
Abstract
Primary biliary cirrhosis (PBC) is characterized histologically by the presence of chronic non-suppurative destructive cholangitis of the small interlobular bile duct, leading to chronic progressive cholestasis. Most PBC patients are asymptomatic and have a reasonable prognosis, but a few develop esophageal varices or jaundice, rapidly leading to liver failure within a short period. As multiple factors appear to be involved in the onset of PBC, its clinical course may be complicated. Therefore, the use of an animal model would be valuable for clarifying the pathogenesis of PBC. Here, we review recent data of selected PBC models, particularly spontaneous models, xenobiotic immunized models, and infection-triggered models. There are a number of spontaneous models: the NOD.c3c4, dominant-negative TGF-β receptor II, IL-2Rα−/−, Scurfy, and Ae2a,b−/− mice. These animal models manifest distinct clinical and immunological features similar, but also often different, from those of human PBC. It is clear that a combination of genetic predisposition, environmental factors, and immunological dysfunction contribute to the pathogenesis of PBC. The diverse clinical course and complexity of the immunological mechanisms of PBC cannot be fully recapitulated solely any single animal model. The challenge remains to develop a progressive PBC disease model that exhibits fibrosis, and ultimately hepatic failure.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
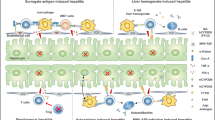
Primary biliary cirrhosis (PBC) is histologically characterized by the presence of chronic non-suppurative destructive cholangitis (CNSDC) of the interlobular small bile duct, leading to chronic progressive cholestasis and liver fibrosis [1, 2]. Serologically, over 95 % of patients with PBC exhibit distinct pattern of positivity for anti-mitochondrial antibodies (AMA) and elevated levels of IgM [3, 4]. The mitochondrial autoantigens have been identified as the E2 subunits of the E2 subunits of pyruvate dehydrogenase (PDC-E2), branched chain 2-oxo acid dehydrogenase (BCOADC-E2), and 2-oxo-glutarate dehydrogenase (OGDC-E2) [5]. In addition to mitochondrial autoantigens, patients with PBC are also reported to be seropositive to nuclear antigens including nuclear pore glycoprotein gp210 and the nuclear body-associated protein sp100 [6, 7]. More recently, two novel PBC autoantigens, kelch-like 12 (KLHL12) and hexokinase 1 (HK1), have been verified by immunoblot and enzyme-linked immunosorbent assay (ELISA) in two independent cohorts of PBC and disease/healthy control patients [8]. Although most PBC patients are asymptomatic and have a reasonable prognosis, a few develop esophageal varices [9] or jaundice, rapidly leading to liver failure within a short period [10]. Multiple SNPs and genetic variants have been reported to be associated with PBC [11–13]. The complexity of the clinical course suggests that the mechanisms of PBC involve the combination of immunological dysfunction, genetic predisposition, and environmental factors, all common themes in autoimmune pathogenesis [14–19]. Attempts to develop animal models have been directed to examine these factors in the pathogenesis of PBC [20–38] (Fig. 1). Here, we review the recent data on models, with emphasis on their immunological and histological characteristics to highlight their significance in PBC (Table 1) and our current understanding on the etiopathological mechanisms of PBC from these animal models.

The onset of PBC and its associated factors. A combination of genetics, environment, and autoimmunity contribute to the etiology and pathogenesis of PBC. Various models that exhibits the clinical, histological, and immunological features of PBC have been generated. Currently, no single animal model can clearly provide answers to the enigma of PBC
Genetically Modified Spontaneous Models
NOD. c3c4 Mice
In 2004, Koarada et al. reported on the discovery of PBC-like characteristics in the NOD.c3c4 double-congenic mouse strain, in which the Idd-resistant alleles from B10 and B6 mice were replaced on chromosome 3,4, respectively, of the non-obese diabetic (NOD) mice [39]. NOD.c3c4 mice developed spontaneous lymphocyte infiltrations around the bile duct, as well as AMA and ANA (PDC-E2 positivity, 56 % for 9–10 weeks; ANA positivity, 80–90 % for 20–25 weeks). Striking similar to patients with PBC, the AMA epitope of NOD.c3c4 mice maps within the PDC-E2 inner lipoyl domain. Moreover, infiltrations of CD3+, CD4+, and CD8+ T cells and PDCA1+ dendritic cells are present in the vicinity of the bile duct epithelium [40], and that in comparison with controls, these mice had a low CD4/CD8 ratio. Histologically, granuloma formation (1/7), eosinophil infiltration (7/7), and fibrosis (1/7) were also present (Table 1). Female NOC.c3c4 mice over 8 months old developed significantly increased titers of IgM dsDNA, IgG dsDNA, and IgG ssDNA, suggesting a more severe autoantibody response when compared with male mice. The liver pathology suggested an autoimmune pathogenesis, as cholangitis was induced by transfer of splenocytes from mice with progressive disease to young naive NOD.c3c4 mice subjected to irradiation treatment. In contrast to human PBC, NOD.c3c4 mice developed biliary polycystic lesions in almost all cases by 30 weeks and beyond, and the presence of CNSDC became unclear. Additionally, 25–50 % of the mice in this model developed liver failure due to exacerbation of the bile duct obstruction. Biochemically, cholestasis was not evident, and extrahepatic bile duct obstruction consistent with that in primary sclerosing cholangitis (PSC). Additionally, anti-Sm antibodies, specific to those in SLE, were observed in more than 50 % of these models.
Using microarrays, Nakagome et al. performed gene expression profiling of biliary epithelial cells from NOD.c3c4 mice and demonstrated a lack of apoptosis due to decreased expression of Fas antigen. This suggested that long-term exposure of the bile duct epithelium to AMA and failure of immune tolerance are likely responsible for the development of autoimmunity [41]. To examine the role of B cells in autoimmune cholangitis in the NOD.c3c4 mice, Moritoki et al. generated a B cell deficient Igμ(−/−) NOD.c3c4 mice and compared their immunopathology with that of the NOD.c3c4 mice [25]. Igμ(−/−) NOD.c3c4 mice demonstrated decreased number of non-B cells in the liver accompanied by reduced numbers of activated natural killer cells. While the degree of granuloma formation and bile duct damage were comparable to NOD.c3c4 mice. Liver inflammation and biliary cyst formation were significantly attenuated in these B cell-deficient mice. The data suggests that B cells play a critical role in liver inflammation, biliary disease and also cyst formation in the NOD.c3c4 mice.
Dominant-Negative TGF-β Receptor II Mice
The dominant-negative TGF-β receptor II (dnTGF-βRII) mouse model of autoimmune cholangitis was first reported in 2006 [42]. dnTGF-βRII mice overexpress a dominant-negative form of TGF-β receptor type II under the control of the CD4 promoter, resulting in specific abrogation of TGF-β signaling in CD4+ T cells [43]. Notably, however, TGF-β signaling is not completely eliminated in these T cells, which explains why the dnTGF-βRII mice can survive for almost normal life spans
The dnTGF-βRII mice exhibit several major serological and histological characteristics of human PBC (Table 1) [42, 44]. (1) They are 100 % AMA positive with autoantibodies directed against the major mitochondrial autoantigens in human PBC including PDC-E2, BCOADC-E2, and OGDC-E2 [45]. (2) Their liver and serum cytokine levels reflect a Th1 profile. (3) Their liver histology exhibits lymphoid cell infiltration in the portal tracts of mice including CD4+, CD8+, and CD19+ cells as in human PBC. This is accompanied by bile duct injury in 25–50 % of mice up to 22 weeks of age [44].
To examine the role of CD4+ and CD8+ T cells in liver pathology in these mice, Yang et al. [46] performed adoptive transfer studies by transferring dnTGF-βRII mice-derived splenic CD4+ and/or CD8+ T cells into Rag1−/− recipients. Rag1−/− recipients of unfractionated dnTGF-βRII mice splenocytes developed features of liver pathology similar to human PBC, suggesting that splenic T and B cell loss of tolerance are associated with autoimmune cholangitis in the dnTGF-βRII mice. Furthermore, transfer of dnTGF-βRII-derived CD8+ T cells into Rag1−/− recipients resulted in liver-specific autoimmunity, whereas CD4+ T cell transfer led to colitis, indicating that CD8+ T cells are the primary contributors for bile duct destruction in this model. This is very similar to the NOD.c3c4 model wherein CD8 cells alone can cause biliary disease [25].
To further delineate whether autoimmune cholangitis in the dnTGF-βRII mice was secondary to antigen-specific autoreactive CD8+ T cells, or due to antigen nonspecific effects of dnTGF-βRII accumulating in the liver, Kawata et al. generated OT-I/dnTGF-βRII/Rag1−/− and OT-II/dnTGF-βRII/Rag1−/− mice in which the entire T cell repertoire was replaced with ovalbumin (OVA)-specific CD8+ or CD4+ T cells, respectively [23]. Importantly, neither the parental OT-I/dnTGF-βRII/Rag1−/− mice and/or OT-II/ dnTGF-βRII/Rag1−/− mice developed cholangitis. However, data from adoptive transfer demonstrated that only transfer of CD8+ T cells from dnTGF-βRII mice but not CD8+ T cells from OT-I/Rag1−/− mice or from OT-I/dnTGF-βRII/Rag1−/− mice transferred disease. These observations were not due to an absence of CD4+ T cell help since a combination of CD8+ T cells from OT-I/dnTGF-βRII/Rag1−/− and CD4+ T cells from OT II/dnTGF-βRII/Rag1−/− or CD8+ T cells from OT-I/dnTGF-βRII/Rag1−/− with CD4+ T cells from OT-II/Rag1−/− mice failed to transfer disease. Altogether, the data showed that defective TGF-βRII signaling and antigen-specific clonal CD8+ T cells that target biliary cells are required for induction of autoimmune cholangitis [23].
Although the presence of high titers of AMAs is present in 95 % of patients with PBC, there is no direct correlation between AMAs and pathogenesis [47, 48]. To investigate the role of AMAs in disease pathology in the dnTGF-βRII mice model of PBC, dnTGF-βRII mice were crossed with B cell-deficient mice (Igμ−/−) and were evaluated for the development of liver inflammation, as well as the severity of accompanying colitis. Surprisingly, Igμ−/− dnTGF-βRII mice developed a more severe cholangitis and colitis compared to dnTGF-βRII mice, indicating a suppressive effect of B cells on the inflammatory response in the dnTGF-βRII mice [49]. The role of B cells in tissue pathology in dnTGF-βRII mice was further studied by evaluating the effects of therapeutic B cell depletion. Young (4–6 weeks) and old (20–22 weeks) dnTGF-βRII mice were injected intraperitoneally with anti-mouse CD20 monoclonal antibody at every 2 weeks, and the disease phenotype compared to control Ab-treated mice [49]. Treatment of young mice demonstrated fully depleted serum AMAs, a lower incidence of liver inflammation, and a fewer number of activated hepatic CD8+ T cells, whereas colon inflammation was significantly exacerbated. In contrast, anti-CD20 treatment of animals with established disease was ineffective, suggesting that B cells play both a positive and negative regulatory role in the pathogenesis of PBC.
Previous studies have shown that CD1d expression and the frequency of CD1d-restricted NKT cells were increased in the livers of patients with PBC [50]. This observation is of particular interest in PBC because NKT cells can comprise of 30–50 % of the intrahepatic lymphoid cells in normal livers and have been recently shown to impede liver regeneration by both IFNγ- and IL4-dependent mechanisms [19, 27, 51]. To examine the role of CD1d-restricted NKT cells in autoimmune cholangitis in this model, CD1d−/− dnTGF-βRII mice were constructed. CD1d−/− dnTGF-βRII mice had decreased mononuclear cell infiltration in the liver and lower IFNγ serum levels, which ultimately ameliorated liver injury compared to that of dnTGF-βRII mice [44]. Data from this work suggests that CD1d-restricted NKT cells have a primarily proinflammatory phenotype with a Th1 cytokine bias and promote deprivation of TGF-β signaling.
The complexity of the IL-12/IL-23 cytokine milieu in autoimmunity in dnTGF-βRII mice was dissected by generating a series of cytokine knockouts with the dnTGF-βRII mice. These include IFNγ−/−, IL-12p35−/−, IL-12/IL-23p40−/−, IL-23p19−/−, and IL-17A−/− (Table 2).
IL-12 promotes IFNΓ production and the triggering of Th1 cell responses, which contribute to loss of tolerance in several models of autoimmunity [29, 44, 52]. Data on IL-12p40−/− dnTGF-ΒRII mice has established that the IL-12p40 subunit is essential for the development of autoimmune cholangitis. Deletion of IL-12p40 in dnTGF-βRII mice resulted in lower levels of inflammatory cytokines, immune cell infiltrates, and bile duct damage but does not alter AMA levels [52]. However, in mice depleted of IFNγ-mediated signaling (IFNγ−/− dnTGF-βRII mice), cholangitis was not abrogated, suggesting that IFNγ is dispensable for the development of autoimmunity. This is further analyzed by deleting IL-12p35 in dnTGF-βRII mice, which is deficient in two members of the IL-12 family, IL-12 and IL-35. Although the mice demonstrated a distinct cytokine profile shift from Th1 to Th17, deletion of IL-12p35 resulted in liver inflammation and bile duct damage with similar severity as in the dnTGF-βRII mice but with delayed onset [29]. Surprising, significant hepatic periportal fibrosis occurs in 50 % of mice at 24 weeks of age in IL-12p35−/− mice, suggesting the contribution of the Th17 family to the progressive fibrosis in the dnTGF-βRII mice.
IL-23p19−/− dnTGF-βRII mice were constructed to examine whether IL-12p40 mediates protection by the IL-23/Th17 pathways. IL-23p19−/− mice exhibited dramatic improvement in the colitis, but no changes in biliary pathology. Th17 cell populations were reduced whereas IFNγ levels remained unchanged in IL-23p19−/− mice. Altogether, the data indicated that IL-12/Th1 pathway is essential for biliary disease pathogenesis, whereas the IL-23/Th17 pathway mediates colitis [21]. Since IL-17A is a major effector cytokine produced by IL-23-dependent Th17 cells, IL-17A−/− dnTGF-βRII mice was also generated to dissect the role of IL-17A in the pathogenesis of cholangitis in dnTGF-βRII mice and to assess the mechanism of the IL-23-mediated protection from colitis. The data demonstrated that IL-17A−/− dnTGF-βRII mice did not have decreased severity of autoimmune cholangitis or colitis, showing that IL-17A is not important for autoimmune cholangitis or colitis. These data suggest that the IL-23/ Th17 pathway contributes colitis in an IL-17-independent manner [21]. Furthermore, it has also been reported that lack of T cell TGF-β signaling is associated with the down regulation of T cell miRNAs in dnTGF-βRII mice. However, the expression of miR-21 from hepatic effector CD8(+) T cells is significantly higher than in the same subsets isolated from spleen and mesenteric lymph nodes of the. This is of significance that when wild-type T cell subsets are transfected with miR-21, the levels of proinflammatory cytokines TNFα and IFNγ are elevated [20]. Hence, miR-21 could play a critical role in the production of pro-inflammatory cytokines and perhaps liver pathology in the dnTGF-βRII mice.
Taken together, these derivatives of the dnTGF-βRII mice have greatly contributed to our understanding of the immunological basis in the biliary pathology of PBC. Although the dnTGF-βRII mice exhibit features resembling that seen in PBC, it is also important to note that there are some differences from human PBC, such as the lack of a female bias, eosinophilic infiltration, and granuloma formation [53]. Nevertheless, the association of decrease in peripheral regulatory T cells (Tregs) with disease in PBC patients [54] and the role of TGF-β in immunomodulation make the dnTGF-βRII mice a useful model to further examine the complexity of clinical courses in PBC.
IL-2Rα−/− Mice
In 2006, Wakabayashi et al. reported that spontaneous lymphocyte infiltration in the portal area and interlobular bile duct destruction, similar to the features seen in CNSDC, are found in IL-2Rα−/− c57BL/6 mice. In these mice, the IL-2 signal, which is important for differentiation of Tregs is blocked [50]. Interestingly, decreased numbers of Tregs have been also reported in patients with PBC and their first-degree relatives had [55]. In IL-2Rα−/− mice, CD4+ T cells, and particularly CD8+ T cells, are predominant among infiltrating lymphocytes, and the CD4+/CD8+ ratio is also decreased. The levels of inflammatory serum cytokines (Th1 cytokines: IFN-γ, TNF-α, IL-2, IL-12p40) are also increased, similarly to those in PBC. One hundred percent of the IL-2Rα−/− mice develop AMA against PDC-E2 and 80 % develop anti-nuclear antibody (Table 1). To address the effects of hepatic and intestinal T cells in the pathology of IL-2Rα−/− mice, Hsu et al. constructed the IL-2Rα−/− CD4−/− mice, IL-2Rα−/− CD8−/− mice, and IL-2Rα−/− TCR-β−/− mice [49], and examined their gut and liver histology. The results showed that in IL-2Rα−/− CD4−/− mice, bile duct obstruction was progressive, but colitis was milder. In IL-2Rα−/− CD8−/− mice, bile duct obstruction was milder, but colitis was progressive. As was the case in controls, IL-2Rα−/− TCR-β−/− mice showed no portal inflammation, bile duct obstruction, or colitis up to 3 months of age. With regard to the hepatic mononuclear cell population, IL-2Rα−/− CD4−/− mice had increased numbers of T and B cells relative to IL-2Rα−/− CD8−/− mice. These two mouse models showed elevated levels of inflammatory cytokines (IFN-γ, TNF-α, IL-2, IL-12p40). Without Treg control, CD8+ T cells play a role in biliary destruction, whereas CD4+ T cells may induce colon-specific autoimmunity. In another study, the immunobiology of p40(−/−) in IL-2Rα(−/−) mice were studied by comparing the immunopathology of liver and colon in IL-2Rα(+/−), IL-2Rα(−/−), and p40(−/−)IL-2Rα(−/−) mice by histology and immunohistochemistry; p40(−/−)IL-2Rα(−/−) mice manifest more severe portal inflammation with a heavy hepatic CD8(+) T cell infiltrate and bile duct damage, including signs of portal hypertension and liver fibrosis, but a significant reduction in colitis [56]. However, in contrast to PBC, the absence of granulomas or eosinophilic infiltrations, co-presence of anemia and inflammatory bowel disease, lack of hypergammaglobulin, and the short life span in the IL-2Rα−/− mice make this model less attractive.
Scurfy Mice
Foxp3 is a member of the forkhead/winged-helix family of transcription factors required for differentiation of T cells into Tregs. The scurfy mice have a gene mutation in the Foxp3 transcription factor, leading to deficient regulation of T cell function. In 2009, Zhang et al. reported that AMA are present at 3 to 4 weeks in 100 % of the scurfy mice [57]. Furthermore, the degree of lymphocyte infiltration and bile duct obstruction in portal areas, and spontaneous lesions are similar to those in PBC. Among the infiltrating lymphocytes, CD4+ T cells accumulate in periportal areas, and cytotoxic CD8+ T cells accumulate around the bile ducts. In comparison with control mice, the serum immunoglobulins IgG, IgA, and IgM are significantly increased and the levels of inflammatory cytokines (TNF-α, IFN-γ, IL-6, IL-12p40, IL-18, IL-10, IL-23) are elevated (Table 1). However, the short life span (approximately) of 4 weeks poses constraints for extensive longitudinal experimental studies with this model.
Ae2a,b−/− Mice
The Cl−/HCO3 − anion exchanger 2 (AE2) plays a role in acid–base transport and export of biliary bicarbonate, which are involved in intracellular pH regulation. In PBC patients, it has been reported that AE2 is decreased in the liver, blood, and mononuclear cells [58]. Since ursodeoxycholic acid restores AE2 expression and promotes biliary bicarbonate secretion, AE could be involved in the pathogenesis of PBC. In 2008, Salas et al. reported that AE2-knockdown mice (Ae2a,b−/− mice) had elevated levels of IL-12p70 and IFNγ and increased numbers of CD8+ T cells [59]. Interestingly, AMA and increased levels of IgM, IgG, and alkaline phosphatase were also observed in Ae2a,b−/− mice. Histologically, 30 % of the mice showed infiltration of CD4+ and CD8+ T cells in the portal areas and around the damaged bile duct with slight fibrosis around areas of bile duct obstruction (Table 1).
The mechanism of this model has been explained by a change in biliary epithelial cell homeostasis due to AE2 deficiency which increases intracellular pH and promotes the proliferation, differentiation, and activity of lymphocytes, leading to an increase in the targeting sensitivity of CD8+ T cells, causing autoimmune biliary change. The disadvantage of this model is that there are variations in the histological features; many mice show no changes and resemble control mice, and furthermore their breeding is difficult.
Xenobiotic Immunized Animal Models
It has been suggested that the onset of PBC involves not only genetic but also environmental factors [60]. The breaching of tolerance to PDC-E2 is believed to be the initiating step that that leads to active PBC [60–62]. This hypothesis was successfully tested by quantitative structure activity relationship studies on a large panel of chemicals for their reactivity to anti-PDC-E2 [32, 63, 64] and the requirement of the structural integrity of the PDC-E2 lipoyl domain in AMA recognition [65]. Induction of autoimmune cholangitis due to failure of immune tolerance after exposure to xenobiotics indicates that the etiology of PBC involves an environmental factor. This model illustrates the characteristics of PBC in the early stage, and the findings persist, making it attractive in having broad utility.
6-Bromohexanote-BSA Immunized Guinea Pigs
In 2007, Park et al. developed a guinea pig model of PBC by immunizing with the xenobiotic 6-bromohexoanate (6-BH) conjugated to bovine serum albumin (BSA) [66]. These 6-BH-BSA immune guinea pigs developed anti-PDC-E2 antibody, anti-BCOADC-E2 antibody, and anti-OGDC-E2 antibody as early as 4 weeks of age, and 100 % of them remained positive until 12 weeks of age and beyond. At 18 months after immunization, there were moderate lymphocyte infiltration in the portal area, causing bile duct loss and autoimmune cholangitis. Granuloma was also often present. Destruction of the small bile ducts was limited to the intrahepatic bile ducts, and the large bile ducts were not affected. These data suggest that the immune process in PBC is caused by continuous exposure of PDC-E2. However, this model does not show lymphocyte infiltration like that in CNSDC until more than 18 months after immunization and the animals did not develop fibrosis.
2-Octynoic Acid-BSA Immunized Mice
2-Octynoic acid (2-OA) is a xenobiotic that has been widely used as a cosmetic ingredient and for seasoning of general foods. Focusing on the fact that 2-OA, which is an organic compound, shows higher AMA affinity than PDC-E2 antigen, Wakabayashi et al. immunized female C57BL/6 mice with 2-OA conjugated to BSA [67]. 2OA-BSA immunized mice developed AMA as early as 4 weeks after immunization, and positivity was sustained thereafter. Serum TNF-α and IFNγ were also increased from 4 weeks after immunization. These mice showed lymphocyte infiltration and bile duct failure in the portal area and granuloma in the liver tissue 12 weeks after immunization. Immunostaining showed that the infiltrating lymphocytes in the portal area were CD4+ and CD8+ T cells, the latter being the more dominant. The intrahepatic CD4/CD8 ratio was lower than in the controls (Table 1).
Induction of autoimmune cholangitis due to failure of immune tolerance after exposure to xenobiotics indicates that the etiology of PBC involves an environmental factor. This model illustrates the characteristics of PBC in the early stage, and the findings persist, making it attractive in having broad utility.
In 2008, Wakabayashi et al. immunized the NOD congenic strain 1101, which harbors the diabetes-resistant allele (Idd10, Idd18) and does not develop spontaneous cholangitis, with 2OA-BSA [31]. AMA was detected as early as 2 weeks after immunization. The histological findings were similar to those of PBC, including infiltration of CD4+ and CD8+ T cells in the portal areas and granuloma formation at 12 weeks after immunization. The infiltrating cells in the liver and spleen were CD8+ T cell-dominant, and the CD4/CD8 ratio was lower than in the controls. Expression of TNF-α and IFNγ in the portal areas was confirmed by immunostaining.
The role of IL-12-Th1/IL-23-Th17 pathways in autoimmune cholangitis in 2OA-BSA PBC model was systematically examined by using specific cytokine knockout mice, including C57BL/6 mice deleted both Th1 and Th17 (IL-12p40), Th1 cytokine (IL-12p35, IFN-γ), and Th17 cytokine (IL-23p19, IL-17A, IL-17 F, or IL-22) (Table 3) [68]. Each of these cytokine-deficient mice was immunized with 2-OA-BSA and followed the natural history of their immunopathology. While both IL-12/Th1 and IL-23/Th17 are involved in cholangitis, it is the IL-12/Th1 signaling pathway that elicits liver pathology in this xenobiotic induction disease model of PBC. In fact, deletion of IFNγ prevents disease and suppresses autoantibodies. Importantly, deletion of the Th17 cytokines IL-17A and IL-22, but not IL-17 F, reduces biliary damage; IL-17A-knockout mice have also reduced levels of AMAs. IFNγ is significantly decreased in livers of IL-23p19−/−, IL-17A−/−, and IL-22−/− mice compared with controls. However, the ability of T cells to produce IFNγ was not affected in Th17 cytokine-deficient mice. In summary, in the 2-OA-BSA immunized mice model: (1) both IL-12/Th1 and IL-23/Th17 are involved in cholangitis; (2) IL-12/Th1 signaling pathway is critical in eliciting liver pathology; and (3) IL-23/Th17 pathway is involved in perpetuating the IL-12/IFNγ mediated pathology. In addition, the role of B cells in the pathogenesis of PBC was also investigated by depleting B cells using two different monoclonal antibodies, CD20 and CD79. In this study, B cell depletion led to exacerbated cholangitis, with higher T cell infiltrates and inflammatory cytokines, indicating a protective role of B cells in PBC [69].
2OA-BSA immunized C57BL/6 mice were also studied for the potential of CTLA4-based therapy on cholangitis by using CTLA4-Ig. CTLA4-Ig is a soluble recombinant human fusion protein comprised of the extracellular domain of human CTLA4 linked to a modified portion of the Fc domain of human IgG [70, 71]. In mice treated beginning 1 day before 2OA-BSA immunization, CTLA4-Ig completely inhibits the manifestations of cholangitis, including AMA production, intra-hepatic T cell infiltrates, and bile duct damage. However, treatment with CTLA-4 Ig initiated after the development of autoimmune cholangitis in 2OA-BSA immunized mice reduced intra-hepatic T cell infiltrates and biliary cell damage, although AMA levels were not altered [72].
The role of innate immune effector cells, such as natural killer (NK) cells and that NK T cells on modulating disease activity, was examined in this model based on the hypothesis that early events during immunization play an important role in the breakdown of tolerance. Shimoda et al. [73] demonstrated that there were marked suppression of AMA and cytokine production following in vivo depletion of NK and NKT cells in 2OA-BSA immunized mice. However, there was no change in the clinical pathology of portal inflammation compared to controls. Thus, further studies have been reported in which 2OA-BSA immunized with and without the addition of α-galactosylceramide (α-GalCer), an invariant natural killer T cell activator, were followed for AMA and liver pathology. 2-OA-BSA immunized mice exposed to α-GalCer developed a profound exacerbation of their autoimmune cholangitis, such as significant increases in CD8+ T cell infiltrates, portal inflammation, granuloma formation, and bile duct damage. More excitingly, these mice produced increased levels of AMAs and have showed evidence of fibrosis [74]. CD4 and CD8 knockout mice immunized with 2-OA-BSA/PBS or 2-OA-BSA/α-GalCer develop AMA, portal infiltrates, and fibrosis with liver pathology exacerbated in the presence of α-GalCer [75]. Indeed, the data suggest that there can be multiple steps in the natural history of PBC, including a role of NK and NK T cells in initiating the breakdown of tolerance. More recently, it was reported that 2-OA-BSA immunized mice administered with a Th2-biasing agonist (2 s,3 s,4r)-1-O-(a-D-galactopyranosyl)-N-tetracosanoyl-2-amino-1,3,4-nonanetriol (OCH) developed portal inflammation and hepatic fibrosis similar to mice treated with α-GalCer [76]. However, inflammatory portal cell infiltrates and AMA responses are reduced in iNKT cell-deficient CD1d knockout mice treated with OCH. These results suggest that activation iNKT cells can occur via overlapping and/or promiscuous pathways and further highlight the role of innate immunity in the natural history of PBC. Furthermore, the data also provides clues to the mechanisms by which autoimmune biliary diseases could be perpetuated in humans, and the recurrence of PBC following liver transplantation in the absence of major histocompatibility complex (MHC) compatibility could be explained.
Infection Triggered Model
Novosphingobium aromaticivorans-Infected Mice
In 2008, Mattner et al. reported on a model with PBC-like features in which mice (common mouse strains C57BL/6, NOD, SJL) were infected with N. aromaticivorans [77]. N. aromaticivorans is a Gram-negative alphaproteobacterium that possesses xenobiotic-metabolizing properties and are found in soil, water, mucosal surfaces, and human stools. It exhibits molecular homology with the PDC-E2 epitope and expresses the glycosphingolipids recognized by CD1d-restricted NKT cells. These NKT cells then become activated and release Th1 and Th2 cytokines. It has been reported that patients with PBC have antibodies against N. aromaticivorans PDC-E2 [78] and show increased expression of NKT cells and CD1d.
Briefly, infection of mice strains (NOD, C57BL/6 and SJL) with N. aromaticivorans at 5 × 107 cfu intravenously at week 0 and week 2 not only induced signature antibodies against microbial PDC-E2 and mammalian PDC-E2 but also bile duct damage and granuloma. Further work has focused on the NOD.1101 mouse strain because it exhibited particularly severe liver disease similar to PBC. NOD.1101 belongs to a set of NOD subcongenic strains originating from NOD.c3c4 mice and was produced by introgressing Idd loci 10 and 18r2 from B6 (chromosome 3) onto an NOD background [40, 79–82]. In this model, disease induction requires NKT cells, which specifically respond to the N. aromaticivorans cell wall, α-glycuronosylceramides, presented by CD1d molecules. As a result of exposure to N. aromaticivorans, intrahepatic natural NKT cells become reactive (the bacteria are eliminated within a week, as illustrated by negative 16S RNA PCR within 8 weeks), leading to liver-specific bile duct destruction. Combined with the natural liver tropism of NKT cells, the accumulation of N. aromaticivorans in the liver likely explains the liver specificity of destructive responses. Furthermore, once established, liver disease could be adoptively transferred by T cells independently of NKT cells and microbes, illustrating the importance of early microbial activation of NKT cells in the initiation of liver autoimmunity in this model [77]. However, this data on N. aromaticivorans has not been completely recapitulated. Although the failure of recapitulation does not detract from the possibility of an infectious origin, it does suggest that multiple infectious agents may be involved [33].
Escherichia coli-Infected Mice
In 2004, Wang et al. demonstrated that NOD.B6-Idd10/Idd18 mice infected with E. coli developed AMA and severe cholangitis [33], both being more severe than those resulting from N. aromaticivorans infection. It has been reported that there are six E. coli peptide sequences that mimic the human PDC-E2 autoepitope with 6–8 identical amino acid residues [83], which may also account for the E. coli-induced anti-PDCE2 response in the NOD.B6-Idd10/Idd18 mice. The difference in microflora between animal colonies may also partly account for the discrepancies between this study and others [77, 84]. Although the serological antibody reactivity to PDC-E2 is relatively weak in microbially infected (E. coli, N. aromaticivorans) mice when compared to sera from patients with PBC or other models of autoimmune cholangitis, including the dnTGF-βRII mice and xenobiotic 2OA-BSA BSA conjugate immunized C57BL/6 mice, the initiation of anti-PDC-E2 during early stage E. coli infection is sufficient to break tolerance and lead to PBC-like liver pathology in E. coli-infected mice.
Summary
It is becoming clear that the etiology of PBC is associated with genetics and environment and immunological factors [12, 64, 85–87]. Primary biliary cirrhosis suffers from the same difficulties as other autoimmune diseases. Clearly, it is a result of genetic and environmental interactions but thus far the genetic data has been disappointing and therapy continues to lag behind. One can make a similar statement for a variety of other autoimmune diseases as well in which animal models and/or genetic analysis of human data has failed to lead to a major breakthrough [88–93]. Although the spontaneous genetic models and their derivatives models could explain the involvement of an autoimmune element in the pathogenesis of PBC, with some models demonstrating either exacerbation or amelioration of cholangitis in mice, it remains unclear whether activation of these immunocompetent cells itself would play a significant role or would be sufficient enough to account for the onset of human PBC. PBC has a diverse clinical course that cannot be solely explained by the presence of autoimmune cholangitis. Thus, human PBC is a group of diverse clinical conditions, ranging from very slowly progressing to rapidly progressing types. The picture is further complicated in its female predominance [94, 95], possible contribution of epigenetics [87, 96, 97]. Furthermore, there is also evidence that the BECs are not merely innocent victims [98–101]. Phenotypically, it is ideal to have a histologically progressive animal model that exhibits PBC-like cholangitis and serological autoantibodies. Slow progression has been reproduced in many of these models. We have described some of the elements that can be involved in the pathogenesis of PBC in animal models, such as immunocompetent cells and increased levels of inflammatory cytokines, a decrease of regulatory T cells, and failure of bile duct epithelial cell homeostasis (Fig. 2). The initial mechanism of bile duct destruction seems to be mostly reproduced by currently available models [24]. Therefore, the remaining task is to develop a female predominant model of progressive autoimmune cholangitis disease exhibiting fibrosis and subsequent hepatic failure. Finally, since human PBC has distinct clinical subgroups, development of animal models reflecting each of these subgroups will be necessary in order to further dissect the pathological basis of this enigma disease. We submit that PBC should be among the easiest of autoimmune diseases to dissect because of its homogeneity and characteristic presentation and disease course, but yet, as with the clinical management of other autoimmune diseases, there is still much to learn [102–105].

The association of cholangitis and immunological factors in animal models of PBC. Multiple elements including increased levels of inflammatory cytokines, decreased numbers of regulatory T cells, activation of immune cells, and abnormal bile duct epithelial cell homeostasis have been demonstrated to contribute in the pathogenesis of cholangitis in animal models of PBC. These animal models provide clues to the mechanisms of bile duct destruction in PBC
References
Nakanuma Y, Ohta G (1979) Histometric and serial section observations of the intrahepatic bile ducts in primary biliary cirrhosis. Gastroenterology 76:1326–1332
Bowlus CL, Gershwin ME (2014) The diagnosis of primary biliary cirrhosis. Autoimmun Rev 13:441–444
Coppel RL, McNeilage LJ, Surh CD et al (1988) Primary structure of the human M2 mitochondrial autoantigen of primary biliary cirrhosis: dihydrolipoamide acetyltransferase. Proc Natl Acad Sci U S A 85:7317–7321
Gershwin ME, Mackay IR, Sturgess A, Coppel RL (1987) Identification and specificity of a cDNA encoding the 70 kd mitochondrial antigen recognized in primary biliary cirrhosis. J Immunol 138:3525–3531
Oertelt S, Rieger R, Selmi C et al (2007) A sensitive bead assay for antimitochondrial antibodies: chipping away at AMA-negative primary biliary cirrhosis. Hepatology 45:659–665
Duarte-Rey C, Bogdanos D, Yang CY et al (2012) Primary biliary cirrhosis and the nuclear pore complex. Autoimmun Rev 11:898–902
Yang CY, Leung PS, Yang GX et al (2012) Epitope-specific anti-nuclear antibodies are expressed in a mouse model of primary biliary cirrhosis and are cytokine-dependent. Clin Exp Immunol 168:261–267
Norman GL, Yang CY, Ostendorff HP et al (2015) Anti-kelch-like 12 and anti-hexokinase 1: novel autoantibodies in primary biliary cirrhosis. Liver Int Off J Int Assoc Study Liver 35:642–651
Gores GJ, Wiesner RH, Dickson ER, Zinsmeister AR, Jorgensen RA, Langworthy A (1989) Prospective evaluation of esophageal varices in primary biliary cirrhosis: development, natural history, and influence on survival. Gastroenterology 96:1552–1559
Pares A, Caballeria L, Rodes J (2006) Excellent long-term survival in patients with primary biliary cirrhosis and biochemical response to ursodeoxycholic acid. Gastroenterology 130:715–720
Dong M, Li J, Tang R et al (2015) Multiple genetic variants associated with primary biliary cirrhosis in a Han Chinese Population. Clin Rev Allergy Immunol
Kar SP, Seldin MF, Chen W et al (2013) Pathway-based analysis of primary biliary cirrhosis genome-wide association studies. Genes Immun 14:179–186
Tang R, Chen H, Miao Q et al (2015) The cumulative effects of known susceptibility variants to predict primary biliary cirrhosis risk. Genes Immun
Boonstra K, Beuers U, Ponsioen CY (2012) Epidemiology of primary sclerosing cholangitis and primary biliary cirrhosis: a systematic review. J Hepatol 56:1181–1188
Selmi C, Mayo MJ, Bach N et al (2004) Primary biliary cirrhosis in monozygotic and dizygotic twins: genetics, epigenetics, and environment. Gastroenterology 127:485–492
Invernizzi P, Ransom M, Raychaudhuri S et al (2012) Classical HLA-DRB1 and DPB1 alleles account for HLA associations with primary biliary cirrhosis. Genes Immun 13:461–468
Pillai S (2013) Rethinking mechanisms of autoimmune pathogenesis. J Autoimmun 45:97–103
Walker LS (2013) Treg and CTLA-4: two intertwining pathways to immune tolerance. J Autoimmun 45:49–57
Hudspeth K, Pontarini E, Tentorio P et al (2013) The role of natural killer cells in autoimmune liver disease: a comprehensive review. J Autoimmun 46:55–65
Ando Y, Yang GX, Kenny TP et al (2013) Overexpression of microRNA-21 is associated with elevated pro-inflammatory cytokines in dominant-negative TGF-beta receptor type II mouse. J Autoimmun 41:111–119
Ando Y, Yang GX, Tsuda M et al (2012) The immunobiology of colitis and cholangitis in interleukin-23p19 and interleukin-17A deleted dominant negative form of transforming growth factor beta receptor type II mice. Hepatology 56:1418–1426
Huang W, Kachapati K, Adams D et al (2014) Murine autoimmune cholangitis requires two hits: cytotoxic KLRG1(+) CD8 effector cells and defective T regulatory cells. J Autoimmun 50:123–134
Kawata K, Yang GX, Ando Y et al (2013) Clonality, activated antigen-specific CD8(+) T cells, and development of autoimmune cholangitis in dnTGFbetaRII mice. Hepatology 58:1094–1104
Leung PS, Yang GX, Dhirapong A, Tsuneyama K, Ridgway WM, Gershwin ME (2012) Animal models of primary biliary cirrhosis: materials and methods. Methods Mol Biol 900:291–316
Moritoki Y, Tsuda M, Tsuneyama K et al (2011) B cells promote hepatic inflammation, biliary cyst formation, and salivary gland inflammation in the NOD.c3c4 model of autoimmune cholangitis. Cell Immunol 268:16–23
Moritoki Y, Zhang W, Tsuneyama K et al (2009) B cells suppress the inflammatory response in a mouse model of primary biliary cirrhosis. Gastroenterology 136:1037–1047
Ridgway WM, Gershwin ME (2014) Prometheus unbound: NKT cells inhibit hepatic regeneration. Hepatology 60:1133–1135
Tanaka H, Zhang W, Yang GX et al (2014) Successful immunotherapy of autoimmune cholangitis by adoptive transfer of forkhead box protein 3(+) regulatory T cells. Clin Exp Immunol 178:253–261
Tsuda M, Zhang W, Yang GX et al (2013) Deletion of interleukin (IL)-12p35 induces liver fibrosis in dominant-negative TGFbeta receptor type II mice. Hepatology 57:806–816
Ueno Y, Ambrosini YM, Moritoki Y, Ridgway WM, Gershwin ME (2010) Murine models of autoimmune cholangitis. Curr Opin Gastroenterol 26:274–279
Wakabayashi K, Yoshida K, Leung PS et al (2009) Induction of autoimmune cholangitis in non-obese diabetic (NOD).1101 mice following a chemical xenobiotic immunization. Clin Exp Immunol 155:577–586
Wang J, Yang GX, Tsuneyama K, Gershwin ME, Ridgway WM, Leung PS (2014) Animal models of primary biliary cirrhosis. Semin Liver Dis 34:285–296
Wang JJ, Yang GX, Zhang WC et al (2014) Escherichia coli infection induces autoimmune cholangitis and anti-mitochondrial antibodies in non-obese diabetic (NOD).B6 (Idd10/Idd18) mice. Clin Exp Immunol 175:192–201
Wang YH, Yang W, Yang JB et al (2015) Systems biologic analysis of T regulatory cells genetic pathways in murine primary biliary cirrhosis. J Autoimmun. doi:10.1016/j.jaut.2015.01.011
Yang GX, Wu Y, Tsukamoto H et al (2011) CD8 T cells mediate direct biliary ductule damage in nonobese diabetic autoimmune biliary disease. J Immunol 186:1259–1267
Yang W, Yao Y, Yang YQ et al (2014) Differential modulation by IL-17A of cholangitis versus colitis in IL-2Ralpha deleted mice. PLoS One 9:e105351
Zhang W, Tsuda M, Yang GX et al (2012) Lymphoma-like T cell infiltration in liver is associated with increased copy number of dominant negative form of TGFbeta receptor II. PLoS One 7:e49413
Zhang W, Tsuda M, Yang GX et al (2010) Deletion of interleukin-6 in mice with the dominant negative form of transforming growth factor beta receptor II improves colitis but exacerbates autoimmune cholangitis. Hepatology 52:215–222
Koarada S, Wu Y, Fertig N et al (2004) Genetic control of autoimmunity: protection from diabetes, but spontaneous autoimmune biliary disease in a nonobese diabetic congenic strain. J Immunol 173:2315–2323
Irie J, Wu Y, Wicker LS et al (2006) NOD.c3c4 congenic mice develop autoimmune biliary disease that serologically and pathogenetically models human primary biliary cirrhosis. J Exp Med 203:1209–1219
Nakagome Y, Ueno Y, Kogure T et al (2007) Autoimmune cholangitis in NOD.c3c4 mice is associated with cholangiocyte-specific Fas antigen deficiency. J Autoimmun 29:20–29
Oertelt S, Lian ZX, Cheng CM et al (2006) Anti-mitochondrial antibodies and primary biliary cirrhosis in TGF-beta receptor II dominant-negative mice. J Immunol 177:1655–1660
Gorelik L, Flavell RA (2000) Abrogation of TGFbeta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity 12:171–181
Chuang YH, Lian ZX, Yang GX et al (2008) Natural killer T cells exacerbate liver injury in a transforming growth factor beta receptor II dominant-negative mouse model of primary biliary cirrhosis. Hepatology 47:571–580
Shimoda S, Harada K, Niiro H et al (2011) Interaction between Toll-like receptors and natural killer cells in the destruction of bile ducts in primary biliary cirrhosis. Hepatology 53:1270–1281
Yang GX, Lian ZX, Chuang YH et al (2008) Adoptive transfer of CD8(+) T cells from transforming growth factor beta receptor type II (dominant negative form) induces autoimmune cholangitis in mice. Hepatology 47:1974–1982
Invernizzi P, Crosignani A, Battezzati PM et al (1997) Comparison of the clinical features and clinical course of antimitochondrial antibody-positive and -negative primary biliary cirrhosis. Hepatology 25:1090–1095
Kim WR, Poterucha JJ, Jorgensen RA et al (1997) Does antimitochondrial antibody status affect response to treatment in patients with primary biliary cirrhosis? Outcomes of ursodeoxycholic acid therapy and liver transplantation. Hepatology 26:22–26
Hsu W, Zhang W, Tsuneyama K et al (2009) Differential mechanisms in the pathogenesis of autoimmune cholangitis versus inflammatory bowel disease in interleukin-2Ralpha(−/−) mice. Hepatology 49:133–140
Wakabayashi K, Lian ZX, Moritoki Y et al (2006) IL-2 receptor alpha(−/−) mice and the development of primary biliary cirrhosis. Hepatology 44:1240–1249
Yin S, Wang H, Bertola A et al (2014) Activation of invariant natural killer T cells impedes liver regeneration by way of both IFN-gamma- and IL-4-dependent mechanisms. Hepatology 60:1356–1366
Yoshida K, Yang GX, Zhang W et al (2009) Deletion of interleukin-12p40 suppresses autoimmune cholangitis in dominant negative transforming growth factor beta receptor type II mice. Hepatology 50:1494–1500
Nakamura A, Yamazaki K, Suzuki K, Sato S (1997) Increased portal tract infiltration of mast cells and eosinophils in primary biliary cirrhosis. Am J Gastroenterol 92:2245–2249
Lan RY, Cheng C, Lian ZX et al (2006) Liver-targeted and peripheral blood alterations of regulatory T cells in primary biliary cirrhosis. Hepatology 43:729–737
Aoki CA, Roifman CM, Lian ZX et al (2006) IL-2 receptor alpha deficiency and features of primary biliary cirrhosis. J Autoimmun 27:50–53
Yao Y, Yang W, Yang YQ et al (2014) Distinct from its canonical effects, deletion of IL-12p40 induces cholangitis and fibrosis in interleukin-2Ralpha(−/−) mice. J Autoimmun 51:99–108
Zhang W, Sharma R, Ju ST et al (2009) Deficiency in regulatory T cells results in development of antimitochondrial antibodies and autoimmune cholangitis. Hepatology 49:545–552
Medina JF, Martinez A, Vazquez JJ, Prieto J (1997) Decreased anion exchanger 2 immunoreactivity in the liver of patients with primary biliary cirrhosis. Hepatology 25:12–17
Salas JT, Banales JM, Sarvide S et al (2008) Ae2a, b-deficient mice develop antimitochondrial antibodies and other features resembling primary biliary cirrhosis. Gastroenterology 134:1482–1493
Kurth MJ, Yokoi T, Gershwin ME (2014) Halothane-induced hepatitis: paradigm or paradox for drug-induced liver injury. Hepatology 60:1473–1475
Wang L, Wang FS, Chang C, Gershwin ME (2014) Breach of tolerance: primary biliary cirrhosis. Semin Liver Dis 34:297–317
Zhang J, Zhang W, Leung PS et al (2014) Ongoing activation of autoantigen-specific B cells in primary biliary cirrhosis. Hepatology 60:1708–1716
Chen RC, Naiyanetr P, Shu SA et al (2013) Antimitochondrial antibody heterogeneity and the xenobiotic etiology of primary biliary cirrhosis. Hepatology 57:1498–1508
Leung PS, Wang J, Naiyanetr P et al (2013) Environment and primary biliary cirrhosis: electrophilic drugs and the induction of AMA. J Autoimmun 41:79–86
Wang J, Budamagunta MS, Voss JC et al (2013) Antimitochondrial antibody recognition and structural integrity of the inner lipoyl domain of the E2 subunit of pyruvate dehydrogenase complex. J Immunol 191:2126–2133
Leung PS, Park O, Tsuneyama K et al (2007) Induction of primary biliary cirrhosis in guinea pigs following chemical xenobiotic immunization. J Immunol 179:2651–2657
Wakabayashi K, Lian ZX, Leung PS et al (2008) Loss of tolerance in C57BL/6 mice to the autoantigen E2 subunit of pyruvate dehydrogenase by a xenobiotic with ensuing biliary ductular disease. Hepatology 48:531–540
Kawata K, Tsuda M, Yang GX et al (2013) Identification of potential cytokine pathways for therapeutic intervention in murine primary biliary cirrhosis. PLoS One 8:e74225
Dhirapong A, Lleo A, Yang GX et al (2011) B cell depletion therapy exacerbates murine primary biliary cirrhosis. Hepatology 53:527–535
Rozelle AL, Genovese MC (2007) Efficacy results from pivotal clinical trials with abatacept. Clin Exp Rheumatol 25:S30–S34
Davis PM, Abraham R, Xu L, Nadler SG, Suchard SJ (2007) Abatacept binds to the Fc receptor CD64 but does not mediate complement-dependent cytotoxicity or antibody-dependent cellular cytotoxicity. J Rheumatol 34:2204–2210
Dhirapong A, Yang GX, Nadler S et al (2013) Therapeutic effect of cytotoxic T lymphocyte antigen 4/immunoglobulin on a murine model of primary biliary cirrhosis. Hepatology 57:708–715
Shimoda S, Tsuneyama K, Kikuchi K et al (2012) The role of natural killer (NK) and NK T cells in the loss of tolerance in murine primary biliary cirrhosis. Clin Exp Immunol 168:279–284
Wu SJ, Yang YH, Tsuneyama K et al (2011) Innate immunity and primary biliary cirrhosis: activated invariant natural killer T cells exacerbate murine autoimmune cholangitis and fibrosis. Hepatology 53:915–925
Chang CH, Chen YC, Yu YH et al (2014) Innate immunity drives xenobiotic-induced murine autoimmune cholangitis. Clin Exp Immunol 177:373–380
Chao-Hsuan Chang Y-CC, Zhang W, Leung PSC, Eric Gershwin M, Chuang Y-H (2015) Innate immunity drives the initiation of a murine model of primary biliary cirrhosis. PLOs One
Mattner J, Savage PB, Leung P et al (2008) Liver autoimmunity triggered by microbial activation of natural killer T cells. Cell Host Microbe 3:304–315
Padgett KA, Selmi C, Kenny TP et al (2005) Phylogenetic and immunological definition of four lipoylated proteins from Novosphingobium aromaticivorans, implications for primary biliary cirrhosis. J Autoimmun 24:209–219
Lyons PA, Hancock WW, Denny P et al (2000) The NOD Idd9 genetic interval influences the pathogenicity of insulitis and contains molecular variants of Cd30, Tnfr2, and Cd137. Immunity 13:107–115
Wicker LS, Leiter EH, Todd JA et al (1994) Beta 2-microglobulin-deficient NOD mice do not develop insulitis or diabetes. Diabetes 43:500–504
Koarada S, Wu Y, Yim YS, Wakeland EW, Ridgway WM (2004) Nonobese diabetic CD4 lymphocytosis maps outside the MHC locus on chromosome 17. Immunogenetics 56:333–337
Podolin PL, Denny P, Armitage N et al (1998) Localization of two insulin-dependent diabetes (Idd) genes to the Idd10 region on mouse chromosome 3. Mamm Genome 9:283–286
Bogdanos DP, Baum H, Grasso A et al (2004) Microbial mimics are major targets of crossreactivity with human pyruvate dehydrogenase in primary biliary cirrhosis. J Hepatol 40:31–39
Mohammed JP, Fusakio ME, Rainbow DB et al (2011) Identification of Cd101 as a susceptibility gene for Novosphingobium aromaticivorans-induced liver autoimmunity. J Immunol 187:337–349
Hirschfield GM, Gershwin ME (2013) The immunobiology and pathophysiology of primary biliary cirrhosis. Annu Rev Pathol 8:303–330
Liaskou E, Hirschfield GM, Gershwin ME (2014) Mechanisms of tissue injury in autoimmune liver diseases. Semin Immunopathol 36:553–568
Selmi C, Cavaciocchi F, Lleo A et al (2014) Genome-wide analysis of DNA methylation, copy number variation, and gene expression in monozygotic twins discordant for primary biliary cirrhosis. Front Immunol 5:128
Kundu-Raychaudhuri S, Chen YJ, Wulff H, Raychaudhuri SP (2014) Kv1.3 in psoriatic disease: PAP-1, a small molecule inhibitor of Kv1.3 is effective in the SCID mouse psoriasis—xenograft model. J Autoimmun 55:63–72
Zhao M, Liu S, Luo S et al (2014) DNA methylation and mRNA and microRNA expression of SLE CD4+ T cells correlate with disease phenotype. J Autoimmun 54:127–136
Fan Y, Gualtierotti G, Tajima A et al (2014) Compromised central tolerance of ICA69 induces multiple organ autoimmunity. J Autoimmun 53:10–25
Fuchs S, Aricha R, Reuveni D, Souroujon MC (2014) Experimental Autoimmune Myasthenia Gravis (EAMG): from immunochemical characterization to therapeutic approaches. J Autoimmun 54:51–59
Sthoeger Z, Sharabi A, Mozes E (2014) Novel approaches to the development of targeted therapeutic agents for systemic lupus erythematosus. J Autoimmun 54:60–71
Berrih-Aknin S (2014) Myasthenia gravis: paradox versus paradigm in autoimmunity. J Autoimmun 52:1–28
Lleo A, Oertelt-Prigione S, Bianchi I et al (2013) Y chromosome loss in male patients with primary biliary cirrhosis. J Autoimmun 41:87–91
Sun Y, Haapanen K, Li B, Zhang W, Van de Water J, Gershwin ME (2014) Women and primary biliary cirrhosis. Clin Rev Allergy Immunol
Mitchell MM, Lleo A, Zammataro L et al (2011) Epigenetic investigation of variably X chromosome inactivated genes in monozygotic female twins discordant for primary biliary cirrhosis. Epigenetics Off J DNA Methylation Soc 6:95–102
Wang Q, Selmi C, Zhou X et al (2013) Epigenetic considerations and the clinical reevaluation of the overlap syndrome between primary biliary cirrhosis and autoimmune hepatitis. J Autoimmun 41:140–145
Kawata K, Kobayashi Y, Gershwin ME, Bowlus CL (2012) The immunophysiology and apoptosis of biliary epithelial cells: primary biliary cirrhosis and primary sclerosing cholangitis. Clin Rev Allergy Immunol 43:230–241
Lleo A, Zhang W, McDonald WH et al (2014) Shotgun proteomics: identification of unique protein profiles of apoptotic bodies from biliary epithelial cells. Hepatology 60:1314–1323
Rong G, Zhong R, Lleo A et al (2011) Epithelial cell specificity and apotope recognition by serum autoantibodies in primary biliary cirrhosis. Hepatology 54:196–203
Rong GH, Yang GX, Ando Y et al (2013) Human intrahepatic biliary epithelial cells engulf blebs from their apoptotic peers. Clin Exp Immunol 172:95–103
Trivedi PJ, Adams DH (2013) Mucosal immunity in liver autoimmunity: a comprehensive review. J Autoimmun 46:97–111
Perez VL, Saeed AM, Tan Y, Urbieta M, Cruz-Guilloty F (2013) The eye: a window to the soul of the immune system. J Autoimmun 45:7–14
Podda M, Selmi C, Lleo A, Moroni L, Invernizzi P (2013) The limitations and hidden gems of the epidemiology of primary biliary cirrhosis. J Autoimmun 46:81–87
Imam MH, Talwalkar JA, Lindor KD (2013) Clinical management of autoimmune biliary diseases. J Autoimmun 46:88–96
Acknowledgments
This study was supported in part by Health and Labour Sciences Research Grants for Research on Measures for Intractable Diseases (from the Ministry of Health, Labour and Welfare of Japan), a Grant-in-Aid for Challenging Exploratory Research (26670376) from JSPS, National Institutes of Health grants DK39588, DK090019, and DK067003.
Conflict of Interest
The authors have no conflicts of interest to disclose.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Katsumi, T., Tomita, K., Leung, P.S.C. et al. Animal Models of Primary Biliary Cirrhosis. Clinic Rev Allerg Immunol 48, 142–153 (2015). https://doi.org/10.1007/s12016-015-8482-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-015-8482-y