
Abstract
The clinical picture of psoriatic arthritis (PsA) is heterogeneous, potentially involving numerous organs and tissues, such as skin and joint. From a clinical point of view, discrete tissue PsA features develop and respond to treatments apparently independently. The pathogenic events occurring in the various tissues are only partially understood. Although the vast majority of known genetic predisposing factors are shared between patients with skin psoriasis (PSO) and those affected by PsA, some tissue-specific variants have been identified. Furthermore, current data suggest that the TNF pathway and IL-23/Th17 pathways may be differentially activated in distinct tissue sites. In this review, we briefly describe current knowledge on the pathogenesis of PsA in terms of genetic predisposition, environmental factors and immunology, advancing our hypothesis to explain why a common immunologic process can express itself with significant differences in various tissues.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
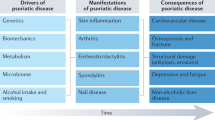
Psoriatic arthritis (PsA) is a systemic chronic inflammatory disease, a member of the wider group of spondyloarthropathies (SpA). Its clinical features encompass inflammatory synovitis (arthritis), enthesitis, dactylitis, tendonitis and cutaneous psoriasis (PSO). Several other clinical manifestations are associated with PsA, such as onychopathy, uveitis, inflammatory bowel disease and metabolic syndrome. PsA imposes an important burden in terms of quality of life and reduced life expectancy, due mainly to increased cardiovascular morbidity and mortality [1]. The integrated pathogenesis of these heterogeneous clinical features is still not well understood but should yield substantial clinical benefits in future once resolved.
Epidemiology of psoriatic arthritis
Performing epidemiological studies on PsA is a challenging task for two main reasons. Firstly, unique criteria to define PsA are imperfect for use in the cohort setting. The CASPAR criteria are the most commonly used having been designed for their utility in clinical trials; they were designed for classification and not diagnostic purposes. Other sets of criteria were used in the past, and accordingly, the population of subjects fulfilling each set can be different. Secondly, the prevalence of PsA is not geographically uniform; thus, results of studies can vary greatly depending on which population is analysed.
In Europe, prevalence of PsA ranges between 0.05 and 0.21%. Similar rates are reported in the US population, but lower prevalence is described in Asia and South America. The incidence rate is variable across populations and may have changed in time. Most recent studies report an incidence of 3.6 to 7.2 cases per 100,000 person-years [2].
PSO (in the absence of recognised musculoskeletal disease) is a more common disease with more than 2% of the US population affected [3]. When the PsA burden is calculated among PSO patients, its prevalence may be as high as 41%, although there is great variability reported due to the reasons described above. Moreover, a proportion of patients with PSO likely have subclinical PsA features, such as enthesitis and low-grade synovitis, which further complicate the picture. As far as specific PsA features are concerned, enthesitis and dactylitis are common. Onycopathy is yet more common; it was shown to be more prevalent in PsA rather than PSO only patients [2].
Pathogenesis of psoriatic arthritis
Like most inflammatory and autoimmune diseases, the pathogenesis of PsA is complex and multifaceted. Although genetic predisposition and environmental factors are widely accepted as “pieces of the puzzle,” their respective proportional contribution is still unclear. Moreover, whether PsA and PSO can be considered a unique entity with different clinical expressions or two distinct disorders with some overlapping features is a long-debated question concerning which final agreement has not yet been reached. Abundant evidence in favour of each hypothesis is available, though we tend to favour a single spectrum of immune and metabolic mechanistic pathways whose contribution is defined by discrete tissue-driven pathogenic expression. The latter may vary between individuals on the basis of as yet ill-defined factors that could be genetic, epigenetic or environmental.
Genetics
There is no doubt that genetic factors play a pivotal role in the development of PSO and PsA. Resulting from the evolution of genomics and transcriptomic techniques, along with a gratifying reduction in costs, studies comprising extensive analysis are becoming more frequent. Another precious source of data to evaluate the weight of genetic background on the probability of developing a disease (i.e. heritability) are studies performed on twins, ideally comparing concordance rates between monozygotic and dizygotic twins. Most recent data suggest heritability of PSO and PsA to be about 66–68 and 23%, respectively [4,5,6].
Many different gene variants have been associated with PSO and/or PsA in various studies, and the number of publications on PSO significantly outnumbers those on PsA; therefore, many of the variants known to carry a risk for PSO have not been as thoroughly studied in PsA. A comprehensive description of all the genetic variants is beyond the purpose of this review, and more details can be found elsewhere [6,7,8,9,10]. A list of previously described variants is provided in Table 1. We will focus on those with the strongest evidence, aligning these to the possible role of the corresponding proteins within the observed complex immunological milieu of PSO and PsA.
The most well-known and potentially impactful gene in terms of PSO predisposition described so far is PSORS1, also known as HLA-C*0602. It is a major histocompatibility complex (MHC)-I allele and it is considered to account for over 35% of the genetic risk [11]. Surprisingly, although PSORS1 is also associated with PsA, it has been shown to have a protective effect against PsA development in patients already affected by PSO [6]. Other HLA variants have been associated with both diseases, and interestingly, some suggest specific associations with clinical subphenotypes and manifestations of PsA, such as dactylitis and enthesitis [12]. However, the large majority of genes detected in genome-wide association studies (GWAS) are non-HLA and singularly account for a very small degree of risk but represent a convenient starting point to detect possible pathogenic pathways to be further dissected with a targeted approach. Linking HLA with innate immunity, endoplasmic reticulum-associated amino-peptidase 1 (ERAP1) is involved in peptide cleavage and presentation on MHC-I molecules; its variants were shown to be involved in PSO but probably not PsA. Moreover, in consideration of the key role of type I interferons (IFN) and tumour necrosis factor (TNF)-α in the pathogenesis of PSO and PsA, as we will describe below, it is not surprising that some of the gene variants associated with PsA and PSO implicate these pathways. Interferon-induced helicase C domain-containing protein 1 (IFIH1) and tyrosine kinase 2 (TYK2) are implicated in type I IFN transcription and signalling, respectively [13, 14]. TNF-α signalling through its receptors induces activation of nuclear factor kappa B (NFκB). TNF-α-induced protein 3 (TNFAIP3) is involved in termination of NFκB activation [15] and binds TNFAIP3-interacting protein 1 (TNIP1), which has shown to have numerous functions, including NFκB inhibition [16]; tumour necrosis factor receptor-associated factor (TRAF)3 interacting protein 2 (TRAF3IP2) is implicated in the downstream signalling of IL-17 receptor which eventually leads to NFκB activation [17]; NFκB inhibitor alpha (NFκBIA) is an essential NFκB inhibitor [18], and REL is one of NFκB subunits [19].
Beyond TRAF3IP2, other IL-23/Th-17 axis variants are involved in both PsA and PSO, such as the two subunits of IL-23 (IL-23p19 and IL-12p40, the latest shared with IL-12) and IL-23 receptor (IL-23R). Finally, runt-related transcription factor 3 (RUNX3) is essential for expansion and activity of CD8+ cytotoxic T cells (Tc) [20]. It is important to consider that although most of the risk loci are common between PSO and PsA, the same is not true for all variants. Different single nucleotide polymorphisms (SNPs) can provide an increased risk for only one of the two conditions, such as the SNP rs12044149 of IL-23R, which seems to provide a specific risk for PsA but not PSO. The same is true for SNP rs715285 of an intergenic region in chromosome 5q31, although other variants of this locus were previously associated with other autoimmune diseases, such as inflammatory bowel disease (IBD) and juvenile idiopathic arthritis (JIA) [21]. One of the possible consequences of this is that these genetic differences, most of which still need to be identified, may partially account for the interindividual variability in treatment response to targeted agents such as anti-TNF and anti-IL-23 or indeed to the varied clinical phenotypic manifestations and penetrance of disease over time.
Microbiota
The role of microbiota in human biology and immunity has acquired much attention in the last few years. The symbiotic relationship between humans and their bacterial flora is well established; numerous metabolic processes carried out by intestinal bacteria are necessary to produce indispensable compounds, such as vitamins, which the human body would not be able to produce alternatively [22].
The immune system has a close physiological interaction with these bacteria to prevent invasion of tissues and an inflammatory response against them. Strong evidence is arising in support of a reciprocal relationship between commensal bacteria and the immune system, where the former have the ability to shape the latter. Thus, it is not surprising that microbiota can influence the biology of inflammatory and autoimmune diseases [23].
There is evidence of an altered commensal intestinal flora in patients with PSO and PsA, characterised by loss of diversity compared to healthy individuals; more specifically, a reduction of Akkermansia and Ruminococcus in PsA (similarly to IBD) and Coprococcus in both has been described. Nonetheless, most of the evidence derives from animal models. HLA-B27 and β2-microglobulin transgenic rats spontaneously develop an inflammatory disease characterised by IBD-like intestinal inflammation, psoriasiform skin, arthritis and sacroileitis; similarly, genetically predisposed ANKENT and SKG mice develop features of SpA following specific stimuli. Interestingly, the development of the disease is microbiota-dependent; thus, when these mice are grown in germ-free conditions, the disease is significantly milder in terms of severity and sometimes completely abolished [22, 23]. Furthermore, a disruption of the balance of Firmicutes and Bacteroides was observed in PSO patients and may partially account for cardiovascular comorbidities. The prevalence of Firmicutes is associated with higher body mass index and higher levels of the proatherogenic compound trimethylamine-N-oxide (TAMO) [24].
However, the mechanism linking microbiota and disease initiation is still elusive. In consideration of the usually long pre-clinical phase of most autoimmune inflammatory diseases, its function is likely involved in one or more processes taking place at that stage, such as immune system tolerance induction, antigen presentation and costimulation of immune cell activation [23]. It may also shape the T cell repertoire or accrue epigenetic changes that can alter the threshold for maintenance of tolerance. Dietary habits are known to influence and change the characteristics of intestinal microbiota, but as yet, there is no evidence that specific dietary regimens or use of probiotics have any ameliorating effect on the clinical picture [23].
The skin microbiota of PSO patients has also been investigated, and a relative reduction of Propionibacterium along with an increase of Streptococcus genus in lesional skin has been described. So far, no studies have explored whether skin microbiota may have any influence on the development of arthritis in PSO patients [24].
Immunopathology
Immune dysfunction is central to the pathogenesis of PsA and PSO. Each tissue (skin, synovium, enthesis) however may develop different target manifestations of a common pathologic process. Studies have examined this variously at the transcriptional and cellular level, and finally at the level of clinical response to exquisitely specific immune-targeted therapies.
The trigger of inflammation in PSO has not yet been identified. The key pathological features of psoriatic skin are epidermal hyperplasia, increased basal layer cell turnover and chronic inflammation. The cause of hyperplasia is an increased proliferation rate of keratinocytes (KC), a process resembling non self-limiting wound healing and response to damage. This, along with the well-known Köbner phenomenon, led to the hypothesis that microtrauma could act as the trigger to the inflammatory response [25]. Other studies have implicated infection (local or systemic) or changes in the microbiota in disease susceptibility. Other triggers such as post-vaccine responses, smoking, UV exposure, chemical irritants and significant emotional trauma have been implicated [25,26,27,28].
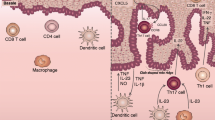
In terms of inflammation, dendritic cells (DC) are prominent as one would expect of a barrier tissue. Their primary role appears to be the activation of T cells both locally and in draining lymph nodes that in turn precipitate T cell maturation and migration back to the dermis and epidermis where they can mediate pathology [29]. Several distinct subtypes of T cell are more abundantly present in psoriatic dermis compared to healthy skin [30, 31]. T helper (Th)1 cells produce IFN-γ, which, in turn, can further stimulate cytokine production by DCs and other myeloid lineage cells, such as interleukin (IL)-23 [32, 33]. IL-23 is a key contributor to Th17 cell differentiation and activation, signalling through its receptor [34]. IL-17 cytokines (of which IL-17A and IL-17F are the best characterised isoforms) are major outputs of Th17 cells; such lymphocytes can also produce and secrete TNF, CCL20 and IL-22 (together with another T cell subtype named Th22). Mast cells are also contributors in terms of IL-22 production in psoriasis [35, 36]. IL-17 can exert a pro-inflammatory effect on KCs, contributing to perpetuation of the inflammatory stimulus, with a positive feedback loop [37], and to recruitment and activation of neutrophils, which are abundant in psoriatic skin [38]. Besides conventional CD3+CD4+CD8− Th17 cells, other T cells have been shown to produce IL-17 in psoriatic skin, such as CD3+CD4−CD8− γδ T cells and CD3+CD4−CD8+ cytotoxic T cells (Tc)17 [39, 40]. The role of IL-22 in disease is less well understood, although pro-inflammatory effects on KCs have been demonstrated [37, 41]. Figure 1 summarises current evidence supporting IL-23/Th17 axis involvement in PsA and PSO.

Current evidence supporting the involvement of the IL-23/Th17 axis in the pathogenic process of PsA derives from various observations comprising genetic studies, animal models, pathological evidence, and the results of clinical trials and real-life observations concerning treatment response [6, 9, 17, 40, 45, 55, 56, 65,66,67,68,69]
The synovial membrane in PsA is less well characterised than the skin, in part reflecting the challenge in accessing tissue compared to skin biopsy. To our knowledge, only one published study has made a comparative analysis of transcriptomes of matched psoriatic skin and synovium samples. In spite of possible pathogenic differences of psoriasis vulgaris and psoriasis in the context of arthritis, this is the best—albeit difficult—method to compare tissues with no bias due to subject heterogeneity. The authors showed that despite PsA skin and synovium share similarities, they also display several clearly distinct features. Microarrays followed by pathway mapping suggest that the TNF pathway, vascular endothelium growth factor (VEGF), transforming growth factor β1 (TGF-β1) and IL-6 signalling are more strongly activated in synovium compared to skin, whereas IL-17 and IL-22 axis activation was more obvious in the skin. Overall, these data were confirmed with single transcripts analysis performed by qPCR [42]. They suggest a priori that consideration should be given to pathogenesis with a reflection on the tissue that is targeted within each clinical manifestation.
The IL-23/Th-17 axis is implicated in PsA pathogenesis not only at the level of genetic associations—several studies show its involvement in synovial tissue inflammation. IL-23 (at low levels) and IL-17 are detected in PsA synovium. The cellular components that express IL-17 include Th and Tc cells, macrophages, neutrophils, mast cells and innate lymphoid cells (ILC)3 [43,44,45,46]. Some cells may serve as producers, whereas others, e.g. mast cells, may reflect primarily receptor-mediated cytokine uptake from the local environment [47].
Another interesting feature shared by both skin and synovium is an increase in vascularisation compared to healthy counterparts, but also to rheumatoid arthritis (RA) synovium. This observation may be partially explained by increased amounts of VEGF found in tissue samples [48]. Hyper-neovascularisation is a characteristic feature of all inflamed tissues, though a difference between PsA and RA is hard to explain. No reasonable explanation as yet exists though the tortuous vessels noted on arthroscopy are diagnostic of PsA compared with RA.
Enthesitis, dactylitis and tendonitis are particular features of PsA and the other spondyloarthritides in general and are less likely to be present in other forms of inflammatory arthropathies such as RA. Interestingly, as with Köbner’s phenomenon in PSO, trauma, even of minor magnitude, has been recognised as a trigger for joint inflammation in PsA patients [49]. Furthermore, enthesitis can often be asymptomatic but still detectable by ultrasonography (US) or magnetic resonance imaging (MRI), especially in patients with clinically evident PSO but no obvious clinical symptoms. From a biomechanical point of view, the enthesis should be seen as one part of a more complex enthesis organ which includes surrounding structures such as bursae, tendon sheaths, enthesis-associated fibrocartilage, fat pads and fasciae (Table 2) [50]. Arising from the description of this structure is the synovio-entheseal concept. Enthesis is avascular at the point of attachment, and synovium provides it with nourishment and lubrication, similarly to articular hyaline cartilage. Entheses are constantly subject to stress, which inevitably leads to microtrauma. The absence of macrophages and inflammatory lymphocytes (e.g. ILCs) at the fibrocartilagineous attachment of healthy enthesis, along with their presence in case of damage, suggests that human synovium-resident cells may migrate towards the enthesis and contribute to tissue repair/inflammation. Alternatively, cells may migrate a priori from blood to the local entheseal microenvironment and thence into the enthesis itself. In patients with a predisposing genetic background, enthesis microdamage, angiogenesis, inflammatory cell migration and tissue repair may represent the initial triggers leading to the clinical syndrome of PsA [51, 52].
Another fascinating site where enthesis and other anatomical structures come together is the distal interphalangeal (DIP) joints. Their peculiar involvement in PsA—unlike RA, in which only synovial joints are affected—was found to be the consequence of enthesitis of extensor digitorum tendon. MRI studies have shown a close link between fingernail, extensor digitorum tendon enthesis and bone of the distal phalanx, suggesting a possible functional and pathologic connection among features such as dactylitis, nail disease, enthesitis and acrolysis as seen in arthritis mutilans [53].
Lessons from the clinic
Treatment of psoriatic disease is complex, not least due to the wide range of clinical features that can potentially manifest, as described above, in addition to broad range of comorbidities. It has long been known that the effect of any given treatment regimen on these distinct clinical manifestations can be very different. As an example, conventional disease-modifying antirheumatic drugs (DMARDs), e.g. methotrexate, are known to have no effect on axial PsA and usually offer only limited effects on enthesitis and dactylitis [54]. Sulphasalazine may exert beneficial effects in the synovium (and as a related drug derivative in the gut as mesalazine) but not in the skin. After the introduction of targeted therapeutics that offer exquisite immunologic specificities, such differences have acquired additional recent interest because response to a medication can be directly linked to a specific underlying biological mechanism. Unfortunately, due to the large number of targeting medications available, comparisons remain difficult. Head-to-head studies are lacking, and most of the published data are the result of post hoc analysis on clinical trials and observational studies, or complex health economic-based comparator methodologies (e.g. MAIC).
Differences of the effect on disease subtypes may be the consequence of different underlying biology and immunology.
TNF inhibitors have conventionally been considered first-line biologics across PsA manifestations, mainly due to the longer experience with their use and stronger data on their efficacy and safety [55].
Currently, five anti-TNF agents (or in some cases their respective biosimilars) have been approved for the treatment of PsA, namely etanercept (ETN), infliximab (IFX), adalimumab (ADA), golimumab (GOL) and certolizumab pegol (CZP). More recently, agents targeting the IL-23/Th17 axis have been extensively studied, and some of them approved for clinical use, such as ustekinumab (UTK), targeting p40 subunit of IL-23 (which is shared with IL-12), secukinumab (SCK) and ixekizumab (IXK), both binding IL-17A [56].
A Bayesian network meta-analysis performed on studies evaluating the effects of ETN, ADA, IFX, UTK and alefacept showed that IFX is the most effective on skin disease, followed by UTK and the other two TNF inhibitors. It is important to underline that most studies using UTK are performed on patients who had previously failed a TNF inhibitor, and therefore, their response to UTK may be weaker a priori compared to biologic-naïve subjects, affecting the results of the study. Furthermore, head-to-head comparison between UTK and ETN, arguably a much stronger evidence base, showed the former to be more effective [3].
In clinical trials, UTK was overall more effective than TNF inhibitors on skin disease, while the opposite was true in terms of joint disease, with lower percentages of patients achieving ACR20, 50 and 75 response among those treated with UTK compared to those expected on the basis of prior studies using TNF inhibitors [57].
The only currently licensed IL-17A targeting treatment is SCK, which showed to be effective on a wide spectrum of PsA and PSO clinical features. Clinical trials in PSO demonstrated effectiveness on skin disease in terms of PASI score reduction, with up to 87% of patients reaching PASI75 and 42% PASI 100 [58,59,60]. In a study comparing the effect of SCK and ETN on skin disease, a significantly higher efficacy of the former over the latter was demonstrated [58, 61, 62]. Moreover, in a recently published phase IIIb head-to-head trial, at 12 and 24 weeks, IXK showed better outcomes on skin and nail disease compared to UTK [63].
As far as synovitis is concerned, SCK showed significant efficacy over placebo as early as 24 weeks after treatment start; the positive effect was maintained for at least 2 years, which may partially account for the good patient compliance observed [64, 65].
Some data are now available on the effect of biologics on enthesopathy and dactylitis, although their interpretation is affected by the numerous different scoring systems for enthesopathy and the lack of a clear definition of dactylitis. Additionally, these clinical features have not been evaluated as primary outcomes of controlled trials. Reported enthesitis and dactylitis amelioration induced by SCK in clinical trial settings looks promising, although these studies were not designed to specifically address this question [65]; there is also good evidence that IFX, GOL, UTK, CZP and apremilast are effective for enthesitis. However, it is not possible to judge whether any of these compounds is superior to the others.60,64 IFX, CZP and UTK seem to be effective on dactylitis as well.60,65 Nonetheless, data on other biologics are missing or provide very little evidence; thus, no definite conclusion can be drawn from these observations.
In our experience, blockage of the IL-23/Th17 axis appears to be more effective on enthesopathy and dactylitis, compared to TNF inhibition, and when these are the main disease features, especially if skin disease is severe too, UTK and SCK should be considered as a possible first-line option.
Conclusion
In recent years, the idea of a shared pathogenesis between PsA and PSO has led to the concept of psoriatic disease, a unifying nomenclature for the two diseases. However, recognising PsA and PSO as a single entity is not straightforward as numerous questions arise, not least driven by our clinical experience when distinct targets are utilised. PsA and PSO share common genetic antecedents and environmental triggers and exhibit striking similarities in immunopathology [66].
Nonetheless, it should be noted that some identified genetic loci variants represent a risk factor only for one of the two clinical manifestations. Furthermore, it is common knowledge that no correlation in terms of disease severity is noted between skin and musculoskeletal disease; as an example nail disease, enthesitis and DIP joint involvement are not always associated, despite that a strong pathogenic and anatomical link among them has been identified, as described above. Similar evidence derives from treatment outcome analysis, which shows that some targeting drugs may be more effective than others on specific clinical features, thus suggesting a different underlying disease mechanism. However, patients’ characteristics differ among clinical trials, and comparisons may be potentially biased.
The published literature on PsA pathogenesis supports several different theories. According to the first, an autoimmune reaction develops in skin, where immune cells mediate a tissue response. Subsequently, autoimmune activation spreads to other tissues, including joints. However, this theory cannot explain why many PSO patients never develop articular disease and why, for example, others show enthesitis and dactylitis as the only manifestations of PsA. Another theory suggests that mechanical stress can induce enthesitis; subsequently, immune cells are attracted in the site of microdamage, causing inflammation of the contiguous structures. Likely, such ideas are non-exclusive. We suggest that intrinsic biological tissue differences may represent the common link and also drive the discrete phenotypic manifestations that arise in the clinic. The physiological host defence activity of the immune system varies among different organs and tissues. Tissues themselves likely direct immune response development and activity. Thus, the immune system in the sterile niche of the anterior chamber of the eye must behave differently compared to the skin and intestine, where constant interaction with the microbial environment is the norm, and to joints, which are subject to mechanical stress but not microbial colonisation. What factors are involved in coordinating different behaviours?
Unfortunately, no definite answer will be available to this question until it is specifically addressed in studies simultaneously investigating the immunobiology of the different tissues involved in psoriatic disease. We commend these to the community forthwith.
References
Ritchlin CT, Colbert RA, Gladman DD (2017) Psoriatic arthritis. N Engl J Med 376(10):957–970. https://doi.org/10.1056/NEJMra1505557
Ogdie A, Weiss P (2015) The epidemiology psoriatic arthritis. Rheum Dis Clin N Am 41(4):545–568. https://doi.org/10.1016/j.rdc.2015.07.001
Lin VW, Ringold S, Devine EB (2012) Comparison of ustekinumab with other biological agents for the treatment of moderate to severe plaque psoriasis: a Bayesian network meta-analysis. Arch Dermatol 148(12):1403–1410. https://doi.org/10.1001/2013.jamadermatol.238
Grjibovski AM, Olsen AO, Magnus P, Harris JR (2007) Psoriasis in Norwegian twins: contribution of genetic and environmental effects. J Eur Acad Dermatol Venereol JEADV 21(10):1337–1134. https://doi.org/10.1111/j.1468-3083.2007.02268.x
Lønnberg AS, Skov L, Skytthe A, Kyvik KO, Pedersen OB, Thomsen SF (2013) Heritability of psoriasis in a large twin sample. Br J Dermatol 169(2):412–441. https://doi.org/10.1111/bjd.12375
O’Rielly DD, Rahman P (2014) Genetics of psoriatic arthritis. Best Pract Res Clin Rheumatol 28(5):673–685. https://doi.org/10.1016/j.berh.2014.10.010
Prinz JC (2017) Autoimmune aspects of psoriasis: heritability and autoantigens. Autoimmun Rev 16(9):970–979. https://doi.org/10.1016/j.autrev.2017.07.011
Stuart PE, Nair RP, Tsoi LC, Tejasvi T, Das S, Kang HM, Ellinghaus E, Chandran V, Callis-Duffin K, Ike R, Li Y, Wen X, Enerbäck C, Gudjonsson JE, Kõks S, Kingo K, Esko T, Mrowietz U, Reis A, Wichmann HE, Gieger C, Hoffmann P, Nöthen MM, Winkelmann J, Kunz M, Moreta EG, Mease PJ, Ritchlin CT, Bowcock AM, Krueger GG, Lim HW, Weidinger S, Weichenthal M, Voorhees JJ, Rahman P, Gregersen PK, Franke A, Gladman DD, Abecasis GR, Elder JT (2015) Genome-wide association analysis of psoriatic arthritis and cutaneous psoriasis reveals differences in their genetic architecture. Am J Hum Genet 97(6):816–836. https://doi.org/10.1016/j.ajhg.2015.10.019
Zhang Z, Yuan J, Tian Z, Xu J, Lu Z (2017) Investigation of 36 non-HLA (human leucocyte antigen) psoriasis susceptibility loci in a psoriatic arthritis cohort. Arch Dermatol Res 309(2):71–77. https://doi.org/10.1007/s00403-016-1706-z
Winchester R, Minevich G, Steshenko V, Kirby B, Kane D, Greenberg DA, FitzGerald O (2012) HLA associations reveal genetic heterogeneity in psoriatic arthritis and in the psoriasis phenotype. Arthritis Rheum 64(4):1134–1144. https://doi.org/10.1002/art.33415
Balendran N, Clough RL, Arguello JR et al (1999) Characterization of the major susceptibility region for psoriasis at chromosome 6p21.3. J Invest Dermatol 113(3):322–328. https://doi.org/10.1046/j.1523-1747.1999.00710.x
FitzGerald O, Haroon M, Giles JT, Winchester R (2015) Concepts of pathogenesis in psoriatic arthritis: genotype determines clinical phenotype. Arthritis Res Ther 17(1):115. https://doi.org/10.1186/s13075-015-0640-3
Gorman JA, Hundhausen C, Errett JS, Stone AE, Allenspach EJ, Ge Y, Arkatkar T, Clough C, Dai X, Khim S, Pestal K, Liggitt D, Cerosaletti K, Stetson DB, James RG, Oukka M, Concannon P, Gale M, Buckner JH, Rawlings DJ (2017) The A946T variant of the RNA sensor IFIH1 mediates an interferon program that limits viral infection but increases the risk for autoimmunity. Nat Immunol 18(7):744–752. https://doi.org/10.1038/ni.3766
Velazquez L, Fellous M, Stark GR, Pellegrini S (1992) A protein tyrosine kinase in the interferon alpha/beta signaling pathway. Cell 70(2):313–322. https://doi.org/10.1016/0092-8674(92)90105-L
Vereecke L, Beyaert R, van Loo G (2009) The ubiquitin-editing enzyme A20 (TNFAIP3) is a central regulator of immunopathology. Trends Immunol 30(8):383–391. https://doi.org/10.1016/j.it.2009.05.007
Ramirez VP, Gurevich I, Aneskievich BJ (2012) Emerging roles for TNIP1 in regulating post-receptor signaling. Cytokine Growth Factor Rev 23(3):109–118. https://doi.org/10.1016/j.cytogfr.2012.04.002
Doyle MS, Collins ES, FitzGerald OM, Pennington SR (2012) New insight into the functions of the interleukin-17 receptor adaptor protein Act1 in psoriatic arthritis. Arthritis Res Ther 14(5):226. https://doi.org/10.1186/ar4071
Boisson B, Puel A, Picard C, Casanova J-L (2017) Human IκBα gain of function: a severe and syndromic immunodeficiency. J Clin Immunol 37(5):397–412. https://doi.org/10.1007/s10875-017-0400-z
Ruben SM, Klement JF, Coleman TA, Maher M, Chen CH, Rosen CA (1992) I-Rel: a novel rel-related protein that inhibits NF-kappa B transcriptional activity. Genes Dev 6(5):745–760. https://doi.org/10.1101/gad.6.5.745
Shan Q, Zeng Z, Xing S, Li F, Hartwig SM, Gullicksrud JA, Kurup SP, van Braeckel-Budimir N, Su Y, Martin MD, Varga SM, Taniuchi I, Harty JT, Peng W, Badovinac VP, Xue HH (2017) The transcription factor Runx3 guards cytotoxic CD8(+) effector T cells against deviation towards follicular helper T cell lineage. Nat Immunol 18(8):931–939. https://doi.org/10.1038/ni.3773
Bowes J, Budu-Aggrey A, Huffmeier U, Uebe S, Steel K, Hebert HL, Wallace C, Massey J, Bruce IN, Bluett J, Feletar M, Morgan AW, Marzo-Ortega H, Donohoe G, Morris DW, Helliwell P, Ryan AW, Kane D, Warren RB, Korendowych E, Alenius GM, Giardina E, Packham J, McManus R, FitzGerald O, McHugh N, Brown MA, Ho P, Behrens F, Burkhardt H, Reis A, Barton A (2015) Dense genotyping of immune-related susceptibility loci reveals new insights into the genetics of psoriatic arthritis. Nat Commun 6(1):6046. https://doi.org/10.1038/ncomms7046
Gill T, Asquith M, Rosenbaum JT, Colbert RA (2015) The intestinal microbiome in spondyloarthritis. Curr Opin Rheumatol 27(4):319–325. https://doi.org/10.1097/BOR.0000000000000187
Scher JU, Littman DR, Abramson SB (2016) Microbiome in inflammatory arthritis and human rheumatic diseases. Arthritis Rheumatol Hoboken NJ 68(1):35–45. https://doi.org/10.1002/art.39259
Yan D, Issa N, Afifi L, Jeon C, Chang HW, Liao W (2017) The role of the skin and gut microbiome in psoriatic disease. Curr Dermatol Rep 6(2):94–103. https://doi.org/10.1007/s13671-017-0178-5
Perera GK, Di Meglio P, Nestle FO (2012) Psoriasis. Annu Rev Pathol 7(1):385–422. https://doi.org/10.1146/annurev-pathol-011811-132448
Boehncke W-H, Schön MP (2015) Psoriasis. Lancet Lond Engl 386(9997):983–994. https://doi.org/10.1016/S0140-6736(14)61909-7
Grozdev I, Korman N, Tsankov N (2014) Psoriasis as a systemic disease. Clin Dermatol 32(3):343–350. https://doi.org/10.1016/j.clindermatol.2013.11.001
Gunes AT, Fetil E, Akarsu S, Ozbagcivan O, Babayeva L (2015) Possible triggering effect of influenza vaccination on psoriasis. J Immunol Res 2015:258430. https://doi.org/10.1155/2015/258430
Gilliet M, Cao W, Liu Y-J (2008) Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat Rev Immunol 8(8):594–606. https://doi.org/10.1038/nri2358
Albanesi C, De Pità O, Girolomoni G (2007) Resident skin cells in psoriasis: a special look at the pathogenetic functions of keratinocytes. Clin Dermatol 25(6):581–588. https://doi.org/10.1016/j.clindermatol.2007.08.013
Pène J, Chevalier S, Preisser L et al (2008) Chronically inflamed human tissues are infiltrated by highly differentiated Th17 lymphocytes. J Immunol Baltim Md 1950 180:7423–7430
Zaba LC, Cardinale I, Gilleaudeau P, Sullivan-Whalen M, Suárez-Fariñas M, Fuentes-Duculan J, Novitskaya I, Khatcherian A, Bluth MJ, Lowes MA, Krueger JG (2007) Amelioration of epidermal hyperplasia by TNF inhibition is associated with reduced Th17 responses. J Exp Med 204(13):3183–3194. https://doi.org/10.1084/jem.20071094
Zaba LC, Fuentes-Duculan J, Eungdamrong NJ, Abello MV, Novitskaya I, Pierson KC, Gonzalez J, Krueger JG, Lowes MA (2009) Psoriasis is characterized by accumulation of immunostimulatory and Th1/Th17 cell-polarizing myeloid dendritic cells. J Invest Dermatol 129(1):79–88. https://doi.org/10.1038/jid.2008.194
McGeachy MJ, Chen Y, Tato CM et al (2009) The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat Immunol 10(3):314–324. https://doi.org/10.1038/ni.1698
Duhen T, Geiger R, Jarrossay D, Lanzavecchia A, Sallusto F (2009) Production of interleukin 22 but not interleukin 17 by a subset of human skin-homing memory T cells. Nat Immunol 10(8):857–863. https://doi.org/10.1038/ni.1767
Mashiko S, Bouguermouh S, Rubio M, Baba N, Bissonnette R, Sarfati M (2015) Human mast cells are major IL-22 producers in patients with psoriasis and atopic dermatitis. J Allergy Clin Immunol 136(2):351–359.e1. https://doi.org/10.1016/j.jaci.2015.01.033
Nograles KE, Zaba LC, Guttman-Yassky E, Fuentes-Duculan J, Suárez-Fariñas M, Cardinale I, Khatcherian A, Gonzalez J, Pierson KC, White TR, Pensabene C, Coats I, Novitskaya I, Lowes MA, Krueger JG (2008) Th17 cytokines interleukin (IL)-17 and IL-22 modulate distinct inflammatory and keratinocyte-response pathways. Br J Dermatol 159:1092–1102. https://doi.org/10.1111/j.1365-2133.2008.08769.x
Witowski J, Pawlaczyk K, Breborowicz A et al (2000) IL-17 stimulates intraperitoneal neutrophil infiltration through the release of GRO alpha chemokine from mesothelial cells. J Immunol Baltim Md 1950 165:5814–5821
Cai Y, Shen X, Ding C, Qi C, Li K, Li X, Jala VR, Zhang HG, Wang T, Zheng J, Yan J (2011) Pivotal role of dermal IL-17-producing γδ T cells in skin inflammation. Immunity 35(4):596–610. https://doi.org/10.1016/j.immuni.2011.08.001
Teunissen MBM, Yeremenko NG, Baeten DLP, Chielie S, Spuls PI, de Rie MA, Lantz O, Res PCM (2014) The IL-17A-producing CD8+ T-cell population in psoriatic lesional skin comprises mucosa-associated invariant T cells and conventional T cells. J Invest Dermatol 134(12):2898–2907. https://doi.org/10.1038/jid.2014.261
Boniface K, Bernard F-X, Garcia M et al (2005) IL-22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J Immunol Baltim Md 1950 174:3695–3702
Belasco J, Louie JS, Gulati N, Wei N, Nograles K, Fuentes-Duculan J, Mitsui H, Suárez-Fariñas M, Krueger JG (2015) Comparative genomic profiling of synovium versus skin lesions in psoriatic arthritis. Arthritis Rheumatol Hoboken NJ 67(4):934–944. https://doi.org/10.1002/art.38995
Costello PJ, Winchester RJ, Curran SA, et al (2001) Psoriatic arthritis joint fluids are characterized by CD8 and CD4 T cell clonal expansions appear antigen driven. J Immunol Baltim Md 1950 166:2878–2886
Kruithof E, Baeten D, De Rycke L et al (2005) Synovial histopathology of psoriatic arthritis, both oligo- and polyarticular, resembles spondyloarthropathy more than it does rheumatoid arthritis. Arthritis Res Ther 7(3):R569–R580. https://doi.org/10.1186/ar1698
van Baarsen LG, Lebre MC, van der Coelen D et al (2014) Heterogeneous expression pattern of interleukin 17A (IL-17A), IL-17F and their receptors in synovium of rheumatoid arthritis, psoriatic arthritis and osteoarthritis: possible explanation for nonresponse to anti-IL-17 therapy? Arthritis Res Ther 16(4):426. https://doi.org/10.1186/s13075-014-0426-z
Leijten EFA, van Kempen TS, Boes M, Michels-van Amelsfort JMR, Hijnen D, Hartgring SAY, van Roon JAG, Wenink MH, Radstake TRDJ (2015) Brief report: enrichment of activated group 3 innate lymphoid cells in psoriatic arthritis synovial fluid. Arthritis Rheumatol Hoboken NJ 67(10):2673–2678. https://doi.org/10.1002/art.39261
Noordenbos T, Blijdorp I, Chen S, Stap J, Mul E, Cañete JD, Lubberts E, Yeremenko N, Baeten D (2016) Human mast cells capture, store, and release bioactive, exogenous IL-17A. J Leukoc Biol 100(3):453–462. https://doi.org/10.1189/jlb.3HI1215-542R
Zhang LY, Ogdie AR, Schumacher HR (2012) Light and electron microscopic features of synovium in patients with psoriatic arthritis. Ultrastruct Pathol 36(4):207–218. https://doi.org/10.3109/01913123.2011.651523
Thorarensen SM, Lu N, Ogdie A, Gelfand JM, Choi HK, Love TJ (2017) Physical trauma recorded in primary care is associated with the onset of psoriatic arthritis among patients with psoriasis. Ann Rheum Dis 76(3):521–525. https://doi.org/10.1136/annrheumdis-2016-209334
Benjamin M, McGonagle D (2009) The enthesis organ concept and its relevance to the spondyloarthropathies. Adv Exp Med Biol 649:57–70. https://doi.org/10.1007/978-1-4419-0298-6_4
McGonagle D, Lories RJU, Tan AL, Benjamin M (2007) The concept of a “synovio-entheseal complex” and its implications for understanding joint inflammation and damage in psoriatic arthritis and beyond. Arthritis Rheum 56(8):2482–2491. https://doi.org/10.1002/art.22758
McGonagle D, Benjamin M, Tan AL (2009) The pathogenesis of psoriatic arthritis and associated nail disease: not autoimmune after all? Curr Opin Rheumatol 21(4):340–347. https://doi.org/10.1097/BOR.0b013e32832c6ab9
Tan AL, Benjamin M, Toumi H, Grainger AJ, Tanner SF, Emery P, McGonagle D (2007) The relationship between the extensor tendon enthesis and the nail in distal interphalangeal joint disease in psoriatic arthritis—a high-resolution MRI and histological study. Rheumatol Oxf Engl 46(2):253–256. https://doi.org/10.1093/rheumatology/kel214
Coates LC, Kavanaugh A, Mease PJ, Soriano ER, Laura Acosta-Felquer M, Armstrong AW, Bautista-Molano W, Boehncke WH, Campbell W, Cauli A, Espinoza LR, FitzGerald O, Gladman DD, Gottlieb A, Helliwell PS, Husni ME, Love TJ, Lubrano E, McHugh N, Nash P, Ogdie A, Orbai AM, Parkinson A, O’Sullivan D, Rosen CF, Schwartzman S, Siegel EL, Toloza S, Tuong W, Ritchlin CT (2016) Group for research and assessment of psoriasis and psoriatic arthritis 2015 treatment recommendations for psoriatic arthritis. Arthritis Rheumatol Hoboken NJ 68:1060–1071. https://doi.org/10.1002/art.39573
Gossec L, Coates LC, de Wit M, Kavanaugh A, Ramiro S, Mease PJ, Ritchlin CT, van der Heijde D, Smolen JS (2016) Management of psoriatic arthritis in 2016: a comparison of EULAR and GRAPPA recommendations. Nat Rev Rheumatol 12(12):743–750. https://doi.org/10.1038/nrrheum.2016.183
Alunno A, Carubbi F, Cafaro G, Pucci G, Battista F, Bartoloni E, Giacomelli R, Schillaci G, Gerli R (2015) Targeting the IL-23/IL-17 axis for the treatment of psoriasis and psoriatic arthritis. Expert Opin Biol Ther 15(12):1727–1737. https://doi.org/10.1517/14712598.2015.1084284
Acosta Felquer ML, Coates LC, Soriano ER, Ranza R, Espinoza LR, Helliwell PS, FitzGerald O, McHugh N, Roussou E, Mease PJ (2014) Drug therapies for peripheral joint disease in psoriatic arthritis: a systematic review. J Rheumatol 41(11):2277–2285. https://doi.org/10.3899/jrheum.140876
Langley RG, Elewski BE, Lebwohl M, Reich K, Griffiths CE, Papp K, Puig L, Nakagawa H, Spelman L, Sigurgeirsson B, Rivas E, Tsai TF, Wasel N, Tyring S, Salko T, Hampele I, Notter M, Karpov A, Helou S, Papavassilis C, ERASURE Study Group, FIXTURE Study Group (2014) Secukinumab in plaque psoriasis—results of two phase 3 trials. N Engl J Med 371(4):326–338. https://doi.org/10.1056/NEJMoa1314258
Pariser D, Frankel E, Schlessinger J, Poulin Y, Vender R, Langley RG, Meng X, Guana A, Nyirady J (2017) Efficacy of secukinumab in the treatment of moderate to severe plaque psoriasis in the north American subgroup of patients: pooled analysis of four phase 3 studies. Dermatol Ther. https://doi.org/10.1007/s13555-017-0211-4
Paul C, Lacour J-P, Tedremets L, Kreutzer K, Jazayeri S, Adams S, Guindon C, You R, Papavassilis C, the JUNCTURE study group (2015) Efficacy, safety and usability of secukinumab administration by autoinjector/pen in psoriasis: a randomized, controlled trial (JUNCTURE). J Eur Acad Dermatol Venereol JEADV 29(6):1082–1090. https://doi.org/10.1111/jdv.12751
Griffiths CEM (2010) Comparing biological therapies in psoriasis: implications for clinical practice. J Eur Acad Dermatol Venereol JEADV 24(Suppl 6):10–14. https://doi.org/10.1111/j.1468-3083.2010.03831.x
Gottlieb A, Menter A, Mendelsohn A, Shen YK, Li S, Guzzo C, Fretzin S, Kunynetz R, Kavanaugh A (2009) Ustekinumab, a human interleukin 12/23 monoclonal antibody, for psoriatic arthritis: randomised, double-blind, placebo-controlled, crossover trial. Lancet Lond Engl 373(9664):633–640. https://doi.org/10.1016/S0140-6736(09)60140-9
Reich K, Pinter A, Lacour JP, Ferrandiz C, Micali G, French LE, Lomaga M, Dutronc Y, Henneges C, Wilhelm S, Hartz S, Paul C, on behalf of the IXORA-S investigators (2017) Comparison of ixekizumab with ustekinumab in moderate-to-severe psoriasis: 24-week results from IXORA-S, a phase III study. Br J Dermatol 177(4):1014–1023. https://doi.org/10.1111/bjd.15666
McInnes IB, Mease PJ, Kirkham B et al (2015) Secukinumab, a human anti-interleukin-17A monoclonal antibody, in patients with psoriatic arthritis (FUTURE 2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Lond Engl 386(9999):1137–1146. https://doi.org/10.1016/S0140-6736(15)61134-5
McInnes IB, Mease PJ, Ritchlin CT et al (2017) Secukinumab sustains improvement in signs and symptoms of psoriatic arthritis: 2 year results from the phase 3 FUTURE 2 study. Rheumatol Oxf Engl 56(11):1993–2003. https://doi.org/10.1093/rheumatology/kex301
Khmaladze I, Kelkka T, Guerard S, Wing K, Pizzolla A, Saxena A, Lundqvist K, Holmdahl M, Nandakumar KS, Holmdahl R (2014) Mannan induces ROS-regulated, IL-17A-dependent psoriasis arthritis-like disease in mice. Proc Natl Acad Sci U S A 111(35):E3669–E3678. https://doi.org/10.1073/pnas.1405798111
Sherlock JP, Joyce-Shaikh B, Turner SP, Chao CC, Sathe M, Grein J, Gorman DM, Bowman EP, McClanahan TK, Yearley JH, Eberl G, Buckley CD, Kastelein RA, Pierce RH, LaFace DM, Cua DJ (2012) IL-23 induces spondyloarthropathy by acting on ROR-γt+ CD3+CD4-CD8- entheseal resident T cells. Nat Med 18(7):1069–1076. https://doi.org/10.1038/nm.2817
Ruutu M, Thomas G, Steck R, Degli-Esposti MA, Zinkernagel MS, Alexander K, Velasco J, Strutton G, Tran A, Benham H, Rehaume L, Wilson RJ, Kikly K, Davies J, Pettit AR, Brown MA, McGuckin MA, Thomas R (2012) β-glucan triggers spondylarthritis and Crohn’s disease-like ileitis in SKG mice. Arthritis Rheum 64(7):2211–2222. https://doi.org/10.1002/art.34423
van der Fits L, Mourits S, Voerman JSA, Kant M, Boon L, Laman JD, Cornelissen F, Mus AM, Florencia E, Prens EP, Lubberts E (2009) Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J Immunol Baltim Md 1950 182(9):5836–5845. https://doi.org/10.4049/jimmunol.0802999
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The manuscript does not contain clinical studies or patient data.
Conflict of interest
IBM has received honoraria or research funding from Novartis, Lilly, Celgene, Janssen, Abbvie, BMS, UCB and Pfizer all of whom are developing or have medicines involved in the treatment of psoriatic arthritis.
Rights and permissions
About this article

Cite this article
Cafaro, G., McInnes, I.B. Psoriatic arthritis: tissue-directed inflammation?. Clin Rheumatol 37, 859–868 (2018). https://doi.org/10.1007/s10067-018-4012-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10067-018-4012-7