
Abstract
Systemic lupus erythematosus (SLE) is a chronic relapsing–remitting autoimmune disease affecting several organs. Although the management of lupus patients has improved in the last years, several aspects still remain challenging. More sensitive and specific biomarkers for an early diagnosis as well as for monitoring disease activity and tissue damage are needed. Genome-wide association and gene mapping studies have supported the genetic background for SLE susceptibility. However, the relatively modest risk association and the studies in twins have suggested a role for environmental and epigenetic factors, as well as genetic–epigenetic interaction. Accordingly, there is evidence that differences in DNA methylation, histone modifications, and miRNA profiling can be found in lupus patients versus normal subjects. Moreover, impaired DNA methylation on the inactive X-chromosome was suggested to explain, at least in part, the female prevalence of the disease. Epigenetic markers may be help in fulfilling the unmet needs for SLE by offering new diagnostic tools, new biomarkers for monitoring disease activity, or to better characterize patients with a silent clinical disease but with an active serology. Anti-DNA, anti-phospholipid, and anti-Ro/SSA autoantibodies are thought to be pathogenic for glomerulonephritis, recurrent thrombosis and miscarriages, and neonatal lupus, respectively. However, tissue damage occurs occasionally or, in some patients, only in spite of the persistent presence of the antibodies. Preliminary studies suggest that epigenetic mechanisms may explain why the damage takes place in some patients only or at a given time.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Systemic lupus erythematosus (SLE) is the prototype of the systemic autoimmune diseases characterized by protean clinical manifestations affecting almost any organ of our body. It is a chronic disorder with a gender prevalence displaying a relapsing–remitting course.
The heterogeneity of the manifestations can be, at least in part, related to the several pathogenic mechanisms that are taking place at different moments during the course of the disease. It is widely accepted that a genetic background plays a role in affecting both the innate and the adaptive immunity. However, the disease complexity within individuals and the heterogeneity among individuals, even genetically identical individuals, speak in favor of a stochastic execution of the inherited program as well as of the importance for environmental variables. End tissue damage is eventually supported by mechanisms that play an additional role at the local level (Fig. 1) [1–3].

Systemic lupus erythematosus occurs in phases: The pathogenesis of lupus is mediated by several mechanisms that take place in phases over time
Clinical and translational research has advanced the therapeutic approach, resulting into better patient outcomes. Five-year survival in patients with SLE has improved from 50 % in the 1950s to over 90 % currently. However, the prompt recognition of the disease is still an issue and the mortality remains high compared with the general population. Moreover, tissue damage due to both the disease and the treatments tends to accumulate over time, making the management of lupus patients a challenging issue for physicians.
Epigenetic modulation is emerging as an important mechanism to understand how the susceptibility genes for SLE may interact with environmental factors to cause a full-blown disease. We will review the most recent advances in SLE epigenetic in order to discuss whether such new information may help us in addressing the unmet needs in our patients.
Genetics of SLE
The predisposition to develop SLE has been confirmed in genome-wide association (GWAS) studies performed in various ethnic populations since 2008. In particular, the disease is associated with inheritance of MHC class II and class III loci [1–4]. Two haplotypes of the MHC class II HLA-DRB1 (DRB1*1501 and DRB1*0301) display the strongest association with lupus. GWA studies reported additional genes and single nucleotide polymorphisms (SNPs) in the MHC class III locus that have been linked to SLE. These include early complement components, the MutShomolg 5 (MSHH5), super viralicidic activity 2-like (SKIV2L), integrin alpha (ITGAM, CD11b), integrin beta chain beta2 (ITGB2, CD18), and Fc γ receptor (FcγR) genes [5, 6].
Further, non-HLA genes have been associated to SLE; most of them encode gene products that are theoretically involved in disease pathogenesis. Additional candidates are located within non-coding regions and regulate gene expression via transcriptional and post-transcriptional mechanisms (Table 1).
Epigenetics in SLE
GWAS and gene mapping studies have identified more than 50 loci associated with SLE susceptibility. However, each locus displays a relatively modest risk association when considered individually. The odds ratio (OR) is no greater than 2.8, and the cumulative effect size of these loci accounts for a small fraction of disease heritability (Table 1). Exceptions to low penetrance for susceptibility SLE loci have been reported for genes encoding for early complement components, particularly C1q, and for the TREX1 gene that encodes for nucleases pivotal in degrading cytosolic DNA. Mutations in C1q and TREX1 are rare, but both display a strong risk for lupus (OR = 10 and 25, respectively) [2, 3].
Moreover, the rate of concordance in monozygotic twins is up to 40 %, suggesting that environmental and epigenetic factors, as well as genetic–epigenetic interaction, play a critical role in the susceptibility for SLE.
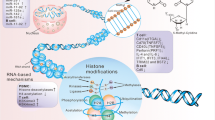
Epigenetic processes refer to heritable modifications that regulate gene expression and affect cellular functions without any change in the genomic sequence. DNA methylation, histone modification, and altered miRNA profiling are widely accepted as the key epigenetic mechanisms playing a role in SLE. Table 2 reports the main evidence for these epigenetic mechanisms in the development of the lupus disease, supporting their role in determining the susceptibility to the disease. While differences in DNA methylation [7–15], histone modifications [19–21], and miRNA profiling [22–26] were found between SLE patients and normal subjects, the specificity of these findings, i.e., in comparison with non-lupus systemic autoimmune diseases, has not been addressed in a systematic way. Accordingly, their value, as diagnostic tools, is still matter of evaluation.
There is a link between genetic and epigenetic factors in the pathogenesis of lupus as shown by the association of susceptibility genes and epigenetic mechanisms in supporting the development of the disease [39]. In conclusion, epigenetic and genetic–epigenetic interaction may account for lupus susceptibility in a significant way. Indeed, there have been multiple observations which suggest an essential role of epigenetic changes in DNA on the effector mechanisms that lead to tissue pathology in lupus [27, 40, 41].
The different epigenetic mechanisms may interact with each other [28]. For example, the transcription factor cAMP-responsive element modulator (CREM)α downregulates IL-2 expression in lupus T cells through both histone deacetylation and CpG-DNA methylation [42]. Comparable interaction was also reported for some miRNAs which target DNA methyltransferases (DNMT)1 directly or indirectly through the modulation of ERK signalling [29, 30].
SLE is a typical female disease with peak disease prevalence between menarche and menopause. There is evidence that estrogen increases the risk of SLE in genetically susceptible women by increasing type 1 interferon (IFN) production and survival of auto-reactive B lymphocytes. On the other hand, progesterone seems to counteract these effects, suggesting that the balance between estrogen and progesterone may account for the female prevalence and for the disease flares during pregnancy and chronic estrogen exposure. However, oral contraceptives containing low-dose estrogen, progesterone, or both and hormone replacement therapy in older patients with milder disease did not show a significant increase in disease flares. These findings suggest that sex hormones can explain the female predominance only in part, indicating that other gender-associated differences contribute to the high female predominance [43, 44]. Epigenetic modifications have been reported to explain the female prevalence for SLE. Lupus patients display impaired DNA methylation on the inactive X-chromosome; as a consequence, reactivation of genes typically suppressed on the inactive X-chromosomes of female lupus patients was suggested to contribute to the female predominance of SLE [45]. Further evidence for the role of the naturally inactivated sex chromosome in SLE susceptibility comes from the observation that SLE is much more frequent in Klinfelter’s syndrome, a condition in which, phenotypically, male individuals possess an additional X-chromosome [46].
All together, these findings may explain the susceptibility for developing lupus and for the female prevalence of the disease. Whether epigenetic modifications can be also helpful in characterizing the disease activity or the risk for specific organ damage will be discussed in the next sections.
Unmet Needs in SLE
The management of lupus patients improved dramatically over the last 10 years as supported by the reduction of the mortality rate at 5 years from 50 % in the 1950s to 90 % nowadays. Both the advance in the knowledge of the pathogenesis as well as the better use of old drugs and the use of new ones contributed to such a progress. However, several aspects of SLE still remain challenging. Diagnosis is usually delayed by the fact that clinical manifestations at the presentation of the disease are highly heterogeneous and not specific. This is a critical issue since it is well known that a prompt diagnosis may allow treating the disease in a more efficient way with lower doses of drugs and much less potential side effects. Once established, the disease should be carefully monitored in order to tune the therapy in the most efficient and safe manner. However, disease monitoring remains difficult due to the low sensitivity of current disease activity markers. Hence, both the early diagnosis and the disease monitoring require biomarkers much more sensitive and specific than the current ones. The management of refractory renal, cutaneous, and neuropsychiatric disease remains unsatisfactory since end-stage renal failure, scarring cutaneous lesions, and neurological damage represent unsolved complications in a significant proportion of patients. Tissue damage due to disease pathogenic mechanisms and to treatment, especially corticosteroid-associated damage, tends to accumulate over time. In this regard, cardiovascular disease secondary to accelerated atherosclerosis has emerged as an important contributor to the morbidity and mortality in longstanding disease. The prolonged use of corticosteroids and/or an uncontrolled active disease (with systemic inflammation) both play a role in accelerated atherosclerosis and tissue damage. The control of the disease is still not optimal in spite of the successes recently obtained. This suggests that the available therapeutic tools reached their maximal effect and that new drugs should be evaluated. Unfortunately, clinical trials display several difficulties in reaching the established end-points because of the heterogeneity of the disease, the limitation of the outcome measures, and the lack of a uniform control group [47]. Table 3 reports the list of the unmet needs in SLE.
Potential Usefulness of Epigenetics to Fulfil Unmet Need in SLE
New Diagnostic and Prognostic Biomarkers
A biomarker is defined as a characteristic that is objectively measured and evaluated as an indicator of normal biologic processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention [48].
SLE is a very heterogeneous disease with different clinical subtypes depending on a given organ involvement (i.e., kidney or CNS damage) or the presence of peculiar risk factors such as specific autoantibodies. This is the case of anti-P ribosomal autoantibodies for CNS involvement or anti-C1q autoantibodies for an active renal disease or anti-phospholipid antibodies (aPL) for recurrent thrombosis and miscarriages. However, the sensitivity and specificity of the commonest autoantibody biomarkers for lupus subtypes are not strong enough with the only exception for aPL. Indeed, there is a general agreement that a persistent medium/high titer of aPL positivities for more than two diagnostic assays (anti-cardiolipin, anti-beta2 glycoprotein I, and lupus anticoagulant) are really predictive for the clinical manifestations of the anti-phospholipid syndrome (APS) and justify a more aggressive primary prophylaxis.
The Issue of Serologically Active Clinically Quiescent Patients
Anti-double stranded (ds)DNA antibodies are recognized as highly specific diagnostic marker for SLE and are also included among the classification criteria of the American College of Rheumatology. Fluctuations of anti-dsDNA antibody titers have been reported to correlate with disease activity and particularly with glomerulonephritis supporting their pathogenic role. Serum complement consumption has been regarded as an additional parameter for disease activity, according to the proposed role of complement activation by immune complexes in the pathogenesis of lupus. However, there is recent evidence that they are not specific and sensitive enough to confirm the clinical suspect of an increase in disease activity. For example, there is a small but consistent group of patients who evolve to persistent serological activity, as supported by elevated anti-dsDNA antibody levels and/or hypocomplementemia despite clinical quiescence: the so-called serologically active clinically quiescent (SACQ) patients. A reciprocal group of patients clinically active but serologically quiescent was also described [49, 50]. These two groups altogether represent a challenge for the conventional theory of lupus as an immune complex-mediated disease. Moreover, they also raise the issue of how to address these patients from a clinical and therapeutic point of view since the traditional biomarkers used to monitor the disease go missing.
New Epigenetic Biomarkers
Looking for new diagnostic/prognostic markers for lupus, epigenetic marks have been recently suggested as possible candidate tools for the diagnosis of the disease as well as for monitoring its evolution and/or its response to therapy. This is true not only of SLE, but also in other autoimmune diseases [51–55].
Differences in DNA methylation and expression changes in genes relevant to SLE pathogenesis have been reported in monozygotic twins discordant for lupus [16], raising the issue whether the degree of DNA methylation may be a potential biomarker for disease activity. Although no relationship was found between DNA methylation and clinical activity or damage indexes in the twins, few studies in small series of SLE patients reported an inverse relationship between global DNA methylation, DNMT1 levels, and Systemic Lupus Erythematosus Disease Activity Score (SLEDAI) score [17, 18]. The suggestion that DNA methylation may correlate with disease activity is intriguing, but much sounder data with larger series and with groups more homogenous in the concomitant treatment are necessary to confirm the finding and to rule out any drug interference.
There is growing evidence that aberrant expression of distinct groups of both immune cell-derived and circulating miRNAs can be found in lupus patients in comparison with healthy controls. It is not possible to identify a distinct profile pattern of deregulated miRNAs in SLE likely because of the heterogeneity of the inclusion criteria as well as the detection techniques in the different studies. However, most of the data are consistent with miRNA abnormalities that may target components of a common pathogenic pathway such as the type I IFN cascade [1]. Since deregulated type I IFN-inducible gene expression pattern—IFN signature—is associated with disease activity, miRNA profiling could represent a potential useful biomarker for monitoring the disease. Accordingly, low miR-146 and high IFN expression correlated with disease activity but very few studies addressed the correlation with specific scoring systems [31]. In addition, some studies reported an association between both cellular and soluble miRNA profiles and levels of anti-dsDNA antibodies in animal models or renal involvement in lupus patients [22, 32–38].
The studies reported in the present review clearly support the association of lupus with epigenetic variations; however, the question whether they represent the cause of the disease or simply the effect of the ongoing autoimmune response or of the treatment is still open. Inhibiting T cell ERK pathway signalling in an animal model results in decreased DNMT1 expression, overexpression of methylation sensitive genes similar to lupus patients, development of anti-dsDNA antibodies, and IFN signature. Such a finding is the strongest argument for a causal role of DNA methylation in lupus, to date [8]. On the other hand, the wide modification of the miRNA profile in cells activated by pro-inflammatory cytokines speaks in favor of epigenetic modifications induced by the inflammation triggered by the autoimmune response [56]. At the same time, we do not know whether the chronic use of anti-inflammatory and immunosuppressive drugs may be responsible for any epigenetic modification.
Can Epigenetic Mechanisms Explain the Onset of Tissue Damage?
Glomerulonephritis is one of the most important complications in the course of SLE and a risk for end stage renal disease; it requires aggressive treatment to achieve remission and relapses are frequent, putting the patients at risk for drug side effects [57]. Autoantibodies to components of chromatin, which include dsDNA, histones, and nucleosomes, are generally thought to play a key role in kidney damage. According to one model, autoantibodies cross-react with intrinsic glomerular structures such as components of membranes, matrices, or exposed non-chromatin ligands released from cells. Antibody-binding triggers inflammation and eventually induces tissue damage. Another model suggests glomerular deposition of autoantibodies in complex with chromatin, thereby inducing classic immune complex-mediated tissue damage [58].
Only a limited proportion of lupus patients develop renal damage, in spite of the presence of theoretically pathogenic anti-chromatin/DNA autoantibodies for a long time. In the same manner, anti-DNA antibodies are produced much before the glomerulonephritis can be detected in animal models [2, 3]. Hence, it has been suggested that further local kidney factors may play a role in addition to the three key pathways related to the autoimmune response to chromatin auto-antigen: defective clearance of apoptotic cells, engagement of toll-like receptors, type I IFN, NFkB signalling, and T and B dysfunctions [2, 3]. It has been recently reported that APOL1 G1/G2 alleles strongly impact the risk of lupus nephritis end-stage renal disease (ESRD) as well as the time of progression to ESRD in African-Americans, suggesting that APOL1 is associated with renal susceptibility to damage [59, 60]. The hypothesis that APOL1 may influence the end-organ damage is supported by the clinical observation that kidneys from diseased donors with two APOL1 nephropathy alleles fail more rapidly than those from diseased donors with 0 or 1 risk allele [61]. Moreover, data from murine congenic dissection and reconstitution studies do suggest that genes from multiple functional categories would act in concert to promote lupus nephritis. In particular, genes that regulate functions in the end-organs, such as the kidneys, lead to fully developed SLE [3].
In this regard, a possible role for renal DNaseI enzyme activity has been recently hypothesized to explain the progression of murine lupus nephritis. DNaseI is required for chromatin breakdown during apoptosis as well as necrosis; accordingly, a loss of this enzyme activity may lead to accumulation of apoptotic chromatin fragments in glomeruli. As a consequence, the amount of anti-chromatin antibodies/chromatin complexes increases and exceeds the threshold for triggering a local inflammatory response.
The reduced chromatin fragmentation has been suggested to display biological consequences beyond the classic immune complex-mediated damage since it appears to affect also responses of the innate immunity. In fact, TLR7-9 and Clec4e have been found upregulated in BW mice at the same time when chromatin–IgG complex deposition in the glomerular basement membrane (GBM) and loss of DNasI activity are demonstrated [62]. TLR7-9 is involved in the processing of DNA–protein complexes while incomplete clearance and degradation of apoptotic material may transform it into necrotic cell debris which contains SAP130, the ligand for the inflammation-related receptor Clec4e [58]. The signalling triggered by SAP130-Clec4e may promote pro-inflammatory cytokine production and upregulation of metalloproteases that altogether can facilitate the chromatin fragment–IgG complex deposition in both the mesangial matrix and the GBM. A comparable correlation between loss of DNasI and progression of glomerulonephritis has been found in human kidneys [63–65].
Looking for possible mechanisms responsible for the DNaseI gene expression, the same authors suggested the role of transcriptional interference [66] as well as the role of an abnormal miRNA profile. These mechanisms may affect DNaseI production through gene silencing independently or even in combination. In line with such hypothesis, different miRNA profiles were found in kidney of lupus-prone mice with glomerulonephritis as well as in biopsies of lupus patients [35, 67, 68].
Since the levels of DNaseI in the urine seems to reflect its modulation in the tissues, it is useful to speculate that both DNaseI dosage and/or specific miRNA profiles may represent possible new tools to predict the evolution of kidney damage in lupus patients.
The mechanisms reported above do not exclude additional pathogenic pathways affecting the kidney and unrelated to anti-chromatin antibody response [2].
aPL and APS as Additional Targets for Epigenetic Biomarkers
aPL are widely accepted as a strong risk biomarker for recurrent thrombotic events and miscarriages in SLE patients. The risk for such manifestations is proportional to the autoantibody levels since medium/high aPL titers and more than one positivity in the diagnostic assays have been reported as the most predictive factors for developing APS [69].
The presence of aPL is required for developing the syndrome, but the antibodies alone are not sufficient. For example, thrombotic events can occur only occasionally and the need of a second “hit” was suggested to explain this condition. In other words, the antibody (first hit) induces a thrombophilic condition but clotting takes place in the presence of another thrombophilic condition (second hit) [70]. Inflammatory stimuli of different origin have been identified as candidate second hits. There is growing evidence that they act by increasing the presence of the main autoantigen target for aPL (i.e., beta2 glycoprotein I – β2GPI) in the vascular tissues, at least in part, by upregulating the expression of the cell membrane receptors for β2GPI [71, 72]. Once upregulated, β2GPI can be recognized by aPL in amounts large enough to activate complement that eventually causes clot formation [70]. It is widely accepted that β2GPI-dependent aPL activate the cells involved in the coagulation cascade (i.e., endothelium, circulating monocytes, and platelets) by recognizing the molecule present on their cell membrane. The antibody binding induces a pro-inflammatory and pro-coagulant phenotype that plays a major role in APS thrombosis [70, 73].
Although the “two hit hypothesis” represents a convincing explanation, it is still unclear what are the true mechanisms beyond the fact that clotting takes place only occasionally in spite of the persistent presence of most of the pathogenic players and the frequent occurrence of inflammatory stimuli potentially able to trigger the APS cascade. For example, up to 40 % of SLE patients are persistently positive for aPL, even at high titre; however, less than one third of them actually display the clinical events. A similar group of aPL-positive asymptomatic carriers can also be found in subjects with no any underlying autoimmune disease and followed for a long period of time [74].
It has been suggested that epigenetic mechanisms may take place by lowering the threshold for the coagulation cascade. For example, miR-19b and miR-20a were reported to downregulate tissue factor (TF) expression on peripheral monocytes; in particular, miR-20a was found to bind TF mRNA, suggesting a direct regulating effect. TF is the starting component of the extrinsic coagulation cascade, and there is evidence for its involvement in APS pathogenesis because of its upregulated expression in circulating monocytes from patients [70, 73]. Levels of miR-19b and miR-20a were both decreased in monocytes from APS patients, and their expression was inversely correlated with the expression of TF on the cell membrane. This finding indicates for the first time that an epigenetic mechanism may take place in regulating one of the thrombophilic pathways thought to play a role in APS pathogenesis. Unfortunately, a comparable decreased expression of miR-19b and miR-20a was also found in monocytes from SLE aPL-negative patients, indicating that the phenomenon is not specific for APS only [74]. Nevertheless, the study paves the way for exploring new epigenetic biomarkers predictive for a stronger thrombophilic state. In this regard, in vitro experiments showed that inhibition of miR-20a in the monocyte lineage THP-1 cells provokes a 50 % increase in their pro-coagulant activity after lipopolysaccharide (LPS) stimulation [74]. Since LPS is known to represent a good model of a “second hit” in APS models and to modulate a large expression of miRNAs in a NFκB-dependent manner [75], it is possible to speculate that an infectious stimulus may affect TF expression in monocytes through changes in their miRNA profiles. We can hypothesize that a similar mechanism can also take place in endothelial cells, the other cell type in which aPL have been demonstrated to upregulate TF expression [72]. Hence, abnormalities in miRNA profiles related to the thrombophilic phenotype of cells involved in the coagulation cascade could represent a further tool to better stratify the whole thrombophilic risk of aPL positive patients.
Neonatal Lupus as a Target for Epigenetic Biomarkers
Neonatal lupus (NL) refers to a clinical spectrum of cutaneous, cardiac, and systemic abnormalities observed in new born infants from mothers positive for autoantibodies against Ro/SSA, La/SSB, and, less commonly, U1-ribonucleoprotein (U1-RNP). The most common presentation is a non-scarring, non-atrophic skin lesion resembling sub-acute cutaneous lupus erythematosus. The lesions may develop at birth or even later during the first weeks of life. Cardiac, hematological, hepatobiliary, central nervous, and pulmonary systems may also be involved. It occurs in about 1 to 2 % of babies born to mothers with autoimmune disease, primarily SLE and Sjögren’s syndrome, and antibodies to Ro/SSA and/or La/SSB. However, many cases can occur in children of mothers positive for the same autoantibodies, but with no signs of lupus or other autoimmune disease at the time of the baby’s birth; about one half of these mothers develop autoimmune disease (more commonly Sjögren syndrome than SLE) [76]. The most serious complication of NL is complete heart block (CHB) and about 10% have an associated cardiomyopathy at the initial diagnosis or develop it later [77].
NL is a prototype example of a passively transferred autoimmune disease due to the trans-placental passage of anti-RoSSA and anti-La/SSB. However, the pathogenesis of the disease probably involves more than simple transplacental passage of antibodies since only few babies born from mothers positive for the autoantibodies are affected, and there is discordance of the disease even in monozygotic twins [77]. Hence, maternal antibodies to Ro/SSA and/or La/SSB, although a powerful risk factor for CHB, are not the only determinant of the development of NL, and further mechanisms have been investigated.
Studies on epitope specificity of anti-Ro autoantibodies showed that antibodies recognizing the peptide aa200-239 of Ro 52 (p200) were pathogenic in experimental models and more predictive for the risk to develop the syndrome. However, the reactivity to p200 has been found dominant but not uniform in women whose children have CHB. Since exposure to this antibody specificity was observed with a similar frequency in children without CHB born to mothers with anti-Ro 52, additional factors were suggested to be necessary for the disease expression [78, 79]. For example, evidence from animal models suggests that antibodies targeting L-type calcium channels may also contribute to the development of cardiac-NL [80].
Heart block develops in only a minority of subsequent pregnancies, despite the persistence of maternal antibodies, suggesting that fetal factors are playing a role. The influence of specific HLA alleles and a polymorphism in the promoter region of the gene for TNF-alpha (−308A, associated with higher TNF-alpha production) was reported in some series [81–83]. However, studies performed in twins did not confirm the association with TNF-alpha polymorphisms, and no difference in TNF-alpha secretion was demonstrated in their peripheral blood mononuclear cells. In addition, a pro-fibrotic TGF beta1 genotype was detected in the twin with CHB and not in the healthy twin in one series but not in another one [84]. This finding is in line with the hypothesis that a pro-fibrotic response to damaged tissues by cardiac macrophages can play a role in favoring the appearance of CHB [85].
Maternal–fetal microchimerism may also contribute to CHB in NL. In fact, an increased number of female, presumably maternal, cells were found in the myocardium in all four of the males with heart block and in two of four controls [86]. Some of the maternal cells in the myocardium expressed differentiation markers of myoctes, while others had surface markers of hematopoietic cells. It has been suggested that these cells may act as a target for an allogenic response as well as effector cells triggering an inflammation and eventually a tissue damage in cooperation with anti-Ro/SSA antibodies [86]. This finding was only partially confirmed in twins’ studies suggesting that such a hypothesis cannot explain all the cases [87].
Finally, the polymorphisms of the FcγRIIA and IIIB were also investigated in the same twins looking for a possible association with CHB. Unfortunately, the data were not conclusive [88].
In conclusion, the reason(s) why the same antibodies can be able to induce NL in few babies only is still a matter of research. The hypothesis that individual fetal factors may be responsible for the disease even in monozygotic twins is quite suggestive for a role of epigenetic mechanisms. Whether epigenetic events may affect the expression of the right myocardial auto-antigen in a given patient or whether the abnormal presence of female cells in males may favor the effect of unsuppressed genes in the X chromosome, all may represent topics for future epigenetic studies.
References
Deng Y, Tsao BP (2014) Advances in lupus genetics and epigenetics. Curr Opin Rheumatol 26:482–492
Marion TN, Postlethwaite AE (2014) Chance, genetics, and the heterogeneity of disease and pathogenesis in systemic lupus erythematosus. Semin Immunopathol 36:495–517
Mohan C, Putterman C (2015) Genetics and pathogenesis of systemic lupus erythematosus and lupus nephritis. Nat Rev Nephrol 11:329–341
Costa-Reis P, Sullivan KE (2013) Genetics and epigenetics of systemic lupus erythematosus. Curr Rheumatol Rep 15:369
Connolly JJ, Hakonarson H (2012) Role of cytokines in systemic lupus erythematosus: recent progress from GWAS and sequencing. J Biomed Biotechnol 2012:798924
Cui Y, Sheng Y, Zhang X (2013) Genetic susceptibility to SLE: recent progress from GWAS. J Autoimmun 41:25–33
Coit P, Jeffries M, Altorok N et al (2013) Genome-wide DNA methylation study suggests epigenetic accessibility and transcriptional poising of interferon-regulated genes in naive CD4+ T cells from lupus patients. J Autoimmun 43:78–84
Sawalha AH, Jeffries M, Webb R et al (2008) Defective T-cell ERK signaling induces interferon-regulated gene expression and overexpression of methylation-sensitive genes similar to lupus patients. Genes Immun 9:368–378
Cornacchia E, Golbus J, Maybaum J, Strahler J, Hanash S, Richardson B (1988) Hydralazine and procainamide inhibit T cell DNA methylation and induce autoreactivity. J Immunol 140:2197–2200
Deng C, Lu Q, Zhang Z et al (2003) Hydralazine may induce autoimmunity by inhibiting extracellular signal-regulated kinase pathway signaling. Arthritis Rheum 48:746–756
Lieberman MW, Beach LR, Palmiter RD (1983) Ultraviolet radiation-induced metallothionein-I gene activation is associated with extensive DNA demethylation. Cell 35:207–214
Quddus J, Johnson KJ, Gavalchin J et al (1993) Treating activated CD4+ T cells with either of two distinct DNA methyltransferase inhibitors, 5-azacytidine or procainamide, is sufficient to cause a lupus-like disease in syngeneic mice. J Clin Invest 92:38–53
Zhang Y, Zhao M, Sawalha AH, Richardson B, Lu Q (2013) Impaired DNA methylation and its mechanisms in CD4(+)T cells of systemic lupus erythematosus. J Autoimmun 41:92–99
Hedrich CM, Tsokos GC (2011) Epigenetic mechanisms in systemic lupus erythematosus and other autoimmune diseases. Trends Mol Med 17:714–724
Javierre BM, Richardson B (2011) A new epigenetic challenge: systemic lupus erythematosus. Adv Exp Med Biol 711:117–136
Javierre BM, Fernandez AF, Richter J et al (2010) Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res 20:170–179
Qin HH, Zhu XH, Liang J et al (2013) Associations between aberrant DNA methylation and transcript levels of DNMT1 and MBD2 in CD4 + T cells from patients with systemic lupus erythematosus. Australas J Dermatol 54:90–95
Zhu X, Liang J, Li F, Yang Y, Xiang L, Xu J (2011) Analysis of associations between the patterns of global DNA hypomethylation and expression of DNA methyltransferase in patients with systemic lupus erythematosus. Int J Dermatol 50:697–704
Zhang Z, Song L, Maurer K, Petri MA, Sullivan KE (2010) Global H4 acetylation analysis by ChIP-chip in systemic lupus erythematosus monocytes. Genes Immun 11:124–133
Hedrich CM, Rauen T, Kis-Toth K, Kyttaris VC, Tsokos GC (2012) cAMP-responsive element modulator alpha (CREMalpha) suppresses IL-17F protein expression in T lymphocytes from patients with systemic lupus erythematosus (SLE). J Biol Chem 287:4715–4725
Rauen T, Hedrich CM, Juang YT, Tenbrock K, Tsokos GC (2011) cAMP-responsive element modulator (CREM)alpha protein induces interleukin 17A expression and mediates epigenetic alterations at the interleukin-17A gene locus in patients with systemic lupus erythematosus. J Biol Chem 286:43437–43446
Ceribelli A, Satoh M, Chan EK (2012) MicroRNAs and autoimmunity. Curr Opin Immunol 24:686–691
Deng X, Su Y, Wu H et al (2015) The role of microRNAs in autoimmune diseases with skin involvement. Scand J Immunol 81:153–165
Liu A, La Cava A (2014) Epigenetic dysregulation in systemic lupus erythematosus. Autoimmunity 47:215–219
Miao CG, Yang YY, He X et al (2013) The emerging role of microRNAs in the pathogenesis of systemic lupus erythematosus. Cell Signal 25:1828–1836
Yan S, Yim LY, Lu L, Lau CS, Chan VS (2014) MicroRNA regulation in systemic lupus erythematosus pathogenesis. Immune Netw 14:138–148
Zhao M, Liu S, Luo S et al (2014) DNA methylation and mRNA and microRNA expression of SLE CD4+ T cells correlate with disease phenotype. J Autoimmun 54:127–136
Richardson BC, Patel DR (2014) Epigenetics in 2013. DNA methylation and miRNA: key roles in systemic autoimmunity. Nat Rev Rheumatol 10:72–74
Pan W, Zhu S, Yuan M et al (2010) MicroRNA-21 and microRNA-148a contribute to DNA hypomethylation in lupus CD4+ T cells by directly and indirectly targeting DNA methyltransferase 1. J Immunol 184:6773–6781
Zhao S, Wang Y, Liang Y et al (2011) MicroRNA-126 regulates DNA methylation in CD4+ T cells and contributes to systemic lupus erythematosus by targeting DNA methyltransferase 1. Arthritis Rheum 63:1376–1386
Tang Y, Luo X, Cui H et al (2009) MicroRNA-146A contributes to abnormal activation of the type I interferon pathway in human lupus by targeting the key signaling proteins. Arthritis Rheum 60:1065–1075
Carlsen AL, Schetter AJ, Nielsen CT et al (2013) Circulating microRNA expression profiles associated with systemic lupus erythematosus. Arthritis Rheum 65:1324–1334
Lu J, Kwan BC, Lai FM et al (2012) Glomerular and tubulointerstitial miR-638, miR-198 and miR-146a expression in lupus nephritis. Nephrology (Carlton 17:346–351
Stagakis E, Bertsias G, Verginis P et al (2011) Identification of novel microRNA signatures linked to human lupus disease activity and pathogenesis: miR-21 regulates aberrant T cell responses through regulation of PDCD4 expression. Ann Rheum Dis 70:1496–1506
Te JL, Dozmorov IM, Guthridge JM et al (2010) Identification of unique microRNA signature associated with lupus nephritis. PLoS One 5:e10344
Thai TH, Patterson HC, Pham DH, Kis-Toth K, Kaminski DA, Tsokos GC (2013) Deletion of microRNA-155 reduces autoantibody responses and alleviates lupus-like disease in the Fas(lpr) mouse. Proc Natl Acad Sci U S A 110:20194–20199
Wang G, Tam LS, Kwan BC et al (2012) Expression of miR-146a and miR-155 in the urinary sediment of systemic lupus erythematosus. Clin Rheumatol 31:435–440
Wen Z, Xu L, Chen X et al (2013) Autoantibody induction by DNA-containing immune complexes requires HMGB1 with the TLR2/microRNA-155 pathway. J Immunol 190:5411–5422
Altorok N, Sawalha AH (2013) Epigenetics in the pathogenesis of systemic lupus erythematosus. Curr Opin Rheumatol 25:569–576
Coit P, Renauer P, Jeffries M A, et al (2015) Renal involvement in lupus is characterized by unique DNA methylation changes in naive CD4+ T cells. J Autoimmun
Coit P, Yalavarthi S, Ognenovski M et al (2015) Epigenome profiling reveals significant DNA demethylation of interferon signature genes in lupus neutrophils. J Autoimmun 58:59–66
Rauen T, Hedrich CM, Tenbrock K, Tsokos GC (2013) cAMP responsive element modulator: a critical regulator of cytokine production. Trends Mol Med 19:262–269
Hughes GC, Choubey D (2014) Modulation of autoimmune rheumatic diseases by oestrogen and progesterone. Nat Rev Rheumatol 10:740–751
Schwartzman-Morris J, Putterman C (2012) Gender differences in the pathogenesis and outcome of lupus and of lupus nephritis. Clin Dev Immunol 2012:604892
Lu Q, Wu A, Tesmer L, Ray D, Yousif N, Richardson B (2007) Demethylation of CD40LG on the inactive X in T cells from women with lupus. J Immunol 179:6352–6358
Scofield RH, Bruner GR, Namjou B et al (2008) Klinefelter’s syndrome (47, XXY) in male systemic lupus erythematosus patients: support for the notion of a gene-dose effect from the X chromosome. Arthritis Rheum 58:2511–2517
Lateef A, Petri M (2012) Unmet medical needs in systemic lupus erythematosus. Arthritis Res Ther 14(Suppl 4):S4
Illei GG, Tackey E, Lapteva L, Lipsky PE (2004) Biomarkers in systemic lupus erythematosus: II. Markers of disease activity. Arthritis Rheum 50:2048–2065
Gladman DD, Hirani N, Ibanez D, Urowitz MB (2003) Clinically active serologically quiescent systemic lupus erythematosus. J Rheumatol 30:1960–1962
Steiman AJ, Gladman DD, Ibanez D, Urowitz MB (2010) Prolonged serologically active clinically quiescent systemic lupus erythematosus: frequency and outcome. J Rheumatol 37:1822–1827
Cornec D, Jamin C, Pers JO (2014) Sjogren’s syndrome: where do we stand, and where shall we go? J Autoimmun 51:109–114
Luo Y, Wang Y, Wang Q, Xiao R, Lu Q (2013) Systemic sclerosis: genetics and epigenetics. J Autoimmun 41:161–167
Pillai S (2013) Rethinking mechanisms of autoimmune pathogenesis. J Autoimmun 45:97–103
Lu Q (2013) The critical importance of epigenetics in autoimmunity. J Autoimmun 41:1–5
Zhang P, Zhao M, Liang G et al (2013) Whole-genome DNA methylation in skin lesions from patients with psoriasis vulgaris. J Autoimmun 41:17–24
Javierre BM, Hernando H, Ballestar E (2011) Environmental triggers and epigenetic deregulation in autoimmune disease. Discov Med 12:535–545
Rahman A, Isenberg DA (2008) Systemic lupus erythematosus. N Engl J Med 358:929–939
Seredkina N, Van Der Vlag J, Berden J, Mortensen E, Rekvig OP (2013) Lupus nephritis: enigmas, conflicting models and an emerging concept. Mol Med 19:161–169
Freedman BI, Langefeld CD, Andringa KK et al (2014) End-stage renal disease in African Americans with lupus nephritis is associated with APOL1. Arthritis Rheumatol 66:390–396
Lin CP, Adrianto I, Lessard CJ et al (2012) Role of MYH9 and APOL1 in African and non-African populations with lupus nephritis. Genes Immun 13:232–238
Reeves-Daniel AM, DePalma JA, Bleyer AJ et al (2011) The APOL1 gene and allograft survival after kidney transplantation. Am J Transplant 11:1025–1030
Thiyagarajan D, Fismen S, Seredkina N et al (2012) Silencing of renal DNaseI in murine lupus nephritis imposes exposure of large chromatin fragments and activation of Toll like receptors and the Clec4e. PLoS One 7:e34080
Ben David D, Reznick AZ, Srouji S, Livne E (2008) Exposure to pro-inflammatory cytokines upregulates MMP-9 synthesis by mesenchymal stem cells-derived osteoprogenitors. Histochem Cell Biol 129:589–597
Lim EJ, Lee SH, Lee JG et al (2006) Activation of toll-like receptor-9 induces matrix metalloproteinase-9 expression through Akt and tumor necrosis factor-alpha signaling. FEBS Lett 580:4533–4538
Merrell MA, Ilvesaro JM, Lehtonen N et al (2006) Toll-like receptor 9 agonists promote cellular invasion by increasing matrix metalloproteinase activity. Mol Cancer Res 4:437–447
Shearwin KE, Callen BP, Egan JB (2005) Transcriptional interference—a crash course. Trends Genet 21:339–345
Chafin CB, Reilly CM (2013) MicroRNAs implicated in the immunopathogenesis of lupus nephritis. Clin Dev Immunol 2013:430239
Dai Y, Sui W, Lan H, Yan Q, Huang H, Huang Y (2009) Comprehensive analysis of microRNA expression patterns in renal biopsies of lupus nephritis patients. Rheumatol Int 29:749–754
Meroni PL, Chighizola CB, Rovelli F, Gerosa M (2014) Antiphospholipid syndrome in 2014: more clinical manifestations, novel pathogenic players and emerging biomarkers. Arthritis Res Ther 16:209
Meroni PL, Borghi MO, Raschi E, Tedesco F (2011) Pathogenesis of antiphospholipid syndrome: understanding the antibodies. Nat Rev Rheumatol 7:330–339
Agostinis C, Biffi S, Garrovo C et al (2011) In vivo distribution of beta2 glycoprotein I under various pathophysiologic conditions. Blood 118:4231–4238
Raschi E, Chighizola CB, Grossi C et al (2014) beta2-glycoprotein I, lipopolysaccharide and endothelial TLR4: three players in the two hit theory for anti-phospholipid-mediated thrombosis. J Autoimmun 55:42–50
Giannakopoulos B, Krilis SA (2013) The pathogenesis of the antiphospholipid syndrome. N Engl J Med 368:1033–1044
Teruel R, Perez-Sanchez C, Corral J et al (2011) Identification of miRNAs as potential modulators of tissue factor expression in patients with systemic lupus erythematosus and antiphospholipid syndrome. J Thromb Haemost 9:1985–1992
Bazzoni F, Rossato M, Fabbri M et al (2009) Induction and regulatory function of miR-9 in human monocytes and neutrophils exposed to proinflammatory signals. Proc Natl Acad Sci U S A 106:5282–5287
Rivera TL, Izmirly PM, Birnbaum BK et al (2009) Disease progression in mothers of children enrolled in the Research Registry for Neonatal Lupus. Ann Rheum Dis 68:828–835
Izmirly PM, Buyon JP, Saxena A (2012) Neonatal lupus: advances in understanding pathogenesis and identifying treatments of cardiac disease. Curr Opin Rheumatol 24:466–472
Clancy RM, Buyon JP, Ikeda K et al (2005) Maternal antibody responses to the 52-kd SSA/RO p200 peptide and the development of fetal conduction defects. Arthritis Rheum 52:3079–3086
Scarsi M, Radice A, Pregnolato F et al (2014) Anti-Ro/SSA-p200 antibodies in the prediction of congenital heart block. An Italian multicentre cross-sectional study on behalf of the ‘Forum Interdisciplinare per la Ricerca nelle Malattie Autoimmuni (FIRMA) Group’. Clin Exp Rheumatol 32:848–854
Lindop R, Arentz G, Thurgood LA, Reed JH, Jackson MW, Gordon TP (2012) Pathogenicity and proteomic signatures of autoantibodies to Ro and La. Immunol Cell Biol 90:304–309
Clancy RM, Backer CB, Yin X et al (2004) Genetic association of cutaneous neonatal lupus with HLA class II and tumor necrosis factor alpha: implications for pathogenesis. Arthritis Rheum 50:2598–2603
Clancy RM, Marion MC, Kaufman KM et al (2010) Identification of candidate loci at 6p21 and 21q22 in a genome-wide association study of cardiac manifestations of neonatal lupus. Arthritis Rheum 62:3415–3424
Siren MK, Julkunen H, Kaaja R, Ekblad H, Koskimies S (1999) Role of HLA in congenital heart block: susceptibility alleles in children. Lupus 8:60–67
Cimaz R, Borghi MO, Gerosa M, Biggioggero M, Raschi E, Meroni PL (2006) Transforming growth factor beta1 in the pathogenesis of autoimmune congenital complete heart block: lesson from twins and triplets discordant for the disease. Arthritis Rheum 54:356–359
Ramos PS, Marion MC, Langefeld CD, International Consortium on Systemic Lupus Erythematosus Genetics, Buyon JP, Clancy RM (2012) Brief report: enrichment of associations in genes with fibrosis, apoptosis, and innate immunity functions with cardiac manifestations of neonatal lupus. Arthritis Rheum 64:4060–4065
Stevens AM, Hermes HM, Rutledge JC, Buyon JP, Nelson JL (2003) Myocardial-tissue-specific phenotype of maternal microchimerism in neonatal lupus congenital heart block. Lancet 362:1617–1623
Stevens AM, Hermes HM, Lambert NC, Nelson JL, Meroni PL, Cimaz R (2005) Maternal and sibling microchimerism in twins and triplets discordant for neonatal lupus syndrome-congenital heart block. Rheumatology (Oxford) 44:187–191
Fesslova V, Mannarino S, Salice P et al (2003) Neonatal lupus: fetal myocarditis progressing to atrioventricular block in triplets. Lupus 12:775–778
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Meroni, P.L., Penatti, A.E. Epigenetics and Systemic Lupus Erythematosus: Unmet Needs. Clinic Rev Allerg Immunol 50, 367–376 (2016). https://doi.org/10.1007/s12016-015-8497-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-015-8497-4