after withdrawal of therapy.
Similar to joint involvement, evidence supports spontaneous or therapy-induced remission of enthesitis, even after a short course of treatment [83,84,85].
Enthesitis and Extra-articular Manifestations
Enthesitis represents a shared feature of the SpA spectrum of diseases and is a common manifestation in PsA [47, 67, 86]. As above reported, it is an area subject to high mechanical stress and chronic microtrauma, resulting in the release of proinflammatory mediators leading to synovitis [7, 47, 87]. Hence, enthesitis is considered the initial site of pathologic alterations in PsA, as supported by observations demonstrating that mechanical stress applied to enthesis is sufficient to cause experimentally induced SpA [88]. Magnetic resonance imaging (MRI) studies have also shown a link between enthesitis and synovitis in swollen joints from PsA patients, demonstrating diffuse bone marrow edema at entheseal insertions and a diffuse inflammation involving surrounding soft tissue including the synovium, ligaments, tendons, and the nail root and matrix [19]. Enthesitis is linked to nail disease as well as dactylitis, where enthesitis was demonstrated in the flexor and dorsal tendon and collateral ligaments [89]. Enthesitis usually occurs with local pain, tenderness, and sometimes swelling; the most commonly involved sites are the Achilles and plantar fascia insertions, quadriceps and patellar tendons, iliac crest, rotator cuff, and the epicondyles [90]. Despite clinical enthesitis has been documented in only a third of a PsA population [91], there is evidence that it is often asymptomatic [19, 86], as demonstrated by using ultrasound examination, which allowed to reveal at least one abnormal enthesis in 95.5% of early PsA patients [92].
Bowel Involvement
Subclinical bowel inflammation has been described in up to 60% of SpA patients, and Crohn’s disease (CD) seems to display the strongest association with PsA [93,94,95]. These data support the idea that bowel inflammation is likely to participate in the pathophysiology of PsA [47]. The dysregulation of the gut epithelial barrier could modulate local and systemic inflammation with the activation and expansion of innate immune cells populating the gut mucosa that can migrate toward extra-intestinal sites [93]. These findings are in line with the known role of microbes in the development of certain forms of SpA such as reactive arthritis [96]. Evidence of the contribution of gut microbiota to the development of joint and bowel disease came from animal models mimicking joint and gut involvement in human SpA [22, 97, 98]. Gut dysbiosis may stimulate Th17 cells to release IL-23 inducing the local activation of innate immune cells that may migrate to articular sites, including entheses, leading to inflammation [98].
Further evidence of the autoinflammatory origin of bowel inflammatory disease (IBD) in the course of PsA comes from GWAS in patients with both SpA and IBD that have revealed polymorphisms in signaling pathways implicated in disease development such as the IL-23 pathway. Moreover, NOD-2, the disease-associated mutation occurring in CD, is linked to aberrant intracellular innate immune responses to bacteria [7].
Uveitis
Patients with uveitis usually complain of pain, redness, photophobia, diminished vision, and sensation of dry eye. Uveitis occurring in psoriatic disease patients is anterior, noninfectious, and usually affects both eyes. It occurs in 7–20% of cases of PsO with a higher incidence in patients suffering from PsA and is associated with HLA-B27. In some cases, it may have a chronic and severe course [99,100,101], and patients with PsA tend to have more ocular complications than those with PsO [102].
Apart from anterior uveitis, inflammation of the posterior segment of the eye and panuveitis is not uncommon in psoriatic disease as they occur in 22–44% of cases [99,100,101,102]. In comparison with healthy subjects, patients with PsA and history of anterior uveitis produce higher levels of TNF by monocytes, suggesting a higher innate immune responsiveness [100]. Also, other innate immune responses driven by Th1/Th17 cells could play an important role in the development of ocular inflammation in PsA uveitis. The clinical course (sudden onset with recurrent course) and the above mentioned pathogenetic pathway support the role of autoinflammation in this manifestation [99,100,101].
Autoinflammatory Conditions Resembling Psoriatic Arthritis
Several autoinflammatory diseases characterized by an abnormal IL-1 signal leading to neutrophilic sterile inflammation resemble psoriatic disease. Clinical features of these entities comprise arthritis, recurrent episodes of fever, acne, hidradenitis suppurativa (HS), pyoderma gangrenosum (PG), and high serum levels of inflammatory markers [103].
Several different syndromes have been described, including SAPHO (synovitis, acne, pustulosis, hyperostosis, osteitis), PsAPASH (PsA, PG, acne, and HS), PAPA (PG, acne, and pyogenic arthritis), PASS (PG, acne, and SpA), PASH (PG, acne, and HS), PAPASH (PG, acne, pyogenic arthritis, and HS), and PAC (PG, acne, and ulcerative colitis).
HS is a chronic inflammatory skin disease sharing with generalized pustular PsO high skin expression of IL-36, IL-17, and IL-1 [104]. HS is characterized by recurrent painful nodules, pustules, abscesses, scars, and fistulas occurring in sites opposite to acne, i.e., in the axillary, anogenital, genitofemoral, inguinal, and inframammary areas. IL-1- and TNF-targeted therapies are treatment solutions supporting the role of the innate immunity in HS pathogenesis [103, 105].
The acronym SAPHO was coined in 1987 to designate a syndrome combining musculoskeletal and skin disorders. The autoinflammatory origin of SAPHO is supported by the neutrophilic infiltration of the skin lesions and by the elevated levels of proinflammatory cytokines, such as TNF, IL-1, IL-8, IL-17, and IL-18. The IL-23–Th17 axis was also reported to be involved in SAPHO syndrome. Consistently, patients respond to biological agents targeting TNF, IL-1, and the IL-17–IL-23 axis [106]. SAPHO is strictly related to PsA, and many SAPHO cases meet the PsA classification criteria. The skin lesions observed in SAPHO include palmoplantar pustulosis (which is recognized as a special type of PsO), acne fulminans, acne conglobata, and HS [107]. Other SAPHO syndrome manifestations include hyperostosis, osteitis, and synovitis with characteristic involvement of the anterior chest wall, followed by the axial skeleton, long bones, and peripheral joints. Extra-articular manifestations may be present, such as IBD, pulmonary involvement, venous thrombosis, dura mater hypertrophy, and uveitis [105,106,107].
The deficiency of IL-1R antagonist (DIRA) is an autoinflammatory skin and bone syndrome caused by the recessive mutations in IL-1 N, the gene encoding the IL-1R antagonist, plus five additional IL-1 family members. The result is the lack of a functional protein or its complete absence with overproduction of proinflammatory cytokines and chemokines. The syndrome starts at birth with multifocal osteomyelitis, periostitis, and pustulosis. The rapid clinical response to IL-1R antagonist corroborates the molecular and clinical findings [108].
Evidence for the Role of Autoimmunity
Clinical Manifestations
Different clues suggest the role of autoimmune events in the pathogenesis of synovitis and enthesitis in PsA. First, PsA may be considered a syndrome, in which several clinical manifestations “run together” and, despite the absence of specific disease-related autoantibodies, it somehow resembles classical autoimmune diseases, such as systemic lupus erythematosus (SLE) [5]. Second, the clinical manifestations of PsA could be indistinguishable from RA, mostly in patients with polyarticular involvement and lack of skin lesions, in which synovitis represents the hallmark of the disease. The polyarticular pattern of PsA is found in 30–50% of patients and share with RA some clinical, histological, and pathogenetic aspects, including the cytokine expression pattern and similar response to biological drugs [109]. Moreover, animal models of collagen-induced arthritis, in which an autoimmune response is induced by the administration of collagen antigens, develop both synovitis and enthesitis, with some clinical manifestations typical of PsA in humans. Of note, the induction of inflammation is strictly linked to biomechanical factors at entheseal sites, and it could be speculated that microdamage with subsequent inflammation may trigger an autoimmune response [110]. However, the role of autoimmune events has still to be clarified in a peculiar clinical manifestation of PsA, the axial involvement, which can be sometimes identical to that of AS, even in the treatment response to biological agents [111].
Presence of Autoantibodies
Another challenging element in the pathogenesis of PsA is the presence of serum autoantibodies, especially because for a long time, SpA has been considered a seronegative disease, as opposed to RA [112]. Interestingly, peripheral B cells were found to be lower in PsA patients compared to healthy controls, suggesting their possible role in the complex immune events leading to the disease onset and progression [113]. Moreover, the detection of ectopic lymphoid structures and neogenesis in PsA synovial tissues supports the possibility that self-antigens may locally trigger the formation of autoantibodies [114]. Indeed, antibodies against common peptides expressed by synovium, skin, and entheses, including those directed toward fibrillin, desmocollin, keratin, and the nebulin-related anchoring protein, have been detected in 85% of PsA patients and a significantly lower number of patients with RA, but not in healthy donors [115]. Furthermore, as demonstrated in different types of autoimmune diseases, abnormalities in the physiological process of posttranslational modifications of the proteins, such as citrullination and carbamylation, may lead to the loss of immune tolerance in genetically susceptible individuals. Although rarely, protein citrullination and the positivity of anti-citrullinated protein antibodies (ACPAs) can be found in PsA patients. The presence of ACPAs, also found in SF as above mentioned [64], correlates with a polyarticular subset, female sex, more aggressive and erosive joint involvement, and higher use of systemic therapies [116,117,118]. ACPAs were found in about 5% of cases in a large cohort of PsA patients, especially those with higher swollen joint count and significantly higher rates of erosive changes and dactylitis compared to ACPA-negative patients [119]. In another study, about 12% of PsA patients were positive for ACPAs. Compared to seronegative PsA patients, ACPA-positive patients were more likely affected by a polyarticular pattern, were more frequently treated with biologics and less frequently with traditional disease-modifying anti-rheumatic drugs, suggesting that a misdiagnosis of PsA versus RA plus PsO may occur in these settings [120]. Anyway, what emerges from studies is that the presence of ACPAs is associated with bone destruction, leading to hypothesize an osteo-catabolic effect of these autoantibodies and hence a role in the pathogenesis of bone damage in both PsA and RA patients. Recently, the presence of anti-carbamylated protein (anti-CarP) antibodies was also demonstrated in patients with active PsA who were negative for ACPAs [121]. In this study, the levels of anti-CarP antibodies were significantly increased compared with that of healthy controls and correlated with some parameters of disease activity [121].
Although the negativity for rheumatoid factor (RF) is included in the CASPAR classification criteria for PsA [122], the presence of RF has been observed in about 2% of PsA patients, mainly in those with polyarticular involvement [123].
Finally, a variety of other autoantibodies has been detected in 1–3% of patients with PsO or PsA, including antinuclear antibodies, anti-dsDNA antibodies, anti-small nuclear RNP (anti-snRNP) and cytoplasmic RNP antibodies, and antibodies against epidermal cells and antimicrobial peptide [123,124,125,126,127]. We already mentioned antibodies targeting LL37, which were found to be elevated in PsO and PsA plasma and PsA SF compared to osteoarthritis and correlated with PASI [63].
Table 2 summarizes the clinical features supporting a role for both autoinflammation and autoimmunity in PsA and PsO.
Treatment of Patients with Psoriatic Arthritis
The complex pathogenesis of PsA sustained by the interaction of adaptive and innate immunity has prompted the use of immunosuppressants in this disease. Before the era of biological agents, conventional synthetic disease–modifying anti-rheumatic drugs (csDMARDs) including methotrexate (MTX), sulfasalazine, cyclosporine, and leflunomide have been used, despite few high-quality studies supporting their efficacy, mainly based on the evidence of the relevant role of T cells in the pathogenesis of the disease and the proven efficacy in RA [128,129,130,131]. Among csDMARDs, MTX is most commonly used, and shares with the other csDMARDs a prominent effectiveness in peripheral arthritis, varying degrees of response in PsO and dactylitis, and usually poor effectiveness in enthesitis and axial involvement [132, 133]. Since the year 2000, the outcome of the different domains of PsA has significantly improved due to the availability of biological DMARDs (bDMARDs) neutralizing TNF (effective in all domains of PsA and in the extra-articular manifestations of the disease) and other targets, such as the common p40 subunit of IL-12 and IL-23 (not effective in axial disease), IL-17, and IL-23 (highly effective in PsO and enthesitis). Apart from csDMARDs and bDMARDs, targeted synthetic DMARDs that inhibit phosphodiesterase-4 or Janus kinases simultaneously blocking several cytokines represent other therapeutic options in PsA [132, 133].
Unfortunately, there remain patients who fail to respond to one or more of these treatments, evoking the great complexity of a disease where the possibility of covering the entire spectrum of clinical manifestations remains challenging in most cases.
Conclusions
Evidence derived from epidemiological, clinical, genetic, and immunohistological studies challenges the classification of PsA in a definite setting. In this review, we have provided insights on the mixed pathogenesis of PsA, combining features of the two boundaries of inflammation, i.e., the autoinflammatory and autoimmune conditions. This complexity reflects the variegated clinical phenotype of PsA, which should always be considered in its overall expression, including the extra-articular manifestations and comorbidities. Indeed, the burden of PsA in terms of quality of life and reduced life expectancy is relevant, due mainly to increased cardiovascular morbidity and mortality. The better knowledge of the integrated pathogenesis of the heterogeneous clinical features of PsA is crucial to guarantee substantial benefits for the patients in the years to come.
References
Ritchlin CT (2005) Pathogenesis of psoriatic arthritis. Curr Opin Rheumatol 17:406–412
Gladman DD, Antoni C, Mease P, Clegg DO, Nash P (2005) Psoriatic arthritis: epidemiology, clinical features, course and outcome. Ann Rheum Dis 64:14–17
Scarpa R, Ayala F, Caporaso N, Olivieri I (2006) Psoriasis, psoriatic arthritis, or psoriatic disease? J Rheumatol 33:210–212
Rahmati S, Li Q, Rahman P, Chandran V (2021) Insights into the pathogenesis of psoriatic arthritis from genetic studies. Semin Immunopathol 43:221–234
Lubrano E, Scriffignano S, Perrotta FM (2019) Psoriatic arthritis, psoriatic disease, or psoriatic syndrome? J Rheumatol 46:1428–1430
Scarpa R (2020) Psoriatic syndrome or psoriatic disease? J Rheumatol 47:941
McGonagle D, McDermott MF (2006) A proposed classification of the immunological diseases. PLoS Med 3:e297
Matzinger P (1994) Tolerance, danger, and the extended family. Annu Rev Immunol 12:991–1045
Hile G, Kahlenberg JM, Gudjonsson JE (2020) Recent genetic advances in innate immunity of psoriatic arthritis. Clin Immunol 214:108405
Morhenn VB (1997) Langerhans cells may trigger the psoriatic disease process via production of nitric oxide. Immunol Today 18:433–436
Sun L, Liu W, Zhang L (2019) The role of toll-like receptors in skin host defense, psoriasis, and atopic dermatitis. J Immunol Res 2019:1824624
Hsieh J, Kadavath S, Efthimiou P (2014) Can traumatic injury trigger psoriatic arthritis? A review of the literature. Clin Rheumatol 33:601–608
Dürr UHN, Sudheendra US, Ramamoorthy A (2006) LL-37, the only human member of the cathelicidin family of antimicrobial peptides. Biochim Biophys Acta 1758:1408–1425
Conrad C, Gilliet M (2018) Psoriasis: from pathogenesis to targeted therapies. Clin Rev Allergy Immunol 54:102–113
Xiao C, Luo Y, Zhang C et al (2020) Negative regulation of dendritic cell activation in psoriasis mediated via CD100-plexin-B2. J Pathol 250:409–419
Lories RJ, de Vlam K (2012) Is psoriatic arthritis a result of abnormalities in acquired or innate immunity? Curr Rheumatol Rep 14:375–382
Benjamin M, McGonagle D (2009) The enthesis organ concept and its relevance to the spondyloarthropathies. Adv Exp Med Biol 649:57–70
McGonagle D, Lories RJU, Tan AL, Benjamin M (2007) The concept of a “synovio-entheseal complex” and its implications for understanding joint inflammation and damage in psoriatic arthritis and beyond. Arthritis Rheum 56:2482–2491
McGonagle D (2009) Enthesitis: an autoinflammatory lesion linking nail and joint involvement in psoriatic disease. J Eur Acad Dermatol Venereol 23(Suppl 1):9–13
McGonagle D, Ash Z, Dickie L, McDermott M, Aydin SZ (2011) The early phase of psoriatic arthritis. Ann Rheum Dis 70(Suppl 1):i71–i76
Kehl AS, Corr M, Weisman MH (2016) Review: enthesitis: new insights into pathogenesis, diagnostic modalities, and treatment. Arthritis Rheumatol 68:312–322
Benham H, Rehaume LM, Hasnain SZ et al (2014) Interleukin-23 mediates the intestinal response to microbial β-1,3-glucan and the development of spondyloarthritis pathology in SKG mice. Arthritis Rheumatol 66:1755–1767
Ruutu M, Thomas G, Steck R et al (2012) β-Glucan triggers spondylarthritis and Crohn’s disease-like ileitis in SKG mice. Arthritis Rheum 64:2211–2222
Reinhardt A, Yevsa T, Worbs T et al (2016) Interleukin-23–dependent γ/δ T cells produce interleukin-17 and accumulate in the enthesis, aortic valve, and ciliary body in mice. Arthritis Rheumatol 68:2476–2486
Thorarensen SM, Lu N, Ogdie A, Gelfand JM, Choi HK, Love TJ (2017) Physical trauma recorded in primary care is associated with the onset of psoriatic arthritis among patients with psoriasis. Ann Rheum Dis 76:521–525
Tan AL, Benjamin M, Toumi H et al (2007) The relationship between the extensor tendon enthesis and the nail in distal interphalangeal joint disease in psoriatic arthritis - a high-resolution MRI and histological study. Rheumatology (Oxford) 46:253–256
Wang A, Bai YP (2020) Dendritic cells: the driver of psoriasis. J Dermatol 47:104–113
Ramirez VP, Gurevich I, Aneskievich BJ (2012) Emerging roles for TNIP1 in regulating post-receptor signaling. Cytokine Growth Factor Rev 23:109–118
Jiang Y, Wang W, Zheng X, Jin H (2020) Immune regulation of TNFAIP3 in psoriasis through its association with Th1 and Th17 cell differentiation and p38 activation. J Immunol Res 21:5980190
Stuart PE, Nair RP, Tsoi LC et al (2015) Genome-wide association analysis of psoriatic arthritis and cutaneous psoriasis reveals differences in their genetic architecture. Am J Hum Genet 97:816–836
Jiang X, Tian H, Fan Y et al (2012) Expression of tumor necrosis factor alpha-induced protein 3 mRNA in peripheral blood mononuclear cells negatively correlates with disease severity in psoriasis vulgaris. Clin Vaccine Immunol 19:1938–1942
Herster F, Bittner Z, Archer NK et al (2020) Neutrophil extracellular trap-associated RNA and LL37 enable self-amplifying inflammation in psoriasis. Nat Commun 11:105
Kaplan MJ, Radic M (2012) Neutrophil extracellular traps: double-edged swords of innate immunity. J Immunol 189:2689–2695
Chiang CC, Cheng WJ, Korinek M, Lin CY, Hwang TL (2019) Neutrophils in psoriasis Front Immunol 10:2376
Hagert C, Sareila O, Kelkka T, Jalkanen S, Holmdahl R (2018) The macrophage mannose receptor regulate mannan-induced psoriasis, psoriatic arthritis, and rheumatoid arthritis-like disease models. Front Immunol 9:114
Zhong J, Scholz T, Yau ACY et al (2018) Mannan-induced Nos2 in macrophages enhances IL-17-driven psoriatic arthritis by innate lymphocytes. Sci Adv 4:eaas9864
Lowes MA, Chamian F, Abello MV et al (2005) Increase in TNF-alpha and inducible nitric oxide synthase-expressing dendritic cells in psoriasis and reduction with efalizumab (anti-CD11a). Proc Natl Acad Sci USA 102:19057–19062
Bowes J, Budu-Aggrey A, Huffmeier U et al (2015) Dense genotyping of immune-related susceptibility loci reveals new insights into the genetics of psoriatic arthritis. Nat Commun 6:6046
Fuentes-Duculan J, Bonifacio KM, Hawkes J et al (2017) Autoantigens ADAMTSL5 and LL-37 are significantly upregulated in active psoriasis and associated with dendritic cells and macrophages. Exp Dermatol 26:1075–1082
Ritchlin CT, Colbert RA, Gladman DD (2017) Psoriatic arthritis. N Engl J Med 376:957–970
Carrasco S, Neves FS, Fonseca MH et al (2011) Toll-like receptor (TLR) 2 is upregulated on peripheral blood monocytes of patients with psoriatic arthritis: a role for a Gram-positive inflammatory trigger? Clin Exp Rheumatol 29:958–962
Garcia-Rodriguez S, Arias-Santiago S, Perandrés-López R et al (2013) Increased gene expression of Toll-like receptor 4 on peripheral blood mononuclear cells in patients with psoriasis. J Eur Acad Dermatol Venereol 27:242–250
Oliveira-Toré CF, Moraes AG, Martinez GF et al (2019) Genetic polymorphisms of toll-like receptors 2 and 9 as susceptibility factors for the development of ankylosing spondylitis and psoriatic arthritis. J Immunol Res 2019:1492092
Spadaro A, Scrivo R, Moretti T et al (2004) Natural killer cells and γ/δ T cells in synovial fluid and in peripheral blood of patients with psoriatic arthritis. Clin Exp Rheumatol 22:389–394
Scrivo R, Conigliaro P, Riccieri V et al (2014) Distribution of interleukin-10 family cytokines in serum and synovial fluid of patients with inflammatory arthritis reveals different contribution to systemic and joint inflammation. Clin Exp Immunol 179:300–308
Cannavò SF, Bertino L, Di Salvo E, Papaianni V, Ventura-Spagnolo E, Gangemi S (2019) Possible roles of IL-33 in the innate-adaptive immune crosstalk of psoriasis pathogenesis. Mediators Inflamm 2019:7158014
Generali E, Bose T, Selmi C, Voncken JW, Damoiseaux J (2018) Nature versus nurture in the spectrum of rheumatic diseases: classification of spondyloarthritis as autoimmune or autoinflammatory. Autoimmun Rev 17:935–941
Queiro R, Gonzalez S, Lopez-Larrea C et al (2006) HLA-C locus alleles may modulate the clinical expression of psoriatic arthritis. Arthritis Res Ther 8:R185
Rahmati S, Tsoi L, O’Rielly D, Chandran V, Rahman P (2020) Complexities in genetics of psoriatic arthritis. Curr Rheumatol Rep 22:10
Chandran V, Schentag CT, Brockbank JE et al (2009) Familial aggregation of psoriatic arthritis. Ann Rheum Dis 68:664–667
FitzGerald O, Haroon M, Giles JT, Winchester R (2015) Concepts of pathogenesis in psoriatic arthritis: genotype determines clinical phenotype. Arthritis Res Ther 17:115
Wiśniewski A, Matusiak L, Szczerkowska-Dobosz A, Nowak I, Kuśnierczyk P (2018) HLA-C*06:02-independent, gender-related association of PSORS1C3 and PSORS1C1/CDSN single-nucleotide polymorphisms with risk and severity of psoriasis. Mol Genet Genomics 293:957–966
Rendon A, Schäkel K (2019) Psoriasis pathogenesis and treatment. Int J Mol Sci 20:1475
Dand N, Duckworth M, Baudry D et al (2019) HLA-C*06:02 genotype is a predictive biomarker of biologic treatment response in psoriasis. J Allergy Clin Immunol 143:2120–2130
Fitzgerald O, Winchester R (2014) Editorial: emerging evidence for critical involvement of the interleukin-17 pathway in both psoriasis and psoriatic arthritis. Arthritis Rheumatol 66:1077–1080
Fife DJ, Waller JM, Jeffes EW, Koo JYM (2007) Unraveling the paradoxes of HIV-associated psoriasis: a review of T-cell subsets and cytokine profiles. Dermatol Online J 13:4
Queirós N, Torres T (2018) HIV-associated psoriasis. Actas Dermosifiliogr 109:303–311
Nititham J, Gupta R, Zeng X et al (2017) Psoriasis risk SNPs and their association with HIV-1 control. Hum Immunol 78:179–184
Castillo RL, Racaza GZ, Roa FD (2014) Ostraceous and inverse psoriasis with psoriatic arthritis as the presenting features of advanced HIV infection. Singapore Med J 55:e60-63
Lawson E, Walker-Bone K (2012) The changing spectrum of rheumatic disease in HIV infection. Br Med Bull 103:203–221
Aboulafia DM, Bundow D, Wilske K, Ochs UI (2000) Etanercept for the treatment of human immunodeficiency virus-associated arthritis. Mayo Clin Proc 75:1093–1098
Blauvelt A, Chiricozzi A (2018) The immunologic role of IL-17 in psoriasis and psoriatic arthritis pathogenesis. Clinic Rev Allergy Immunol 55:379–390
Yuan Y, Qiu J, Lin Z-T et al (2019) Identification of novel autoantibodies associated with psoriatic arthritis. Arthritis Rheumatol 71:941–951
Spadaro A, Riccieri V, Scrivo R, Alessandri C, Valesini G (2007) Anti-cyclic citrullinated peptide antibody determination in synovial fluid of psoriatic arthritis. Clin Exp Rheumatol 25:599–604
Menon B, Gullick NJ, Walter GJ et al (2014) Interleukin-17+CD8+ T cells are enriched in the joints of patients with psoriatic arthritis and correlate with disease activity and joint damage progression. Arthritis Rheumatol 66:1272–1281
Scotti L, Franchi M, Marchesoni A, Corrao G (2018) Prevalence and incidence of psoriatic arthritis: a systematic review and meta-analysis. Semin Arthritis Rheum 48:28–34
Veale DJ, Fearon U (2018) The pathogenesis of psoriatic arthritis. Lancet 391:2273–2284
Olivieri I, Padula A, D’Angelo S, Scarpa R (2008) Role of trauma in psoriatic arthritis. J Rheumatol 35:2085–2087
Tönük Ş, Yorgancıoğlu ZR (2019) Biomechanical factors in psoriatic disease: defective repair exertion as a potential cause. hypothesis presentation and literature review. ACR Open Rheumatol 1:452–461
Buckley WR, Raleigh RL (1959) Psoriasis with acro-osteolysis. N Engl J Med 261:539–541
Ng J, Tan AL, McGonagle D (2015) Unifocal psoriatic arthritis development in identical twins following site specific injury: evidence supporting biomechanical triggering events in genetically susceptible hosts. Ann Rheum Dis 74:948–949
Langevitz P, Buskila D, Gladman DD (1990) Psoriatic arthritis precipitated by physical trauma. J Rheumatol 17:695–697
Eder L, Law T, Chandran V et al (2011) Association between environmental factors and onset of psoriatic arthritis in patients with psoriasis. Arthritis Care Res (Hoboken) 63:1091–1097
Pattison E, Harrison BJ, Griffiths CE, Silman AJ, Bruce IN (2008) Environmental risk factors for the development of psoriatic arthritis: results from a case-control study. Ann Rheum Dis 67:672–676
Love TJ, Zhu Y, Zhang Y et al (2012) Obesity and the risk of psoriatic arthritis: a population-based study. Ann Rheum Dis 71:1273–1277
Punzi L, Pianon M, Rizzi E, Rossini P, Todesco S (1997) Prevalence of post-traumatic psoriatic rheumatism. Presse Med 26:420
Scarpa R, Del Puente A, di Girolamo C, della Valle G, Lubrano E, Oriente P (1992) Interplay between environmental factors, articular involvement, and HLA-B27 in patients with psoriatic arthritis. Ann Rheum Dis 51:78–79
Simon D, Faustini F, Kleyer A et al (2016) Analysis of periarticular bone changes in patients with cutaneous psoriasis without associated psoriatic arthritis. Ann Rheum Dis 75:660–666
Salvarani C, Cantini F, Olivieri I et al (1997) Isolated peripheral enthesitis and/or dactylitis: a subset of psoriatic arthritis. J Rheumatol 24:1106–1110
Padula A, Belsito F, Barozzi L et al (1999) Isolated tenosynovitis associated with psoriasis triggered by physical injury. Clin Exp Rheumatol 17:103–104
Cantini F, Niccoli L, Nannini C, Kaloudi O, Bertoni M, Cassarà E (2010) Psoriatic arthritis: a systematic review. Int J Rheum Dis 13:300–317
Cantini F, Niccoli L, Nannini C et al (2008) Frequency and duration of clinical remission in patients with peripheral psoriatic arthritis requiring second-line drugs. Rheumatology (Oxford) 47:872–876
Olivieri I, Scarano E, Gigliotti P, Giasi V, Padula A (2006) Successful treatment of juvenile-onset HLA-B27-associated severe and refractory heel thesitis with adalimumab documented by magnetic resonance imaging. Rheumatology (Oxford) 45:1315–1317
Olivieri I, Scarano E, Padula A, D’Angelo S, Cantini F (2007) Switching tumor necrosis factor alpha inhibitors in HLA-B27-associated severe heel enthesitis. Arthritis Rheum 57:1572–1574
Olivieri I, Giasi V, Scarano E, Gigliotti P, D’Angelo S, Padula A (2009) A brief course of anti-TNF-alpha therapy can cure recurrent episodes of HLA-B27-associated severe and refractory heel enthesitis. Clin Exp Rheumatol 27:1057; author reply 1058
Haroon M, Fitzgerald O (2012) Pathogenetic overview of psoriatic disease. J Rheumatol Suppl 89:7–10
McGonagle D, Gibbon W, Emery P (1998) Classification of inflammatory arthritis by enthesitis. Lancet 352:1137–1140
Jacques P, Lambrecht S, Verheugen E et al (2014) Proof of concept: enthesitis and new bone formation in spondyloarthritis are driven by mechanical strain and stromal cells. Ann Rheum Dis 73:437–445
Bridgewood C, Watad A, Cuthbert RJ, McGonagle D (2018) Spondyloarthritis: new insights into clinical aspects, translational immunology and therapeutics. Curr Opin Rheumatol 30:526–532
Firestein GS, Gabriel SE, McInnes IB, O'Dell JR (2017) Kelley and Firestein’s Textbook of Rheumatology. Tenth edition
McGonagle D, Tan AL (2015) The enthesis in psoriatic arthritis. Clin Exp Rheumatol 33(5 Suppl 93):S36-39
Perrotta FM, Astorri D, Zappia M, Reginelli A, Brunese L, Lubrano E (2016) An ultrasonographic study of enthesis in early psoriatic arthritis patients naive to traditional and biologic DMARDs treatment. Rheumatol Int 36:1579–1583
Rizzo A, Ferrante A, Guggino G, Ciccia F (2017) Gut inflammation in spondyloarthritis. Best Pract Res Clin Rheumatol 31:863–876
Li WQ, Han JL, Chan AT, Qureshi AA (2013) Psoriasis, psoriatic arthritis and increased risk of incident Crohn’s disease in US women. Ann Rheum Dis 72:1200–1205
Charlton R, Green A, Shaddick G et al (2018) Risk of uveitis and inflammatory bowel disease in people with psoriatic arthritis: a population-based cohort study. Ann Rheum Dis 77:277–280
Sibley CH (2016) Autoinflammation and HLA-B27: beyond antigen presentation. Ocul Immunol Inflamm 24:460–469
Rehaume LM, Matigian N, Mehdi AM et al (2019) IL-23 favours outgrowth of spondyloarthritis-associated pathobionts and suppresses host support for homeostatic microbiota. Ann Rheum Dis 78:494–503
Rossini M, Epis OM, Tinazzi I, Grembiale RD, Iagnocco A (2020) Role of the IL-23 pathway in the pathogenesis and treatment of enthesitis in psoriatic arthritis. Expert Opin Biol Ther 20:787–798
Rosenbaum JT, Kim HW (2013) Innate immune signals in autoimmune and autoinflammatory uveitis. Int Rev Immunol 32:68–75
Huhtinen M, Repo H, Laasila K et al (2002) Systemic inflammation and innate immune response in patients with previous anterior uveitis. Br J Ophthalmol 86:412–417
Fotiadou C, Lazaridou E (2019) Psoriasis and uveitis: links and risks. Psoriasis (Auckl) 9:91–96
Abbouda A, Abicca I, Fabiani C (2017) Psoriasis and psoriatic arthritis-related uveitis: different ophthalmological manifestations and ocular inflammation features. Semin Ophthalmol 32:715–720
Vinkel C, Thomsen SF (2017) Autoinflammatory syndromes associated with hidradenitis suppurativa and/or acne. Int J Dermatol 56:811–818
De Vita V, McGonagle D (2018) Hidradenitis suppurativa as an autoinflammatory keratinization disease. J Allergy Clin Immunol 141:1953
Gadelha RL, Paiva RDSR, Palitot EB, Costa JEFD (2020) PsAPASH: a rare and recent autoinflammatory syndrome associated with hidradenitis suppurativa. An Bras Dermatol 95:203–206
Liu S, Tang M, Cao Y, Li C (2020) Synovitis, acne, pustulosis, hyperostosis, and osteitis syndrome: review and update. Ther Adv Musculoskelet Dis 12:1759720X20912865
Olivieri I, Giasi V, D’Angelo S, Palazzi C, Padula A (2014) The SAPHO syndrome. In: Zouboulis C, Katsambas A, Kligman A, eds. Pathogenesis and treatment of acne and rosacea: Springer-Verlag Berlin Heidelberg Springer:579–584
Aksentijevich I, Masters SL, Ferguson PJ et al (2009) An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N Engl J Med 360:2426–2437
Lubrano E, Mesina F, Caporali R (2018) Clinical remission in rheumatoid arthritis and psoriatic arthritis. Clin Exp Rheumatol 36:900–910
Gracey E, Burssens A, Cambré I et al (2020) Tendon and ligament mechanical loading in the pathogenesis of inflammatory arthritis. Nat Rev Rheumatol 16:193–207
Lubrano E, Parsons WJ, Perrotta FM (2016) Assessment of response to treatment, remission, and minimal disease activity in axial psoriatic arthritis treated with tumor necrosis factor inhibitors. J Rheumatol 43:918–923
Moll JM, Haslock I, Macrae IF, Wright V (1974) Associations between ankylosing spondylitis, psoriatic arthritis, Reiter’s disease, the intestinal arthropathies, and Behcet’s syndrome. Medicine (Baltimore) 53:343–364
Conigliaro P, Triggianese P, Perricone C et al (2014) Restoration of peripheral blood natural killer and B cell levels in patients affected by rheumatoid and psoriatic arthritis during etanercept treatment. Clin Exp Immunol 177:234–243
Cañete JD, Santiago B, Cantaert T et al (2007) Ectopic lymphoid neogenesis in psoriatic arthritis. Ann Rheum Dis 66:720–726
Dolcino M, Lunardi C, Ottria A, Tinazzi E, Patuzzo G, Puccetti A (2014) Crossreactive autoantibodies directed against cutaneous and joint antigens are present in psoriatic arthritis. PLoS One 16:e115424
Chimenti MS, Caso F, Alivernini S et al (2019) Amplifying the concept of psoriatic arthritis: the role of autoimmunity in systemic psoriatic disease. Autoimmun Rev 18:565–575
Pasquetti P, Morozzi G, Galeazzi M (2009) Very low prevalence of anti-CCP antibodies in rheumatoid factor-negative psoriatic polyarthritis. Rheumatology (Oxford) 48:315–316
Perez-Alamino R, Garcia-Valladares I, Cuchacovich R, Iglesias-Gamarra A, Espinoza LR (2014) Are anti-CCP antibodies in psoriatic arthritis patients a biomarker of erosive disease? Rheumatol Int 34:1211–1216
Behrens F, Koehm M, Thaçi D et al (2016) Anti-citrullinated protein antibodies are linked to erosive disease in an observational study of patients with psoriatic arthritis. Rheumatology (Oxford) 55:1791–1795
Popescu C, Zofotă S, Bojincă V, Ionescu R (2013) Anti-cyclic citrullinated peptide antibodies in psoriatic arthritis–cross-sectional study and literature review. J Med Life 6:376–382
Chimenti MS, Triggianese P, Nuccetelli M et al (2015) Auto-reactions, autoimmunity and psoriatic arthritis. Autoimmun Rev 14:1142–1146
Taylor W, Gladman D, Helliwell P, Marchesoni A, Mease P, Mielants H; CASPAR Study Group (2006) Classification criteria for psoriatic arthritis: development of new criteria from a large international study. Arthritis Rheumatol 54:2665–2673
Johnson SR, Schentag CT, Gladman DD (2005) Autoantibodies in biological agent naive patients with psoriatic arthritis. Ann Rheum Dis 64:770–772
Reeves WH, Fisher DE, Wisniewolski R, Gottlieb AB, Chiorazzi N (1986) Psoriasis and Raynaud’s phenomenon associated with autoantibodies to U1 and U2 small nuclear ribonucleoproteins. N Engl J Med 315:105–111
Reeves W (1991) Autoimmune mechanisms in psoriasis. Semin Dermatol 10:217–224
Frasca L, Palazzo R, Chimenti MS et al (2018) Anti-LL37 antibodies are present in psoriatic arthritis (PsA) patients: new biomarkers in PsA. Front Immunol 9:1936
Guarneri C, Aguennouz M, Guarneri F, Polito F, Benvenga S, Cannavò SP (2018) Autoimmunity to heterogeneous nuclear ribonucleoprotein A1 in psoriatic patients and correlation with disease severity. J Dtsch Dermatol Ges 16:1103–1107
Breedveld FC, Dayer JM (2000) Leflunomide: mode of action in the treatment of rheumatoid arthritis. Ann Rheum Dis 59:841–849
Cutolo M, Seriolo B, Pizzorni C, Craviotto C, Sulli A (2002) Methotrexate in psoriatic arthritis. Clin Exp Rheumatol 20(6 Suppl 28):S76-80
Salvarani C, Macchioni P, Olivieri I et al (2001) A comparison of cyclosporine, sulfasalazine, and symptomatic therapy in the treatment of psoriatic arthritis. J Rheumatol 28:2274–2282
Salvarani C, Olivieri I, Cantini F, Macchioni L, Boiardi L (1998) Psoriatic arthritis. Curr Opin Rheumatol 10:299–305
Gossec L, Baraliakos X, Kerschbaumer A et al (2020) EULAR recommendations for the management of psoriatic arthritis with pharmacological therapies: 2019 update. Ann Rheum Dis 79:700–712
Coates LC, Kavanaugh A, Mease PJ et al (2016) Group for Research and Assessment of Psoriasis and Psoriatic Arthritis 2015 treatment recommendations for psoriatic arthritis. Arthritis Rheumatol 68:1060–1071
Author information
Authors and Affiliations
Ethics declarations
Informed Consent
Not applicable.
Research Involving Human Participants and/or Animals
Not applicable.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Scrivo, R., D’Angelo, S., Carriero, A. et al. The Conundrum of Psoriatic Arthritis: a Pathogenetic and Clinical Pattern at the Midpoint of Autoinflammation and Autoimmunity. Clinic Rev Allerg Immunol 65, 72–85 (2023). https://doi.org/10.1007/s12016-021-08914-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-021-08914-w