
Abstract
The three common themes that underlie the induction and perpetuation of autoimmunity are genetic predisposition, environmental factors, and immune regulation. Environmental factors have gained much attention for their role in triggering autoimmunity, with increasing evidence of their influence as demonstrated by epidemiological studies, laboratory research, and animal studies. Environmental factors known to trigger and perpetuate autoimmunity include infections, gut microbiota, as well as physical and environmental agents. To address these issues, we will review major potential mechanisms that underlie autoimmunity including molecular mimicry, epitope spreading, bystander activation, polyclonal activation of B and T cells, infections, and autoinflammatory activation of innate immunity. The association of the gut microbiota on autoimmunity will be particularly highlighted by their interaction with pharmaceutical agents that may lead to organ-specific autoimmunity. Nonetheless, and we will emphasize this point, the precise mechanism of environmental influence on disease pathogenesis remains elusive.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
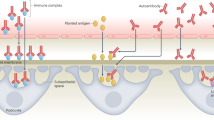
Autoimmune disease can occur in any site, but the three common themes that underlie induction and perpetuation include: genetic predisposition, environmental factors, and immune regulation (1–4) (Fig. 1). Current data suggests that environmental factors, i.e., infections, gut microbiota, toxic chemicals, and dietary components have up to a 70 % contribution to loss of tolerance (5, 6). The number of peer-reviewed papers in PubMed identified with “autoimmune disease” increased from 8890 in 1997 to 67,229 in 2015. Clearly, knowledge on the interaction between environmental factors and the architecture of the immune system is critical is critical to unveil the mechanisms of autoimmunity and future design of treatment modalities. In this review, we will describe current perspectives on environment and autoimmune diseases. Specific examples on selected autoimmune diseases are included to illustrate the significance of environmental agents on the development of autoimmunity. Many of these principles are illustrated in the methods by which autoimmune diseases are classified (7–11). Finally, we will discuss lessons, we learned in our recent studies on the role of environmental etiology on primary biliary cirrhosis (PBC), a prototypic organ-specific autoimmune disease.

Three common themes that underlie induction and perpetuation of autoimmune diseases
Infections
An autoimmune disease may be induced or triggered by infectious agents including viruses, bacteria, fungi, and parasites (12). The levels of evidence are determined using three criteria, in particular: (i) epidemiological studies, (ii) laboratory studies, and (iii) experimental models (13). Examples of virus-induced autoimmune diseases are shown in Table 1 (14–27). Autoimmune diseases which have been reported to be triggered by viruses include rheumatoid arthritis (RA), thyroid diseases, primary biliary cirrhosis (PBC), type I diabetes, and autoimmune hepatitis (AIH). Interestingly, hepatitis A virus (HAV) has been reported to be associated to a fulminant type I diabetes in a 38-year-old man who suffered acute hepatitis A before the onset of diabetes (16). In this case, pancreatic involvement was attributed to an immune response, rather than to a direct cytotoxic effect of HAV. Moreover, additional cases of AIH have been reported to be triggered by HAV (25, 26). Another possible trigger for AIH is the Epstein-Barr virus (EBV); some cases of AIH have been described in strict temporal sequence after an acute EBV infection (25). In two cases, a defect in suppressor/inducer T cells controlling the response to the asialoglycoprotein receptor (ASGPR) had been identified prior to the viral infection, and anti-ASGPR antibodies persisted and increased after the viral illness (26). Examples of bacteria-induced autoimmune disease are shown in Table 2 (28–44). An early report on the presence of an immunological response to Candida albicans in the synovial fluid and peripheral blood lymphocytes suggests that even fungi may induce autoimmune disease (45). However, in many cases, it is not a single infection but rather the “burden of infections” from childhood that is responsible for the induction of autoimmunity (46). The mechanisms and cell types responsible for the onset and progression of these multifactorial diseases remain unclear. Both autoimmune pathogenic and protective immune responses require the cooperative function of the innate, humoral, and cellular arms of the immune system. Thus, B and T lymphocytes and innate antigen-presenting cells (APCs) all contribute to these responses.
Infectious agents are known to trigger an autoimmune disease through several mechanisms: (1) molecular mimicry, (2) epitope spreading, (3) bystander activation and stimulation of pattern recognition receptors, (4) viral persistence and polyclonal activation of B cells, (5) autoinflammatory activation of innate immunity.
Molecular Mimicry
Molecular mimicry, a mechanism by which infections can induce autoimmunity, occurs when foreign antigens share sequence or structural similarities with self-antigens (47). This is due to the fact that immune responses can be directed against peptides with similar charge distribution and overall shape (48). Immune response to microbial antigens could then result in activation of T cells that are cross-reactive with self-antigens. For example, molecular mimicry and cross-reactivity involving Escherichia coli and human subunit E2 epitopes of the pyruvate dehydrogenase complex (PDC) have been considered to trigger of the initiation of E. coli-associated anti-mitochondrial immune response in PBC (49–52). Strong evidence regarding CD4-T cell cross-recognition of E. coli and human mitochondrial autoantigens has been obtained over the past 20 years (53–56), further supporting the concept of molecular mimicry as the driving force of the immunological breakdown characteristic of PBC.
Epitope Spreading
Epitope spreading consists of the development of autoimmune responses to endogenous epitopes secondary to the release of self-antigens during a chronic autoimmune or inflammatory response (57). Epitope spreading can result from a change in protein structure, i.e., changing of an amino acid residue from arginine to citrulline. This may result in an immune reaction not only against the original protein or in its citrullinated form but also against other citrullinated proteins; this is a characteristic of rheumatoid arthritis. Systemic lupus erythematosus (SLE), multiple sclerosis, pemphigus bullous, pemphigoid, and other autoimmune diseases are all influenced by intermolecular and intramolecular B cell epitope spreading. Endocytic processing, antigen presentation, and somatic hypermutation are just some of the molecular mechanisms that assist in driving epitope spreading and broadening the immune response in autoimmune diseases (58). An example is type I diabetes in which autoimmunity first may be triggered against the B9–23 region of insulin (59), and progression to overt disease is mediated by epitope spreading to an array of beta cell antigens.
Epitope spreading is a crucial point for therapeutic strategies. Due to the unknown biological pathways and changes in antigen specificity, it is difficult to design drugs directed at epitope spreading. Instead, historically, the use of several drugs to control and inhibit both B and T cell activities has been the most commonly used approach to autoimmune diseases (60–62).
Bystander Activation and Stimulation of Pattern Recognition Receptors
Bystander activation occurs when microbial infection stimulates toll-like receptors (TLRs) and other pattern recognition receptors on antigen-presenting cells (APCs), leading to the production of pro-inflammatory mediators, which in turn may lead to tissue damage (63). The release of both tissue antigens and bacterial antigens could result in autoreactive T cells and bacterial-specific T cells in the process called bystander activation, a process contributing to autoimmunity. Additionally, virally infected APCs and concomitantly released mediators activate autoreactive Th1 or Th17 cells in a bystander manner (64). It has been shown that gut-derived antigens stimulate liver cells and result in a distinctive immune response via TLRs. TLRs are expressed on Kupffer cells, dendritic cells, hepatic stellate cells, endothelial cells, and hepatocytes. The cross-talk between gut-derived antigens and TLRs on immune cells trigger a distinctive set of mechanisms to induce immunity, contributing to various acute and chronic liver diseases including liver cirrhosis and hepatocellular carcinoma (65).
Viral Persistence and Polyclonal Activation of B Cells
Prolonged infection with a virus, such as EBV, can lead to constant activation and proliferation of T cells, resulting in the production of monoclonal and polyclonal antibodies as well as immune complexes, leading to loss of tolerance (48). Patients with several autoimmune diseases have a high prevalence of anti-hepatitis C virus (HCV) serum antibodies suggesting a pervasive role for HCV in tolerance breakdown (66).
Autoinflammatory Activation of Innate Immunity
Autoinflammatory diseases (AIDs) and autoimmune diseases are characterized by an aberrant chronic activation of the immune system, leading to tissue inflammation and damage. In AIDs, the innate immune system is directly responsible for tissue inflammation, while in autoimmune diseases, the adaptive immunity becomes the main effector of the inflammatory process (67). The immune system is constituted by immune sensors, i.e., soluble and cellular receptors and effector mechanisms, including different cell types. The sensors of innate immunity are named pattern recognition receptors (PRRs). They bind to highly conserved structures of pathogen-associated molecular patterns (PAMPs) or damaged cells (67) and include three classes of receptors: TLRs, nucleotide-binding oligomerization domain-like receptors (NLRs), and retinoic acid-inducible gene-I-like receptors (68). Activation of NLRs and NLR family CARD domain-containing protein 4 (NLRC4) leads to the formation of large protein complexes termed inflammasomes. Inflammasomes play a role in the defense against pathogens through activation of pro-caspase-1 and cleavage of the pro-forms of interleukin (IL)-1β and IL-18 to their respective active forms (68). A dysregulation of an inflammasome complex, in some cases, may lead to the development of autoimmune responses (69). Infections can trigger autoinflammatory processes in genetically predisposed individuals by various mechanisms, including induction of innate immune responses through binding to TLRs and activation of the intracellular inflammasome system. The effector cells of innate immunity include macrophages, dendritic cells, and other APCs. Moreover, vaccinations in genetically susceptible hosts have been found to trigger autoimmune diseases or AIDs in different animal models and to induce autoimmune/autoinflammatory symptoms in humans (70).
The Gut Microbiota
There is growing evidence that the commensal bacteria in the gastrointestinal tract (the gut microbiota) influence the development of autoimmunity (71, 72). This evidence has been reinforced by studies in neonatal subjects. This is illustrated by a study (73) of microbiomes of 33 HLA-matched infants from monthly samples collected from birth until 3 years of age. Study participants were selected on the basis of having HLA risk alleles associated with type 1 diabetes mellitus (73). Of the 33 infants, 11 seroconverted to serum autoantibody positivity during the course of the study, and 4 of them developed type 1 diabetes. The taxonomic composition of the microbiomes varied widely within the same individual; more importantly, unique changes to the microbiomes were shared among the individuals who developed autoantibodies, including an increase in pathobionts and a decrease in bacteria that produce short-chain fatty acids (73).
Gut microbiota can be influenced by several factors: the motility of the gastrointestinal tract and the intake of pharmaceutical medications, including antibiotics, nonsteroidal anti-inflammatory drugs, smoking, and alcohol. These factors can lead to inappropriate gut-brain axis signaling and associated consequences for central nervous system function and ultimately resulting in disease (71). Most interestingly, a recent study showed that an oral bacteria-derived lipopeptide, lipid 654, which is produced by commensal bacteria, is present at significantly lower levels in the serum of patients with multiple sclerosis compared to levels observed in the serum samples of both healthy individuals and patients with Alzheimer’s disease (74). This result identified for the first time a potential mechanism relating gastrointestinal and oral commensal microbiome to a human systemic autoimmune disease.
Several experimental models of autoimmune diseases associated to gut microbiota have been developed, particularly focusing on inflammatory arthritis (75). Naïve RA patients carry high levels of Prevotella copri, a Gram-negative anaerobe relevant in many inflammatory and autoimmune conditions (75). However, the gut microbiome interacts not only with the host but also with other organisms and environmental factors. Indeed, exogenous viruses and the virome, the genomes of all viruses that inhabit a host, interact with the gut microbiota (76, 77) and live within commensal bacteria. Furthermore, the microbiota and immune systems interact: malnutrition affects the innate and adaptive immune systems as well as the microbiota (78). The microbiota acts as a barrier to enteropathogen infection; this barrier function may be disrupted by malnutrition or perturbations in immune system function (79). Finally, there is clear and increasing evidence that changes in the microbiota are associated with autoimmune diseases involving the gastrointestinal mucosa that lies in close contact with luminal contents as exemplified by celiac disease and also autoimmunity targeted toward distant sites, such as type 1 diabetes and RA (80–83). Clearly, much work remains to be done (84).
Environmental Agents
Both physical and chemical environmental agents, because of their ubiquitous nature, have been the subject of active investigations. Of particular significance, change in lifestyle that have led to an increase in exposure to environmental chemicals over several decades has been considered as a putative reason for the increase in autoimmune diseases.
Ultraviolet Light
Exposure to ultraviolet light (UV) light is a factor associated with SLE (85). UV-B induces apoptosis of keratinocytes and other dermal cells, thus releasing self molecules and pro-inflammatory cytokines, triggering systemic inflammation (86). Despite experimental studies showing a significant immunomodulatory role for UV radiation (87, 88), strong epidemiologic data regarding its role in triggering SLE is lacking.
Vitamin D Deficiency
1,25-dihydroxyvitamin D (1,25 OH2 vit D) is a steroid hormone derived from vitamin D which plays an important role in maintaining an adequate levels of serum calcium and phosphorus. Recent findings that several human tissues and cells express the vitamin D receptor (VDR) and 1α-hydroxylase have led to growing interest in extra-skeletal functions of vitamin D (89). Vitamin D plays an essential role in a variety of acute and chronic illness, including autoimmune diseases (90, 91), and several genetic studies have demonstrated an association between thyroid autoimmunity susceptibility and gene polymorphisms of numerous proteins and enzymes associated with vitamin D function and the vitamin D receptor (92). It should be stressed, however, much of the data is still controversial (93–103). One of the reasons of this controversy is study design, particularly inadequate sample sizes (89).
Smoking, Silica, Tropospheric Pollutants, and Solvent/Pesticides
A variety of chemicals have been associated with autoimmune diseases (104–130), including cigarette smoking, silica, and tropospheric pollutants (Table 3) (131). However, the precise mechanisms how chemicals can cause autoimmunity are still unknown and can vary greatly depending on the nature of the physiological interaction and exposure. SLE represents a paradigm for understanding how environmental agents convert normal antigen-specific “helper” T cells into autoreactive, cytotoxic, pro-inflammatory T cells causing lupus in animal models and similar changes in humans (132). An expert panel workshop of the National Institutes of Environmental Health Sciences demonstrated that an autoimmune response following exposure to environmental factors is dependent upon genetic background of the host and can vary widely among species and strains (133).
Accumulating data have implicated that exposure to industrial solvents is associated to the development of autoimmunity (134, 135). Industrials solvents can be classified into two categories: organic and inorganic. Organic solvents (Oss) contain carbon and can be broken down into aliphatic-chain compounds, such as n-hexane, and aromatic compounds, such as benzene or xylene. Common uses for OSs include dry cleaning (e.g., tetrachloroethylene), paint thinner (e.g., toluene, turpentine), nail polish removers, glue solvents (acetone, methyl acetate, ethyl acetate), spot removers (e.g., hexane, petrol ethers), detergents (citrus turpenes), and perfumes (ethanol) (136). A systematic review and meta-analysis including 33 articles found that exposure to OSs is associated with systemic sclerosis, primary systemic vasculitis, and multiple sclerosis individually and also to several autoimmune diseases evaluated when taken together as a single trait (137).
Alcohol
Alcohol has been investigated for its potential role in triggering autoimmunity in several diseases with conflicting results. Interestingly, there are a number of studies suggesting a protective role in at least two autoimmune diseases: SLE and RA. The first report with a statistically significant inverse association and dose-response relationship between alcohol consumption and SLE came from the UK (138). Two further studies in Japan (139) and Sweden (140) corroborated these results. However, this association was only seen in light and moderate drinkers and not in heavy drinkers (140). Other studies, including two case-control studies and a prospective cohort analysis, failed to find any association between alcohol consumption and SLE susceptibility (141–143). These inconsistent findings have been explained on the basis of selection bias and different patterns of alcohol consumption (144). Nevertheless, in a meta-analysis assessing the relationship between alcohol and SLE risk in patients treated for less than 10 years (including six case-control studies and one cohort study), Wang et al. found a significant protective effect of alcohol (145).
A beneficial effect of alcohol has also been found for patients with RA associated with antibodies to citrullinated protein antigen in a meta-analysis comprising six case-control studies (3564 cases, 8477 controls, and three cohort studies (444 RA cases, 84,421 individuals) (146)).
Psoriasis seems to be the only common autoimmune disease for which excess alcohol is a risk factor for its onset (147). This finding has been shown in population studies (148, 149). Moreover, an epidemiological study based on the Finnish nationwide Hospital Discharge Register and Cause of Death over a 22-year period demonstrated that the cause of death in women was due to liver disease and in men was due to both liver disease and alcohol-related psoriasis (150). Alcohol and its metabolites have been shown in experimental studies in vivo to increase critical markers involved in systemic immunoregulation in active psoriasis such as tumor necrosis factor (TNF)-α-converting enzyme (TACE) and TNF-receptor type 1 (151). In theory, alcohol may have a potential risk in triggering autoimmunity in the liver since it involves a complex array of derangements in cellular signaling of hepatic parenchymal and non-parenchymal cells as well as cells of the immune system. Chronic ethanol renders Kupffer cells hyperresponsive to endotoxins, which results in production of cytokines and TNFα via a TLR 4-dependent pathway, leading to inflammation and hepatic necrosis (152).
Drugs
To date more than 100 drugs spanning over ten drug categories have been associated with drug-induced autoimmunity (153). In general, there are two ways to classify these reactions: (i) drug-induced autoimmunity (DIA), an immune-related drug reaction temporally related to continuous drug exposure, which resolves after withdrawal of the offending drug and (ii) autoimmune disease triggered by a drug and perpetuating over time. The difficulty in distinguishing the two conditions is rooted in the understanding of the pathophysiologic mechanisms of drug reactions and the chronological criteria in a long period observation after the drug withdrawal. Two drugs are considered as a paradigm for DIA: procainamide and hydralazine. The incidence of procainamide-induced lupus was approximately 20 % during the first year of therapy (154), and the incidence of hydralazine-induced lupus was approximately 5–8 % (155). Procainamide, an antiarrhythmic agent, can induce lupus-like disease in susceptible individuals by inhibiting cellular DNA methylation (156). Hydralazine, an antihypertensive agent as well as an anti-heart failure agent, when used in combination with nitrates, reduces DNA methylation and protein levels in T cells by deactivating the ERK pathway (157). Interestingly, the new biological drugs especially the inhibitors of TNFα (etanercept, infliximab, adalimumab, certolizumab pegol, golibumab) have been associated with DIA. Biological modulators have been developed for a number of autoimmune diseases including Crohn’s disease, rheumatoid arthritis, Sjogren’s syndrome, and multiple sclerosis (158, 159). In contrast to lupus-like syndrome induced by hydralazine and procainamide, the occurrences of anti-dsDNA antibodies and hypocomplementemia are more common in anti-TNF-induced lupus, although the mechanism is not fully understood (160).
Autoimmune hepatitis (AIH) is an example of an autoimmune disease, which can be triggered by a number of drugs (161, 162). Table 4 includes examples of drugs and toxins implicated in DIA-like hepatitis (163). Minocycline and nitrofurantoin are such examples. Minocycline, an antibiotic commonly used for acne, can induce AIH within 2 years after starting drug (range 3 days–6 years) with the typical hallmarks of autoimmunity (non-organ-specific autoantibodies, hypogammaglobulinemia, and characteristic histologic changes (lymphoplasmacytic inflammation and necrosis) (164, 165)). Nitrofurantoin, an antibiotic used in the treatment of urinary tract infections can cause an acute hepatocellular reaction, which can progress in chronic hepatitis in one third of cases with the classical histological changes of AIH (166, 167). The clinical characteristics at presentation of both DIA-like hepatitis and classical AIH are similar; however, there is a higher prevalence of cirrhosis in classical AIH compared to DIA-like hepatitis, but it primarily differs by the absence of relapse after corticosteroid withdrawal (163).
Hormones
Sex hormones, particularly estrogens, have been shown to play a role in autoimmunity through supporting survival of autoreactive T cells and influencing cytokine profile in the natural killer T cells (168, 169). A marked beneficial effect of pregnancy has been observed in RA, whereas several other rheumatic diseases such as ankylosing spondylitis and SLE demonstrated either no protective effect or an aggravation of symptoms during pregnancy (170). Differences emerging in regard to modulation of disease symptoms during pregnancy appear to be related to response to hormones, cytokine profile and immune responses, and downstream interactions of molecular pathways associated with inflammation (170). Patients with some autoimmune diseases such as PBC or systemic sclerosis have significantly higher numbers of pregnancies compared to controls (171–173). Interestingly, there is an increased susceptibility of RA in the first-year postpartum (174, 175), suggesting also a possible effect of fetal microchimerism.
Progesterone, an immunomodulatory sex steroid, is also important in autoimmune diseases. For example, SLE is associated with early menarche where there is an increased exposure to endogenous sex steroids, and exogenous sex steroids (contraceptive pill or hormone replace therapy) are associated with an approximately 1.5- to 3.0-fold increased risk of SLE (176). Data from animal studies also support a role of progesterone and estrogens in SLE models (177, 178). Sex hormones can potentially modulate the expression of autoimmunity, with androgens suppressing and estrogens accelerating disease.
In the collagen-induced arthritis model, pretreatment with progesterone had little effect on joint swelling or serum TNFα and prostaglandin E2 but appeared to reduce the beneficial effect of estrogen treatment on these parameters (179, 180). Interestingly, neither progesterone nor estrogen treatment altered anti-collagen autoantibody responses, suggesting that hormones were not modulating autoimmunity but rather acting primarily at the level of joint inflammation (181). Finally, sex hormones, particularly estrogens, can be found in a variety of foods (182). Examples include daidzein in soybeans, genistein in vegetables, zearalenone in corn, or 17β-estradiol in poultry meat. Moreover, estrogen-mimicking chemicals are found in many household items such as detergents, surfactants, and plastics (182). Several estrogen-containing pesticides (methoxychlor, chlordane, hexachlorbenzene, pentachlorphenol, aldicarb) may act as triggers for autoimmune diseases.
Personal Care Products and Cosmetics
Personal care products such as shampoo, hair dyes, and cosmetics contain natural and synthetic chemicals. The interest in hair dyes and other hair products in induction of autoimmunity is based on similarities of some constituents of these products (acrylamides) to medications involved in drug-induced lupus. However, hair dyes failed to demonstrate an association with the development of SLE in two case-control studies (139, 183) and in a cohort study (184) (Table 5). Nail polish emerged as a risk factor for PBC and for SLE (185, 186). In a study by Gershwin et al. (185), 1032 patients with PBC and 1041 controls matched for sex, age, race, and geographical location were administered a modified version of the US National Health and Nutrition Examination Study questionnaire including 180 questions and 300 subquestions regarding demographics, lifestyle, personal and familial medical history, and reproductive and occupational history. Family history of PBC, history of urinary tract infections, past smoking, use of hormone replacement therapies, and frequent use of nail polish emerged as risk factors significantly associated with PBC. Although the odds ratio for increased frequency of nail polish use was not impressive, this finding is intriguing in view of the xenobiotic hypothesis proposed for the development PBC with specific halogenated compounds that could increase the immunogenicity of mitochondrial proteins and induce AMA in animal models (187, 188). A study by Cooper et al. (186) was conducted in Canada using 258 cases with SLE and 263 controls matched for sex, age, and area of residence. Relatively strong, but imprecise associations were observed in people who worked with paints, dyes or film developing, and work that included applying nail polish or nail applications.
Interestingly, lipstick contains some components which have been associated to autoimmune phenomena: eosin, phthalate, and 2-octynoic acid. Eosin is a red dye implicated in both photosensitivity and lupus flares (189). Phthalate can induce anti-DNA antibody responses and SLE-like syndrome in an experimental model (190–192). 2-Octynoic acid is a xenobiotic that can modify the immunodominant E2 component of pyruvate dehydrogenase complex (PDC-E2) and induce AMA responses in PBC (193). A more recent work extending from our 2-octynoic acid data suggests that a broad class of electrophilic drugs including acetaminophen and other commonly used nonsteroidal anti-inflammatory drugs may contribute to xenobiotic-induced mimicry and loss of tolerance to PDC-E2 seen in PBC (194–196).
Pristane and Naturally Occurring Hydrocarbons
Tetramethylpentadecane is a naturally occurring hydrocarbon oil commonly known as pristane which is found in small quantities in many plants and thought to be derived primarily from the metabolism of phytol, a ubiquitous ester of chlorophyll (197). Relatively high levels are also found in various marine organisms, including algae and zooplanktonic copepods, and pristane is strongly concentrated in the livers of sharks (197). Pristane occurs also in crude oils and is a common constituent of mineral oil, a byproduct of the fractional distillation of petroleum containing straight- and branched-chain paraffinic, naphthenic, and aromatic hydrocarbons. Medicinal (pharmaceutical or food grade) mineral oils, which are free of aromatic and unsaturated compounds, are used as laxatives, protective coatings for food, and cosmetics (198). For instance, canned sardines contain up to 370 mg/kg and white bread up to 550 mg/kg of mineral oil (197). Pristane-induced lupus is a murine model of SLE. Renal disease and autoantibody production depend on signaling through the interferon (IFN)-I receptor (198). The major source of IFN-I is immature monocytes bearing high levels of the surface marker Ly6C. Interferon production is mediated exclusively by signaling through TLR7 and the adapter protein MyD88. It is likely that endogenous TLR7 ligands such as components of small nuclear ribonucleoprotein complexes are involved in triggering disease (199).
The PBC Lesson
There is extensive literature on primary biliary cirrhosis, including animal models, that illustrate the importance of genetics and environment, and indeed, PBC is considered the model autoimmune disease {Beuers, 2015 #336; Chang, 2015 #337; Floreani, 2015 #334; Katsumi, 2015 #338; Kurth, 2014 #331; Lleo, 2014 #332; Sun, 2015 #335; Wang, 2015 #339; Ando, 2013 #342; Deng, 2013 #340; Floreani, 2015 #333; Tanaka, 2014 #329; Wang, 2015 #328; Lleo, 2013 #343; Leung, 2013 #344; Hudspeth, 2013 #345; Kawata, 2013 #346; Ridgway, 2014 #347; (173)}.
Epidemiological studies support the hypothesis that environmental factors play a role in the etiology and pathogenesis of PBC in genetically susceptible individuals (185). Numerous microbial agents, mainly bacteria, but also viruses, parasites, and fungi, have been investigated as possible agents involved in PBC (200), but most studies have failed to demonstrate a clear association of a microbial agent with disease and report only circumstantial evidence that could not be independently recapitulated. Indeed, most studies supporting the role of infectious agents in the pathogenesis of PBC are based on the linear or conformational mimicry between microbial proteins and human mitochondrial antigens (201–203). Notwithstanding a substantial shared sequence homology, in a fewer cases, a cross reactivity by 2OADC-specific autoantibodies and/or T cells has been also demonstrated (56). This is the case for Escherichia coli, Novosphingobium aromaticivorans, Salmonella minnesota, Pseudomonas aeruginosa, Haemophilus influenzae, Yersinia enterocolitica, Streptococcus intermedius, Lactobacillus delbrueckii, Paracoccus denitrificans, Mycoplasm, Mycobacterium gordonae, Borrelia burgdorferi, Trypanosoma, and Ascaridia galli (200). Moreover, microbial antigens or DNA have been found in liver specimens, gallbladder bile, and fecal samples of patients with PBC as for N. aromaticivorans, Propionibacterium acnes, and the Epstein-Barr virus (200).
PBC is also the first autoimmune cholangitis studied with different spatial analysis since cluster distribution has been observed (204). A 3-year study was conducted in the city of Sheffield (1977–1979), and a closer inspection demonstrated an apparent clustering of cases with a suspected relationship with one water reservoir (205). Nevertheless, analysis of the water showed no significant relevant differences between the reservoir serving areas with a high prevalence of PBC and other reservoirs. Twenty-five years later, a spatial clustering was observed in the North-East England (206). A further study in the same region was performed during a defined period (1987–2003) (207). Space-time clustering was observed when excess of cases of a disease were found within limited geographical areas at limited period of time. This finding is suggestive of the involvement of one or more environmental components in the cause of a disease. When a more rigorous statistical method was applied, clustering was most marked for cases diagnosed within 1–4 months of one another, suggesting that transient environmental agents may play a role in the cause of PBC (204). The finding of a seasonal variation in the diagnosis of PBC provides evidence for the involvement of a seasonally varying environmental agent in the etiology of PBC (208). Seasonal variation in PBC is consistent with the involvement of at least one transient environmental agent in etiology: examples of such factors that may be implicated include infections, air pollution, and diet (208). Another interesting study evaluating the relationship between environmental factors and PBC was published in 2006 (209). This study suggested that the number of PBC patients requiring transplantation was increased near superfund toxic waste sites in New York state (210). Additionally, a statistical significant PBC patient cluster, including both patients not listed for transplantation or listed for transplantation, was identified in Staten Island near a superfund waste site contaminated with volatile aromatic hydrocarbons and trichloroethylene. This has led to suggest that inhalation of volatile organic compounds (e.g., benzene) and particle-bound chlorinated hydrocarbons released into the air from these sites is a plausible method of exposure. Although specific environmental compounds causing PBC have not been clearly identified, xenobiotics are now emerging as compounds that could possibly narrow the gap between environmental exposure and pathogenesis (125). The working hypothesis is that modifications of the lipoylated major mitochondrial autoantigen could trigger the production of autoantibodies (193, 211, 212).
Conclusions
Several indirect lines of evidence support the role of environmental factors in triggering autoimmunity in genetically predisposed individuals. No single factor has been identified as prominent. Greater understanding of how different environmental exposures results in different disease phenotypes and varying degrees of severity will help identify the mechanisms and checkpoints that control development of autoimmunity and autoimmune disease (6). In addition, it should be noted that genome-wide association studies have proven extremely disappointing and have not pointed to any “smoking guns”. Indeed, such work has only further highlighted the role of environment and, in particular, specific epigenetic events that may contribute to loss of tolerance.
Abbreviations
- AIDs:
-
Autoinflammatory diseases
- AIH:
-
Autoimmune hepatitis
- APCs:
-
Antigen-presenting cells
- ASGPR:
-
Asialo glycoprotein receptor
- DIA:
-
Drug-induced autoimmunity
- EBV:
-
Epstein-Barr Virus
- HAV:
-
Hepatitis A virus
- HCV:
-
Hepatitis C Virus
- HLA:
-
Human leukocyte antigen
- IFN:
-
Interferon
- IUL:
-
Interleukin
- LSE:
-
Systemic lupus erythematosus
- NLRs:
-
Nucleotide-binding oligomerization domain-like receptors
- NLRC4:
-
NLR family CARD domain-containing protein 4
- Oss:
-
Organic solvents
- PAMPs:
-
Pathogen-associated molecular patterns
- PBC:
-
Primary biliary cirrhosis
- PDC:
-
Pyruvate dehydrogenase complex
- PRRs:
-
Pattern recognition receptor
- RA:
-
Rheumatoid arthritis
- TACE:
-
TNF-a-converting enzyme
- TLRs:
-
Toll-like receptors
- TNF:
-
Tumor necrosis factor
References
Ermann J, Fathman CG (2001) Autoimmune diseases: genes, bugs and failed regulation. Nat Immunol 2:759–761
Cortes JR, Sanchez-Diaz R, Bovolenta ER et al (2014) Maintenance of immune tolerance by Foxp3+ regulatory T cells requires CD69 expression. J Autoimmun 55:51–62
Berrih-Aknin S (2014) Myasthenia gravis: paradox versus paradigm in autoimmunity. J Autoimmun 52:1–28
Bao Y, Cao X (2014) The immune potential and immunopathology of cytokine-producing B cell subsets: a comprehensive review. J Autoimmun 55:10–23
Vojdani A, Pollard KM, Campbell AW (2014) Environmental triggers and autoimmunity. Autoimmun Dis 2014:798029
Pollard KM (2015) Environment, autoantibodies, and autoimmunity. Front Immunol 6:60
Berrih-Aknin S, Frenkian-Cuvelier M, Eymard B (2014) Diagnostic and clinical classification of autoimmune myasthenia gravis. J Autoimmun 48–49:143–148
Karussis D (2014) The diagnosis of multiple sclerosis and the various related demyelinating syndromes: a critical review. J Autoimmun 48–49:134–142
Mosca M, Tani C, Vagnani S, Carli L, Bombardieri S (2014) The diagnosis and classification of undifferentiated connective tissue diseases. J Autoimmun 48–49:50–52
Mouthon L, Dunogue B, Guillevin L (2014) Diagnosis and classification of eosinophilic granulomatosis with polyangiitis (formerly named Churg-Strauss syndrome). J Autoimmun 48–49:99–103
Hajj-Ali RA, Calabrese LH (2014) Diagnosis and classification of central nervous system vasculitis. J Autoimmun 48–49:149–152
Kivity S, Arango MT, Ehrenfeld M et al (2014) Infection and autoimmunity in Sjogren’s syndrome: a clinical study and comprehensive review. J Autoimmun 51:17–22
Selmi C, Leung PS, Sherr DH et al (2012) Mechanisms of environmental influence on human autoimmunity: a National Institute of Environmental Health Sciences expert panel workshop. J Autoimmun 39:272–284
Brouqui P, Raoult D, Conte-Devolx B (1991) Coxsackie thyroiditis. Ann Intern Med 114:1063–1064
Fernandez-Soto L, Gonzalez A, Escobar-Jimenez F et al (1998) Increased risk of autoimmune thyroid disease in hepatitis C vs hepatitis B before, during, and after discontinuing interferon therapy. Arch Intern Med 158:1445–1448
Hwang YC, Jeong IK, Chon S et al (2010) Fulminant type 1 diabetes mellitus associated with acute hepatitis A. Diabet Med 27:366–367
Kawai H, Inui T, Kashiwagi S et al (1992) HTLV-I infection in patients with autoimmune thyroiditis (Hashimoto’s thyroiditis). J Med Virol 38:138–141
Kerr JR, Cartron JP, Curran MD, Moore JE, Elliott JR, Mollan RA (1995) A study of the role of parvovirus B19 in rheumatoid arthritis. Br J Rheumatol 34:809–813
Leite JL, Bufalo NE, Santos RB, Romaldini JH, Ward LS (2010) Herpesvirus type 7 infection may play an important role in individuals with a genetic profile of susceptibility to Graves’ disease. Eur J Endocrinol 162:315–321
Mason AL (2011) The evidence supports a viral aetiology for primary biliary cirrhosis. J Hepatol 54:1312–1314
Mori K, Munakata Y, Saito T et al (2007) Intrathyroidal persistence of human parvovirus B19 DNA in a patient with Hashimoto’s thyroiditis. J Infect 55:e29–e31
Parmar RC, Bavdekar SB, Sahu DR, Warke S, Kamat JR (2001) Thyroiditis as a presenting feature of mumps. Pediatr Infect Dis J 20:637–638
Pratesi F, Tommasi C, Anzilotti C, Chimenti D, Migliorini P (2006) Deiminated Epstein-Barr virus nuclear antigen 1 is a target of anti-citrullinated protein antibodies in rheumatoid arthritis. Arthritis Rheum 54:733–741
Shimon I, Pariente C, Shlomo-David J, Grossman Z, Sack J (2003) Transient elevation of triiodothyronine caused by triiodothyronine autoantibody associated with acute Epstein-Barr-virus infection. Thyroid 13:211–215
Vento S, Cainelli F (2004) Is there a role for viruses in triggering autoimmune hepatitis? Autoimmun Rev 3:61–69
Vento S, Guella L, Mirandola F et al (1995) Epstein-Barr virus as a trigger for autoimmune hepatitis in susceptible individuals. Lancet 346:608–609
Ziring PR, Gallo G, Finegold M, Buimovici-Klein E, Ogra P (1977) Chronic lymphocytic thyroiditis: identification of rubella virus antigen in the thyroid of a child with congenital rubella. J Pediatr 90:419–420
Farquharson D, Butcher JP, Culshaw S (2012) Periodontitis, porphyromonas, and the pathogenesis of rheumatoid arthritis. Mucosal Immunol 5:112–120
Wu HJ, Ivanov II, Darce J et al (2010) Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity 32:815–827
Bech K, Clemmensen O, Larsen JH, Thyme S, Bendixen G (1978) Cell-mediated immunity of Yersinia enterocolitica serotype 3 in patients with thyroid diseases. Allergy 33:82–88
Hill Gaston JS, Lillicrap MS (2003) Arthritis associated with enteric infection. Best Pract Res Clin Rheumatol 17:219–239
McColl GJ, Diviney MB, Holdsworth RF et al (2000) HLA-B27 expression and reactive arthritis susceptibility in two patient cohorts infected with Salmonella Typhimurium. Aust N Z J Med 30:28–32
Hannu T, Mattila L, Siitonen A, Leirisalo-Repo M (2005) Reactive arthritis attributable to Shigella infection: a clinical and epidemiological nationwide study. Ann Rheum Dis 64:594–598
Ebringer A, Rashid T (2006) Rheumatoid arthritis is an autoimmune disease triggered by Proteus urinary tract infection. Clin Dev Immunol 13:41–48
Pope JE, Krizova A, Garg AX, Thiessen-Philbrook H, Ouimet JM (2007) Campylobacter reactive arthritis: a systematic review. Semin Arthritis Rheum 37:48–55
Dominguez-Lopez ML, Cancino-Diaz ME, Jimenez-Zamudio L, Granados-Arreola J, Burgos-Vargas R, Garcia-Latorre E (2000) Cellular immune response to Klebsiella pneumoniae antigens in patients with HLA-B27+ ankylosing spondylitis. J Rheumatol 27:1453–1460
Cope A, Anderson J, Wilkins E (1992) Clostridium difficile toxin-induced reactive arthritis in a patient with chronic Reiter’s syndrome. Eur J Clin Microbiol Infect Dis 11:40–43
Liu ZQ, Deng GM, Foster S, Tarkowski A (2001) Staphylococcal peptidoglycans induce arthritis. Arthritis Res 3:375–380
Fae KC, da Silva DD, Oshiro SE et al (2006) Mimicry in recognition of cardiac myosin peptides by heart-intralesional T cell clones from rheumatic heart disease. J Immunol 176:5662–5670
Sutliff WD, Shepard R, Dunham WB (1953) Acute Leptospira pomona arthritis and myocarditis. Ann Intern Med 39:134–140
Carter JD, Espinoza LR, Inman RD et al (2010) Combination antibiotics as a treatment for chronic Chlamydia-induced reactive arthritis: a double-blind, placebo-controlled, prospective trial. Arthritis Rheum 62:1298–1307
Kim HR, Kim EY, Cerny J, Moudgil KD (2006) Antibody responses to mycobacterial and self heat shock protein 65 in autoimmune arthritis: epitope specificity and implication in pathogenesis. J Immunol 177:6634–6641
Imai D, Holden K, Velazquez EM, Feng S, Hodzic E, Barthold SW (2013) Influence of arthritis-related protein (BBF01) on infectivity of Borrelia burgdorferi B31. BMC Microbiol 13:100
Smyk DS, Bogdanos DP, Kriese S, Billinis C, Burroughs AK, Rigopoulou EI (2012) Urinary tract infection as a risk factor for autoimmune liver disease: from bench to bedside. Clin Res Hepatol Gastroenterol 36:110–121
Hermann E, Mayet WJ, Klein O et al (1991) Candida arthritis: cellular immune responses of synovial fluid and peripheral blood lymphocytes to Candida albicans. Ann Rheum Dis 50:697–701
Kivity S, Agmon-Levin N, Blank M, Shoenfeld Y (2009) Infections and autoimmunity—friends or foes? Trends Immunol 30:409–414
Cusick MF, Libbey JE, Fujinami RS (2012) Molecular mimicry as a mechanism of autoimmune disease. Clin Rev Allergy Immunol 42:102–111
Vojdani A (2014) A potential link between environmental triggers and autoimmunity. Autoimmun Dis 2014:437231
Burroughs AK, Butler P, Sternberg MJ, Baum H (1992) Molecular mimicry in liver disease. Nature 358:377–378
Van de Water J, Ishibashi H, Coppel RL, Gershwin ME (2001) Molecular mimicry and primary biliary cirrhosis: premises not promises. Hepatology 33:771–775
Wang J, Yang GX, Tsuneyama K, Gershwin ME, Ridgway WM, Leung PS (2014) Animal models of primary biliary cirrhosis. Semin Liver Dis 34:285–296
Wang JJ, Yang GX, Zhang WC et al (2014) Escherichia coli infection induces autoimmune cholangitis and anti-mitochondrial antibodies in non-obese diabetic (NOD).B6 (Idd10/Idd18) mice. Clin Exp Immunol 175:192–201
Kita H, Matsumura S, He XS et al (2002) Quantitative and functional analysis of PDC-E2-specific autoreactive cytotoxic T lymphocytes in primary biliary cirrhosis. J Clin Invest 109:1231–1240
Matsumura S, Kita H, He XS et al (2002) Comprehensive mapping of HLA-A0201-restricted CD8 T-cell epitopes on PDC-E2 in primary biliary cirrhosis. Hepatology 36:1125–1134
Shigematsu H, Shimoda S, Nakamura M et al (2000) Fine specificity of T cells reactive to human PDC-E2 163–176 peptide, the immunodominant autoantigen in primary biliary cirrhosis: implications for molecular mimicry and cross-recognition among mitochondrial autoantigens. Hepatology 32:901–909
Shimoda S, Nakamura M, Shigematsu H et al (2000) Mimicry peptides of human PDC-E2 163–176 peptide, the immunodominant T-cell epitope of primary biliary cirrhosis. Hepatology 31:1212–1216
Vanderlugt CJ, Miller SD (1996) Epitope spreading. Curr Opin Immunol 8:831–836
Cornaby C, Gibbons L, Mayhew V, Sloan CS, Welling A, Poole BD (2015) B cell epitope spreading: mechanisms and contribution to autoimmune diseases. Immunol Lett 163:56–68
Prasad S, Kohm AP, McMahon JS, Luo X, Miller SD (2012) Pathogenesis of NOD diabetes is initiated by reactivity to the insulin B chain 9–23 epitope and involves functional epitope spreading. J Autoimmun 39:347–353
Vanderlugt CL, Miller SD (2002) Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nat Rev Immunol 2:85–95
Dhirapong A, Lleo A, Yang GX et al (2011) B cell depletion therapy exacerbates murine primary biliary cirrhosis. Hepatology 53:527–535
Dhirapong A, Yang GX, Nadler S et al (2013) Therapeutic effect of cytotoxic T lymphocyte antigen 4/immunoglobulin on a murine model of primary biliary cirrhosis. Hepatology 57:708–715
Getts DR, Chastain EM, Terry RL, Miller SD (2013) Virus infection, antiviral immunity, and autoimmunity. Immunol Rev 255:197–209
Pane JA, Webster NL, Coulson BS (2014) Rotavirus activates lymphocytes from non-obese diabetic mice by triggering toll-like receptor 7 signaling and interferon production in plasmacytoid dendritic cells. PLoS Pathog 10:e1003998
Nakamoto N, Kanai T (2014) Role of toll-like receptors in immune activation and tolerance in the liver. Front Immunol 5:221
Agmon-Levin N, Ram M, Barzilai O et al (2009) Prevalence of hepatitis C serum antibody in autoimmune diseases. J Autoimmun 32:261–266
Zen M, Gatto M, Domeneghetti M et al (2013) Clinical guidelines and definitions of autoinflammatory diseases: contrasts and comparisons with autoimmunity—a comprehensive review. Clin Rev Allergy Immunol 45:227–235
Martinon F, Mayor A, Tschopp J (2009) The inflammasomes: guardians of the body. Annu Rev Immunol 27:229–265
Kahlenberg JM, Kaplan MJ (2014) The inflammasome and lupus: another innate immune mechanism contributing to disease pathogenesis? Curr Opin Rheumatol 26:475–481
Shoenfeld Y, Agmon-Levin N (2011) ‘ASIA’—autoimmune/inflammatory syndrome induced by adjuvants. J Autoimmun 36:4–8
Vieira SM, Pagovich OE, Kriegel MA (2014) Diet, microbiota and autoimmune diseases. Lupus 23:518–526
Van Praet JT, Donovan E, Vanassche I et al (2015) Commensal microbiota influence systemic autoimmune responses. EMBO J 34:466–474
Sargent J (2015) Autoimmunity: T1DM and the gut microbiome. Nat Rev Endocrinol 11:193
Farrokhi V, Nemati R, Nichols FC et al (2013) Bacterial lipodipeptide, lipid 654, is a microbiome-associated biomarker for multiple sclerosis. Clin Transl Immunol 2:e8
Brusca SB, Abramson SB, Scher JU (2014) Microbiome and mucosal inflammation as extra-articular triggers for rheumatoid arthritis and autoimmunity. Curr Opin Rheumatol 26:101–107
Foxman EF, Iwasaki A (2011) Genome-virome interactions: examining the role of common viral infections in complex disease. Nat Rev Microbiol 9:254–264
Moon C, Stappenbeck TS (2012) Viral interactions with the host and microbiota in the intestine. Curr Opin Immunol 24:405–410
Velasquez-Manoff M (2015) Gut microbiome: the peacekeepers. Nature 518:S3–S11
Kau AL, Ahern PP, Griffin NW, Goodman AL, Gordon JI (2011) Human nutrition, the gut microbiome and the immune system. Nature 474:327–336
Greer RL, Morgun A, Shulzhenko N (2013) Bridging immunity and lipid metabolism by gut microbiota. J Allergy Clin Immunol 132:253–262, quiz 263
Greiner TU, Hyotylainen T, Knip M, Backhed F, Oresic M (2014) The gut microbiota modulates glycaemic control and serum metabolite profiles in non-obese diabetic mice. PLoS One 9:e110359
Rogier R, Koenders MI, Abdollahi-Roodsaz S (2015) Toll-like receptor mediated modulation of T cell response by commensal intestinal microbiota as a trigger for autoimmune arthritis. J Immunol Res 2015:527696
Sargent J (2014) Diabetes. Altered gut microbial networks linked to islet cell autoimmunity. Nat Rev Endocrinol 10:313
McLean MH, Dieguez D Jr, Miller LM, Young HA (2015) Does the microbiota play a role in the pathogenesis of autoimmune diseases? Gut 64:332–341
Barbhaiya M, Costenbader KH (2014) Ultraviolet radiation and systemic lupus erythematosus. Lupus 23:588–595
Caricchio R, McPhie L, Cohen PL (2003) Ultraviolet B radiation-induced cell death: critical role of ultraviolet dose in inflammation and lupus autoantigen redistribution. J Immunol 171:5778–5786
Coleman DJ, Garcia G, Hyter S et al (2014) Retinoid-X-receptors (alpha/beta) in melanocytes modulate innate immune responses and differentially regulate cell survival following UV irradiation. PLoS Genet 10:e1004321
Metwally D, Sayed K, Abdel Hay R and Rashed L (2014) Reduction in tissue plasmin: a new mechanism of action of narrowband ultraviolet B in psoriasis. Clin Exp Dermatol
Yang CY, Leung PS, Adamopoulos IE, Gershwin ME (2013) The implication of vitamin D and autoimmunity: a comprehensive review. Clin Rev Allergy Immunol 45:217–226
Grober U, Spitz J, Reichrath J, Kisters K, Holick MF (2013) Vitamin D: update 2013: from rickets prophylaxis to general preventive healthcare. Dermatoendocrinol 5:331–347
Gorman S, Hart PH (2012) The current state of play of rodent models to study the role of vitamin D in UV-induced immunomodulation. Photochem Photobiol Sci 11:1788–1796
D’Aurizio F, Villalta D, Metus P, Doretto P, Tozzoli R (2015) Is vitamin D a player or not in the pathophysiology of autoimmune thyroid diseases? Autoimmun Rev 14:363–369
Lin WY, Wan L, Tsai CH, Chen RH, Lee CC, Tsai FJ (2006) Vitamin D receptor gene polymorphisms are associated with risk of Hashimoto’s thyroiditis in Chinese patients in Taiwan. J Clin Lab Anal 20:109–112
Stefanic M, Papic S, Suver M, Glavas-Obrovac L, Karner I (2008) Association of vitamin D receptor gene 3′-variants with Hashimoto’s thyroiditis in the Croatian population. Int J Immunogenet 35:125–131
Yazici D, Yavuz D, Tarcin O, Sancak S, Deyneli O, Akalin S (2013) Vitamin D receptor gene ApaI, TaqI, FokI and BsmI polymorphisms in a group of Turkish patients with Hashimoto’s thyroiditis. Minerva Endocrinol 38:195–201
Stefanic M, Karner I, Glavas-Obrovac L et al (2005) Association of vitamin D receptor gene polymorphism with susceptibility to Graves’ disease in Eastern Croatian population: case-control study. Croat Med J 46:639–646
Horst-Sikorska W, Ignaszak-Szczepaniak M, Marcinkowska M, Kaczmarek M, Stajgis M, Slomski R (2008) Association analysis of vitamin D receptor gene polymorphisms with bone mineral density in young women with Graves’ disease. Acta Biochim Pol 55:371–380
Inoue N, Watanabe M, Ishido N et al (2014) The functional polymorphisms of VDR, GC and CYP2R1 are involved in the pathogenesis of autoimmune thyroid diseases. Clin Exp Immunol 178:262–269
Pani MA, Regulla K, Segni M et al (2002) A polymorphism within the vitamin D-binding protein gene is associated with Graves’ disease but not with Hashimoto’s thyroiditis. J Clin Endocrinol Metab 87:2564–2567
Kurylowicz A, Ramos-Lopez E, Bednarczuk T, Badenhoop K (2006) Vitamin D-binding protein (DBP) gene polymorphism is associated with Graves’ disease and the vitamin D status in a Polish population study. Exp Clin Endocrinol Diabetes 114:329–335
Collins JE, Heward JM, Nithiyananthan R et al (2004) Lack of association of the vitamin D receptor gene with Graves’ disease in UK Caucasians. Clin Endocrinol (Oxf) 60:618–624
Maalej A, Petit-Teixeira E, Chabchoub G et al (2008) Lack of association of VDR gene polymorphisms with thyroid autoimmune disorders: familial and case/control studies. J Clin Immunol 28:21–25
Pani MA, Regulla K, Segni M et al (2002) Vitamin D 1alpha-hydroxylase (CYP1alpha) polymorphism in Graves’ disease, Hashimoto’s thyroiditis and type 1 diabetes mellitus. Eur J Endocrinol 146:777–781
Jaakkola JJ, Gissler M (2005) Maternal smoking in pregnancy as a determinant of rheumatoid arthritis and other inflammatory polyarthropathies during the first 7 years of life. Int J Epidemiol 34:664–671
van der Helm-van Mil AH, Verpoort KN, le Cessie S, Huizinga TW, de Vries RR, Toes RE (2007) The HLA-DRB1 shared epitope alleles differ in the interaction with smoking and predisposition to antibodies to cyclic citrullinated peptide. Arthritis Rheum 56:425–432
Sugiyama D, Nishimura K, Tamaki K et al (2010) Impact of smoking as a risk factor for developing rheumatoid arthritis: a meta-analysis of observational studies. Ann Rheum Dis 69:70–81
Arnson Y, Shoenfeld Y, Amital H (2010) Effects of tobacco smoke on immunity, inflammation and autoimmunity. J Autoimmun 34:J258–J265
Conrad K, Mehlhorn J, Luthke K, Dorner T, Frank KH (1996) Systemic lupus erythematosus after heavy exposure to quartz dust in uranium mines: clinical and serological characteristics. Lupus 5:62–69
Costenbader KH, Chang SC, De Vivo I, Plenge R, Karlson EW (2008) Genetic polymorphisms in PTPN22, PADI-4, and CTLA-4 and risk for rheumatoid arthritis in two longitudinal cohort studies: evidence of gene-environment interactions with heavy cigarette smoking. Arthritis Res Ther 10:R52
Costenbader KH, Kim DJ, Peerzada J et al (2004) Cigarette smoking and the risk of systemic lupus erythematosus: a meta-analysis. Arthritis Rheum 50:849–857
Ghaussy NO, Sibbitt W Jr, Bankhurst AD, Qualls CR (2003) Cigarette smoking and disease activity in systemic lupus erythematosus. J Rheumatol 30:1215–1221
Hatron PY, Plouvier B, Francois M, Baclet JL, Deconninck P, Devulder B (1982) Association of lupus erythematosus and silicosis. Rev Med Interne 3:245–246
Jones RN, Turner-Warwick M, Ziskind M, Weill H (1976) High prevalence of antinuclear antibodies in sandblasters’ silicosis. Am Rev Respir Dis 113:393–395
Karlson EW, Chang SC, Cui J et al (2010) Gene-environment interaction between HLA-DRB1 shared epitope and heavy cigarette smoking in predicting incident rheumatoid arthritis. Ann Rheum Dis 69:54–60
Kiyohara C, Washio M, Horiuchi T et al (2009) Cigarette smoking, N-acetyltransferase 2 polymorphisms and systemic lupus erythematosus in a Japanese population. Lupus 18:630–638
Klareskog L, Padyukov L, Alfredsson L (2007) Smoking as a trigger for inflammatory rheumatic diseases. Curr Opin Rheumatol 19:49–54
Klockars M, Koskela RS, Jarvinen E, Kolari PJ, Rossi A (1987) Silica exposure and rheumatoid arthritis: a follow up study of granite workers 1940–81. Br Med J (Clin Res Ed) 294:997–1000
Kobayashi S, Okamoto H, Iwamoto T et al (2008) A role for the aryl hydrocarbon receptor and the dioxin TCDD in rheumatoid arthritis. Rheumatology (Oxford) 47:1317–1322
Lee DM, Phillips R, Hagan EM, Chibnik LB, Costenbader KH, Schur PH (2009) Quantifying anti-cyclic citrullinated peptide titres: clinical utility and association with tobacco exposure in patients with rheumatoid arthritis. Ann Rheum Dis 68:201–208
Olsson AR, Skogh T, Axelson O, Wingren G (2004) Occupations and exposures in the work environment as determinants for rheumatoid arthritis. Occup Environ Med 61:233–238
Rosenman KD, Moore-Fuller M, Reilly MJ (1999) Connective tissue disease and silicosis. Am J Ind Med 35:375–381
Sanchez-Roman J, Wichmann I, Salaberri J, Varela JM, Nunez-Roldan A (1993) Multiple clinical and biological autoimmune manifestations in 50 workers after occupational exposure to silica. Ann Rheum Dis 52:534–538
Silman AJ, Newman J, MacGregor AJ (1996) Cigarette smoking increases the risk of rheumatoid arthritis. Results from a nationwide study of disease-discordant twins. Arthritis Rheum 39:732–735
Simard JF, Costenbader KH, Liang MH, Karlson EW, Mittleman MA (2009) Exposure to maternal smoking and incident SLE in a prospective cohort study. Lupus 18:431–435
Smyk D, Mytilinaiou MG, Rigopoulou EI, Bogdanos DP (2010) PBC triggers in water reservoirs, coal mining areas and waste disposal sites: from Newcastle to New York. Dis Markers 29:337–344
Steenland K, Brown D (1995) Mortality study of gold miners exposed to silica and nonasbestiform amphibole minerals: an update with 14 more years of follow-up. Am J Ind Med 27:217–229
Steenland K, Sanderson W, Calvert GM (2001) Kidney disease and arthritis in a cohort study of workers exposed to silica. Epidemiology 12:405–412
Stolt P, Kallberg H, Lundberg I et al (2005) Silica exposure is associated with increased risk of developing rheumatoid arthritis: results from the Swedish EIRA study. Ann Rheum Dis 64:582–586
Turner S, Cherry N (2000) Rheumatoid arthritis in workers exposed to silica in the pottery industry. Occup Environ Med 57:443–447
Freemer MM, King TE Jr, Criswell LA (2006) Association of smoking with dsDNA autoantibody production in systemic lupus erythematosus. Ann Rheum Dis 65:581–584
Selmi C (2012) Cutting-edge issues in autoimmunity and allergy of the digestive system. Clin Rev Allergy Immunol 42:265–268
Somers EC, Richardson BC (2014) Environmental exposures, epigenetic changes and the risk of lupus. Lupus 23:568–576
Germolec D, Kono DH, Pfau JC, Pollard KM (2012) Animal models used to examine the role of the environment in the development of autoimmune disease: findings from an NIEHS Expert Panel Workshop. J Autoimmun 39:285–293
Gilbert KM, Reisfeld B, Zurlinden TJ, Kreps MN, Erickson SW, Blossom SJ (2014) Modeling toxicodynamic effects of trichloroethylene on liver in mouse model of autoimmune hepatitis. Toxicol Appl Pharmacol 279:284–293
Lerner A, Matthias T (2015) Changes in intestinal tight junction permeability associated with industrial food additives explain the rising incidence of autoimmune disease. Autoimmun Rev 14:479–489
Gourley M, Miller FW (2007) Mechanisms of disease: environmental factors in the pathogenesis of rheumatic disease. Nat Clin Pract Rheumatol 3:172–180
Barragan-Martinez C, Speck-Hernandez CA, Montoya-Ortiz G, Mantilla RD, Anaya JM, Rojas-Villarraga A (2012) Organic solvents as risk factor for autoimmune diseases: a systematic review and meta-analysis. PLoS One 7:e51506
Hardy CJ, Palmer BP, Muir KR, Sutton AJ, Powell RJ (1998) Smoking history, alcohol consumption, and systemic lupus erythematosus: a case-control study. Ann Rheum Dis 57:451–455
Bengtsson AA, Rylander L, Hagmar L, Nived O, Sturfelt G (2002) Risk factors for developing systemic lupus erythematosus: a case-control study in southern Sweden. Rheumatology (Oxford) 41:563–571
Washio M, Horiuchi T, Kiyohara C et al (2006) Smoking, drinking, sleeping habits, and other lifestyle factors and the risk of systemic lupus erythematosus in Japanese females: findings from the KYSS study. Mod Rheumatol 16:143–150
Nagata C, Fujita S, Iwata H et al (1995) Systemic lupus erythematosus: a case-control epidemiologic study in Japan. Int J Dermatol 34:333–337
Ghaussy NO, Sibbitt WL Jr, Qualls CR (2001) Cigarette smoking, alcohol consumption, and the risk of systemic lupus erythematosus: a case-control study. J Rheumatol 28:2449–2453
Formica MK, Palmer JR, Rosenberg L, McAlindon TE (2003) Smoking, alcohol consumption, and risk of systemic lupus erythematosus in the Black Women’s Health Study. J Rheumatol 30:1222–1226
Takvorian SU, Merola JF, Costenbader KH (2014) Cigarette smoking, alcohol consumption and risk of systemic lupus erythematosus. Lupus 23:537–544
Wang J, Pan HF, Ye DQ, Su H, Li XP (2008) Moderate alcohol drinking might be protective for systemic lupus erythematosus: a systematic review and meta-analysis. Clin Rheumatol 27:1557–1563
Scott IC, Tan R, Stahl D, Steer S, Lewis CM, Cope AP (2013) The protective effect of alcohol on developing rheumatoid arthritis: a systematic review and meta-analysis. Rheumatology (Oxford) 52:856–867
Adamzik K, McAleer MA, Kirby B (2013) Alcohol and psoriasis: sobering thoughts. Clin Exp Dermatol 38:819–822
Poikolainen K, Reunala T, Karvonen J, Lauharanta J, Karkkainen P (1990) Alcohol intake: a risk factor for psoriasis in young and middle aged men? BMJ 300:780–783
Qureshi AA, Dominguez PL, Choi HK, Han J, Curhan G (2010) Alcohol intake and risk of incident psoriasis in US women: a prospective study. Arch Dermatol 146:1364–1369
Poikolainen K, Karvonen J, Pukkala E (1999) Excess mortality related to alcohol and smoking among hospital-treated patients with psoriasis. Arch Dermatol 135:1490–1493
Farkas A, Kemeny L (2010) Psoriasis and alcohol: is cutaneous ethanol one of the missing links? Br J Dermatol 162:711–716
Nath B, Szabo G (2009) Alcohol-induced modulation of signaling pathways in liver parenchymal and nonparenchymal cells: implications for immunity. Semin Liver Dis 29:166–177
Xiao X, Chang C (2014) Diagnosis and classification of drug-induced autoimmunity (DIA). J Autoimmun 48–49:66–72
Rubin RL (2005) Drug-induced lupus. Toxicology 209:135–147
Yokogawa N, Vivino FB (2009) Hydralazine-induced autoimmune disease: comparison to idiopathic lupus and ANCA-positive vasculitis. Mod Rheumatol 19:338–347
Scheinbart LS, Johnson MA, Gross LA, Edelstein SR, Richardson BC (1991) Procainamide inhibits DNA methyltransferase in a human T cell line. J Rheumatol 18:530–534
Lu Q, Wu A, Richardson BC (2005) Demethylation of the same promoter sequence increases CD70 expression in lupus T cells and T cells treated with lupus-inducing drugs. J Immunol 174:6212–6219
Kinch MS and Merkel J (2015) An analysis of FDA-approved drugs for inflammation and autoimmune diseases. Drug Discov Today
Sada PR, Isenberg D, Ciurtin C (2015) Biologic treatment in Sjogren’s syndrome. Rheumatology (Oxford) 54:219–230
Lisnevskaia L, Murphy G, Isenberg D (2014) Systemic lupus erythematosus. Lancet 384:1878–1888
Liberal R, Mieli-Vergani G, Vergani D (2013) Clinical significance of autoantibodies in autoimmune hepatitis. J Autoimmun 46:17–24
Invernizzi P (2013) Liver auto-immunology: the paradox of autoimmunity in a tolerogenic organ. J Autoimmun 46:1–6
Czaja AJ (2011) Drug-induced autoimmune-like hepatitis. Dig Dis Sci 56:958–976
Lawrenson RA, Seaman HE, Sundstrom A, Williams TJ, Farmer RD (2000) Liver damage associated with minocycline use in acne: a systematic review of the published literature and pharmacovigilance data. Drug Saf 23:333–349
Elkayam O, Yaron M, Caspi D (1999) Minocycline-induced autoimmune syndromes: an overview. Semin Arthritis Rheum 28:392–397
Amit G, Cohen P, Ackerman Z (2002) Nitrofurantoin-induced chronic active hepatitis. Isr Med Assoc J 4:184–186
Hatoff DE, Cohen M, Schweigert BF, Talbert WM (1979) Nitrofurantoin: another cause of drug-induced chronic active hepatitis? A report of a patient with HLA-B8 antigen. Am J Med 67:117–121
Verthelyi D (2001) Sex hormones as immunomodulators in health and disease. Int Immunopharmacol 1:983–993
Gourdy P, Araujo LM, Zhu R et al (2005) Relevance of sexual dimorphism to regulatory T cells: estradiol promotes IFN-gamma production by invariant natural killer T cells. Blood 105:2415–2420
Ostensen M, Villiger PM, Forger F (2012) Interaction of pregnancy and autoimmune rheumatic disease. Autoimmun Rev 11:A437–A446
Parikh-Patel A, Gold E, Utts J, Gershwin ME (2002) The association between gravidity and primary biliary cirrhosis. Ann Epidemiol 12:264–272
Cockrill T, del Junco DJ, Arnett FC et al (2010) Separate influences of birth order and gravidity/parity on the development of systemic sclerosis. Arthritis Care Res (Hoboken) 62:418–424
Floreani A, Infantolino C, Franceschet I et al (2015) Pregnancy and primary biliary cirrhosis: a case-control study. Clin Rev Allergy Immunol 48:236–242
Lansink M, de Boer A, Dijkmans BA, Vandenbroucke JP, Hazes JM (1993) The onset of rheumatoid arthritis in relation to pregnancy and childbirth. Clin Exp Rheumatol 11:171–174
Nelson JL, Ostensen M (1997) Pregnancy and rheumatoid arthritis. Rheum Dis Clin North Am 23:195–212
Costenbader KH, Feskanich D, Stampfer MJ, Karlson EW (2007) Reproductive and menopausal factors and risk of systemic lupus erythematosus in women. Arthritis Rheum 56:1251–1262
Hughes GC (2012) Progesterone and autoimmune disease. Autoimmun Rev 11:A502–A514
Talal N (1978) Natural history of murine lupus. Modulation by sex hormones. Arthritis Rheum 21:S58–S63
Ganesan K, Balachandran C, Manohar BM, Puvanakrishnan R (2012) Effects of testosterone, estrogen and progesterone on TNF-alpha mediated cellular damage in rat arthritic synovial fibroblasts. Rheumatol Int 32:3181–3188
Ganesan K, Tiwari M, Balachandran C, Manohar BM, Puvanakrishnan R (2008) Estrogen and testosterone attenuate extracellular matrix loss in collagen-induced arthritis in rats. Calcif Tissue Int 83:354–364
Jochems C, Islander U, Erlandsson M et al (2011) Effects of oestradiol and raloxifene on the induction and effector phases of experimental postmenopausal arthritis and secondary osteoporosis. Clin Exp Immunol 165:121–129
Chighizola C, Meroni PL (2012) The role of environmental estrogens and autoimmunity. Autoimmun Rev 11:A493–A501
Petri M, Allbritton J (1992) Hair product use in systemic lupus erythematosus. A case-control study. Arthritis Rheum 35:625–629
Sanchez-Guerrero J, Karlson EW, Colditz GA, Hunter DJ, Speizer FE, Liang MH (1996) Hair dye use and the risk of developing systemic lupus erythematosus. Arthritis Rheum 39:657–662
Gershwin ME, Selmi C, Worman HJ et al (2005) Risk factors and comorbidities in primary biliary cirrhosis: a controlled interview-based study of 1032 patients. Hepatology 42:1194–1202
Cooper GS, Wither J, Bernatsky S et al (2010) Occupational and environmental exposures and risk of systemic lupus erythematosus: silica, sunlight, solvents. Rheumatology (Oxford) 49:2172–2180
Long SA, Quan C, Van de Water J et al (2001) Immunoreactivity of organic mimeotopes of the E2 component of pyruvate dehydrogenase: connecting xenobiotics with primary biliary cirrhosis. J Immunol 167:2956–2963
Leung PS, Quan C, Park O et al (2003) Immunization with a xenobiotic 6-bromohexanoate bovine serum albumin conjugate induces antimitochondrial antibodies. J Immunol 170:5326–5332
Morikawa F, Fukuda M, Naganuma M, Nakayama Y (1976) Phototoxic reaction to xanthene dyes induced by visible light. J Dermatol 3:59–67
Lim SY, Ghosh SK (2003) Autoreactive responses to an environmental factor: 1. Phthalate induces antibodies exhibiting anti-DNA specificity. Immunology 110:482–492
Lim SY, Ghosh SK (2004) Autoreactive responses to an environmental factor. 2. Phthalate-induced anti-DNA specificity is downregulated by autoreactive cytotoxic T cells. Immunology 112:94–104
Lim SY, Ghosh SK (2005) Autoreactive responses to environmental factors: 3. Mouse strain-specific differences in induction and regulation of anti-DNA antibody responses due to phthalate-isomers. J Autoimmun 25:33–45
Amano K, Leung PS, Rieger R et al (2005) Chemical xenobiotics and mitochondrial autoantigens in primary biliary cirrhosis: identification of antibodies against a common environmental, cosmetic, and food additive, 2-octynoic acid. J Immunol 174:5874–5883
Chen RC, Naiyanetr P, Shu SA et al (2013) Antimitochondrial antibody heterogeneity and the xenobiotic etiology of primary biliary cirrhosis. Hepatology 57:1498–1508
Leung PS, Wang J, Naiyanetr P et al (2013) Environment and primary biliary cirrhosis: electrophilic drugs and the induction of AMA. J Autoimmun 41:79–86
Naiyanetr P, Butler JD, Meng L et al (2011) Electrophile-modified lipoic derivatives of PDC-E2 elicits anti-mitochondrial antibody reactivity. J Autoimmun 37:209–216
Grob K, Vass M, Biedermann M, Neukom HP (2001) Contamination of animal feed and food from animal origin with mineral oil hydrocarbons. Food Addit Contam 18:1–10
Reeves WH, Lee PY, Weinstein JS, Satoh M, Lu L (2009) Induction of autoimmunity by pristane and other naturally occurring hydrocarbons. Trends Immunol 30:455–464
Minhas U, Das P, Bhatnagar A (2011) Role of reactive intermediates in the immunopathogenesis of the pristane-induced Balb/c model of lupus. Lupus 20:1421–1425
Selmi C, De Santis M, Cavaciocchi F, Gershwin ME (2010) Infectious agents and xenobiotics in the etiology of primary biliary cirrhosis. Dis Markers 29:287–299
Bogdanos DP, Pares A, Baum H et al (2004) Disease-specific cross-reactivity between mimicking peptides of heat shock protein of Mycobacterium gordonae and dominant epitope of E2 subunit of pyruvate dehydrogenase is common in Spanish but not British patients with primary biliary cirrhosis. J Autoimmun 22:353–362
Juran BD, Lazaridis KN (2014) Environmental factors in primary biliary cirrhosis. Semin Liver Dis 34:265–272
Ortega-Hernandez OD, Levin NA, Altman A, Shoenfeld Y (2010) Infectious agents in the pathogenesis of primary biliary cirrhosis. Dis Markers 29:277–286
Prince MI, Chetwynd A, Diggle P, Jarner M, Metcalf JV, James OF (2001) The geographical distribution of primary biliary cirrhosis in a well-defined cohort. Hepatology 34:1083–1088
Triger DR (1980) Primary biliary cirrhosis: an epidemiological study. Br Med J 281:772–775
McNally RJ, Ducker S, James OF (2009) Are transient environmental agents involved in the cause of primary biliary cirrhosis? Evidence from space-time clustering analysis. Hepatology 50:1169–1174
McNally RJ, James PW, Ducker S, James OF (2011) Seasonal variation in the patient diagnosis of primary biliary cirrhosis: further evidence for an environmental component to etiology. Hepatology 54:2099–2103
Dronamraju D, Odin J, Bach N (2010) Primary biliary cirrhosis: environmental risk factors. Dis Markers 29:323–328
Ala A, Stanca CM, Bu-Ghanim M et al (2006) Increased prevalence of primary biliary cirrhosis near Superfund toxic waste sites. Hepatology 43:525–531
Kurth MJ, Yokoi T, Gershwin ME (2014) Halothane-induced hepatitis: paradigm or paradox for drug-induced liver injury. Hepatology 60:1473–1475
Amano K, Leung PS, Xu Q et al (2004) Xenobiotic-induced loss of tolerance in rabbits to the mitochondrial autoantigen of primary biliary cirrhosis is reversible. J Immunol 172:6444–6452
Gershwin ME, Mackay IR (2008) The causes of primary biliary cirrhosis: convenient and inconvenient truths. Hepatology 47:737–745
Butler P, Valle F, Hamilton-Miller JM, Brumfitt W, Baum H, Burroughs AK (1993) M2 mitochondrial antibodies and urinary rough mutant bacteria in patients with primary biliary cirrhosis and in patients with recurrent bacteriuria. J Hepatol 17:408–414
Sluis-Cremer GK, Hessel PA, Hnizdo E, Churchill AR (1986) Relationship between silicosis and rheumatoid arthritis. Thorax 41:596–601
Finckh A, Cooper GS, Chibnik LB et al (2006) Occupational silica and solvent exposures and risk of systemic lupus erythematosus in urban women. Arthritis Rheum 54:3648–3654
Parks CG, Cooper GS, Nylander-French LA et al (2002) Occupational exposure to crystalline silica and risk of systemic lupus erythematosus: a population-based, case-control study in the southeastern United States. Arthritis Rheum 46:1840–1850
Parks CG, Conrad K, Cooper GS (1999) Occupational exposure to crystalline silica and autoimmune disease. Environ Health Perspect 107(Suppl 5):793–802
Hart JE, Laden F, Puett RC, Costenbader KH, Karlson EW (2009) Exposure to traffic pollution and increased risk of rheumatoid arthritis. Environ Health Perspect 117:1065–1069
Zeft AS, Prahalad S, Lefevre S et al (2009) Juvenile idiopathic arthritis and exposure to fine particulate air pollution. Clin Exp Rheumatol 27:877–884
Bernatsky S, Fournier M, Pineau CA, Clarke AE, Vinet E, Smargiassi A (2011) Associations between ambient fine particulate levels and disease activity in patients with systemic lupus erythematosus (SLE). Environ Health Perspect 119:45–49
Balluz L, Philen R, Ortega L et al (2001) Investigation of systemic lupus erythematosus in Nogales, Arizona. Am J Epidemiol 154:1029–1036
Cooper GS, Parks CG, Treadwell EL, St Clair EW, Gilkeson GS, Dooley MA (2004) Occupational risk factors for the development of systemic lupus erythematosus. J Rheumatol 31:1928–1933
Dahlgren J, Takhar H, Anderson-Mahoney P, Kotlerman J, Tarr J, Warshaw R (2007) Cluster of systemic lupus erythematosus (SLE) associated with an oil field waste site: a cross sectional study. Environ Health 6:8
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Floreani, A., Leung, P.S.C. & Gershwin, M.E. Environmental Basis of Autoimmunity. Clinic Rev Allerg Immunol 50, 287–300 (2016). https://doi.org/10.1007/s12016-015-8493-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-015-8493-8