
Abstract
Background
Human pluripotent stem cells (hPSCs) have started to emerge as a potential tool with application in fields of drug discovery, disease modelling and cell therapy. A variety of protocols for culturing and differentiating pluripotent stem cells into pancreatic β like cells have been published. However, small-scale dynamic suspension culture systems, which could be applied toward systematically optimizing production strategies for cell replacement therapies to accelerate the pace of their discovery and development toward the clinic, are overlooked.
Methods
Human embryonic stem cell (hESC) line H9 was used to establish the novel 48-well dynamic suspension culture system. The effects of various rotational speeds and culture medium volumes on cell morphology, cell proliferation, cell viability and cell phenotype were evaluated. Effect of cell density on the pancreatic differentiation efficiency from H9 cells in 48-well plates was further investigated. In vitro the function of pancreatic β like cells was assessed by measuring glucose-stimulated insulin secretion.
Main Results
A 48-well dynamic suspension culture system for hESC expansion as cell aggregates was developed. With optimized rotational speed and culture medium volume, hESCs maintained normal karyotype, viability and pluripotency. Furthermore, the system can also support the hESC aggregates subsequent differentiation into functional pancreatic β like cells after optimizing initial cell seeding density.
Conclusion
A controllable 48-well suspension culture system in microplates for hESCs maintenance, expansion and pancreatic differentiation was developed, which may provide an efficient platform for high-throughput drug screening.
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pluripotent stem cells stand out due to their indefinite proliferative capacity and pluripotency and have attracted the attention of researchers and brought substantial hope to the regenerative medicine field [1,2,3]. To date, two types of human pluripotent stem cells, namely, also referred to as human embryonic stem cells and induced pluripotent stem cells, have been explored in clinical studies as cell therapies for various diseases, such as age-related macular degeneration [4, 5], acute graft versus host disease [6] and type 1 diabetes [7]. In addition, pluripotent stem cells also play an important role in the fields of drug screening [8,9,10] and disease modelling [9, 11, 12]. Thus, to meet the variable demands for the use of pluripotent stem cells or stem cell-derived organoids, studies have been carried out on how to culture and differentiate stem cells into specific organoids [13,14,15,16]. Currently, the culture systems for hPSCs can be classified into static adherent culture systems and dynamic suspension culture systems. It is widely acknowledged that dynamic suspension culture systems are superior to 2D static adherent culture due to their scalability, monitoring capacity and controllability [17, 18]. To the best of our knowledge, the scale-up of dynamic suspension culture systems always begins with 6-well plates and ultimately ends with a 500 mL or larger bioreactor system for expansion [19,20,21]. However, with regard to high-throughput screening, the established dynamic suspension culture systems appear to be time-consuming and labour-intensive. Small-scale dynamic suspension culture systems could be applied toward systematically optimizing production strategies for cell replacement therapies to accelerate the pace of their discovery and development toward the clinic. Thus, it is necessary to establish a microplate dynamic suspension culture system to fill this gap.
To develop an hPSC dynamic suspension culture system, the selection of cell culture medium, culture vessels and control of cell aggregate sizes must be taken into consideration [18]. The common hPSC medium mTeSR-1 has been used widely in reported hPSC suspension culture systems [22]. For scale-up production, it is critical to select a proper culture vessel that can provide better scalability. Suspension cultured hPSCs, with the help of appropriate shear force, can form spheres and eventually grow into cell clusters of 200–300 μm [23]. The rotational speed directly determines the shear condition of the dynamic suspension culture system, which affects not only the morphology of cell aggregates but also their function [17]. Additionally, the initial cell seeding density has a direct effect on the diameter of cell aggregates, which may affect nutrient and metabolite penetration into the centre of the cell spheres [18]. Accordingly, the cell culture vessel, rotational speed and initial cell seeding number are key factors in the dynamic suspension culture and differentiation systems.
In this study, we attempted to study the influence of rotational speed, culture medium volume and cell density on hESC dynamic suspension culture along with the differentiation efficiency in 48-well plates. An optimized hESC dynamic suspension culture system was established, representing more homogeneous cell aggregates with controllable diameters and maintaining stable pluripotency after three passages. The established hESC dynamic suspension differentiation system showed that pancreatic β like cells were successfully obtained in 48-well plates and exerted normal pancreatic β cells functions, according to their physiological function in vitro of secreting insulin in response to glucose. Thus, we succeeded in establishing an hESC dynamic suspension culture and differentiation system in 48-well plates, which may provide an efficient platform for high-throughput drug screening using hESCs or hESC-derived pancreatic β like cells.
Materials and Methods
Dynamic Suspension Culture of Human Embryonic Stem Cells
The hESC line H9 (WA09/H9, WiCell, USA) was cultured in mTeSR 1™ medium (STEMCELL Technologies) in 500 mL spinner flasks (Corning) placed on a stir plate (Chemglass) rotating at 90 rpm in a humidified 37 °C incubator set at 5% CO2. Undifferentiated cells were maintained by passaging clusters dispersed with Gentle Cell Dissociation Reagent (GCDR, Stem Cell Technologies) and seeded at 0.5 × 106 cells per ml in mTeSR1 supplemented with 10 mM Y27632 (Sigma-Aldrich). Cultures were passaged every 72–96 h [14, 24].
For 48-well culture system, undifferentiated hESCs were cultured at a density of 5.45 × 105 cells per well in chemically defined mTeSR 1™ medium supplemented with 10 μM Y-27632 in a 48-well plate (Corning) placed on orbital shaker (Infors Celltron) for 24 h to allow time for aggregation. Then, the medium was replaced with fresh medium excluding Y-27632 daily. Serial passaging of the cell aggregates was performed every three days. The cell aggregates were incubated at 37 °C for 5 min with Accutase (STEMCELL Technologies) to dissociate into single cells. Cells were filtered through a 37 μm filter (STEMCELL Technologies) and reseeded into a 48-well plate at the same initial density as described above. For every day of cultivation, the cell aggregate morphology was observed and captured with a Leica microscope using the bright field. Total numbers of single cells were counted and cell viability was measured by AO/PI fluorescence assays using a Cell Counter when serial passage of the cell aggregates was performed.
Experiments studying the effects of various rotational speeds and culture medium volumes on cell morphology, cell proliferation, cell viability and cell phenotype were sequentially performed (Fig. 1A). The rotational speeds were first set at 160 rpm and 170 rpm with culture medium volumes of 0.6 mL per well, 0.8 mL per well and 1.0 mL per well. Based on the experimental results, the rotational speed was adjusted to 180 rpm and 190 rpm, while the culture medium volume was changed to 0.4 mL per well, 0.5 mL per well, 0.6 mL per well and 0.7 mL per well.

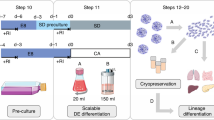
48-well dynamic suspension culture system for pancreatic differentiation from human embryonic stem cells. (A) Flow diagram showing the experimental design of the study. (B) Schematic of the differentiation protocol used for 48-well plate suspension differentiations
Stem Cell-Derived β Cell Differentiation in 48-Well Plates
Differentiation of stem cells into β cells was performed following a protocol described by Douglas A. Melton et al. [14]. The protocol is shown in Fig. 1B and was conducted as follows:
-
Stage 0: H9 cells were dissociated with Accutase and cultured in 0.6 mL mTeSR 1™ medium supplemented with 10 μM Y-27632 in a 48-well plate for 24 h. Then, half the volume of the medium was replaced with fresh mTeSR 1™ medium without Y-27632 daily for another two days. Stage 1: Differentiation was subsequently initiated, and the differentiation phases were divided into six stages (stage 1 ~ stage 6). The period of stage 1 was defined as definitive endoderm (DE) and lasted for three days, and the medium was changed only on the first two days. The basic medium, named S1, was self-configured and supplemented with 100 ng/mL Activin A (R&D Systems, on the first and second day only) and 3 μM Chir 99,021 (Stemgent). The cells in stage 2 were defined as primitive gut tubes (PGTs), derived in S2 medium supplemented with 50 ng/mL KGF (R&D Systems) for three days with the medium changed every other day. Stage 3 occurred on days 7 and 8. The cells were defined as pancreatic progenitor 1 (PP1) cells and maintained in S3 medium with 50 ng/mL KGF, 0.25 μM Sant1, 2 μM Retinoic acid (RA) (Sigma-Aldrich), 200 nM LDN 193189 (Sigma-Aldrich) (day 7 only) and 500 nM PdBu (R&D Systems) daily. The cells in stage 4 were defined as pancreatic progenitor 2 (PP2) cells and were cultured in S3 medium with 50 ng/mL KGF, 0.25 μM Sant1 (Sigma-Aldrich), 100 nM Retinoic acid (Sigma-Aldrich), 10 μM Y27632 and 5 ng/ml Activin A for 5 days, and the medium was changed every other day. The cells in stage 5 were defined as pancreatic endocrine (EN) cells and were maintained in S5 medium with 0.25 μM Sant1, 100 nM RA, 1 μM XXI (EMD Millipore), 10 μM Alk5i II (Sigma-Aldrich), 1 μM T3 (EMD Millipore) and 20 ng/mL betacellulin (Sigma-Aldrich) for 7 days. In addition, the medium was changed every other day. The last stage, defined as SC-β, required S3 medium to support cell cultivation, which was changed every other day.
The optimization parameter of the differentiation system in a 48-well plate was the undifferentiated cell density, set as 0.0545 × 106 cells per well (9 × 104 cells/mL), 0.109 × 106 cells per well (1.8 × 105 cells/mL) and 0.327 × 106 cells per well (5.4 × 105 cells/mL).
To evaluate whether the differentiation system in a 48-well plate was successfully established, H9 cells were seeded at a density of 0.327 × 106 cells per well to complete the direct differentiation process.
In Vitro Glucose-Stimulated Insulin Secretion
In vitro pancreatic β like cells function was assessed by measuring in vitro glucose-stimulated insulin secretion [14]. Clusters were pre-incubated for 1 h in Krebs–Ringer buffer (KRB) containing 2 mM glucose, followed by incubation for 1 h in KRB containing 2 mM glucose (low glucose) followed by 1 h incubation in KRB containing 20 mM glucose (high glucose) and 30 mM KCl. Human insulin levels were quantified using an ultrasensitive Insulin ELISA kit (ALPCO Diagnostics) and were normalized by viable cell counts that were acquired by dispersing clusters with TrypLE Expression (Thermo fisher Scientific) and counted using a cell counter.
Flow Cytometry
For analysing surface and intracellular pluripotency markers, Human and Mouse Pluripotent Stem Cell Analysis Kit (BD Biosciences, 560,477) was used according to the manufacturer’s protocol.
For differentiation-associated markers, cell aggregates in the culture medium were collected by a 37 μm filter and dissociated into single cells with TrypLE™ Express (1X), fixed, permeabilized and stained for various intracellular markers [25]. Antibodies used for flow-cytometry were listed in Supplementary Table 1.
Immunofluorescence Staining
For immunofluorescence staining of H9 cell aggregates, the cell aggregates in 48-well plate were processed using a Pluripotent Stem Cell 4-Marker Immunocytochemistry Kit (Thermo Fisher Scientific, A25526) according to the manufacturer’s instructions. For differentiation-associated markers, antibodies used for immunocytochemistry were listed in Supplementary Table 2. Fluorescence images were captured by confocal laser-scanning microscopy (Leica TCS SP8).
Quantitative RT-PCR
Total RNA was extracted from H9 cells cultured in the 48-well plate and reverse transcribed into cDNA using a FastQuant RT Kit (TIANGEN). The diluted cDNA was used to further analyze the gene expression of pluripotency markers-Nanog, Sox2, Lin28 and Rex1 using QuantiFast SYBR Green RT-PCR Kit (Qiagen). Primer sequences were listed in Supplementary Table 2.
Teratoma Formation Assay
Human H9 cells cultured in the 48-well plate were collected after digested using Accutase. After resuspended with 30% Corning Matrigel Matrix, 3 million H9 cells were injected intramuscularly into the hind limb of NOD/SCID mice (GemPharmatech Co., Ltd.). Teratoma formation was monitored for 7 weeks. Recovered tumors were sectioned and stained with hematoxylin/eosin for further analysis.
Karyotyping
H9 cells cultured in the 48-well plate were seeded in a T25 flask coated with Matrigel. When the H9 cells reached 30% confluence in the T25 flasks, they were transferred to Cell Inspire Bio. Inc. for karyotyping analysis. H9 cells were treated with 100 μg/ml colchicine for 10 min, and the metaphase chromosomes were analyzed after G-banding stain. At least 20 metaphase spreads were counted.
Statistics
The data are presented as the mean ± SD of at least three independent experiments. Statistical significance was assessed using Student’s t test using GraphPad Prism software. Two-tailed p values <0.05 were considered significant.
Results
Establishment of the 48-Well Dynamic Suspension Culture System
In a suspension culture system, it is important for stem cells to form aggregates and maintain spherical morphology. To optimize the protocol for H9 cell suspensions culture in 48-well plates, the effects of rotational speed and culture medium volume were studied.
Rotational speeds of 160 rpm and 170 rpm were explored, with culture medium volumes of 0.6 mL per well, 0.8 mL per well and 1.0 mL per well (cell seeding density: 5.4 × 105 cells/mL). The morphologies of cell aggregates under different culture conditions are presented in Supplementary Fig. 1. At the same rotational speed, H9 cells in larger culture medium volumes formed larger cell aggregates, resulting in poorer mass transfer efficiency. H9 cells formed more homogenous spheres at 170 rpm than at 160 rpm. And, we further adjusted the culture medium volume to 0.4 mL per well, 0.5 mL per well, 0.6 mL per well and 0.7 mL per well, and set the rotational speed to 180 rpm and 190 rpm. As shown in Supplementary Fig. 1, the cells in the suspension culture system tended to form ellipsoidal clusters with various diameters at 190 rpm regardless of the culture medium volume. However, when H9 cells were cultured at a rotational speed of 180 rpm, the results revealed that most of the single cells were formed into homogenous spheres, especially at a volume of 0.6 mL per well. Even after culture for three passages, the H9 cell aggregates maintained their morphology with an average diameter range of 171.9 μm to 234.8 μm (Fig. 2A and D). We assessed the expansion ability of H9 cells in 48-well plates. As shown in Fig. 2B and C, the cell numbers increased over 2-fold in the first two passages, with cell viability over 95%. The fold change shown in Fig. 2C was the value that divided the number of cells harvested by the number of cells inoculated. And, at the third passage, cell proliferation rate slightly decreased compared with that at the first passage. Flow cytometry was applied to analyse the expression of surface markers OCT4 and SSEA4, as shown in Fig. 2E, which was used to evaluate the pluripotency of H9 cells. Comparing the expression levels of OCT4 and SSEA4 in the first passage with the third passage, as shown in Fig. 2F, there was no significant difference, revealing that the pluripotency of the H9 cells was successfully maintained in the established 48-well plate suspension culture system. H9 cells cultured in the 48-well plate expressed pluripotency markers of SOX2, TRA-1-60, SSEA4 and Oct4 (Fig. 2G). qPCR results revealed that H9 cells cultured in the 48-well plate suspension system at passage 3 had a higher or equivalent expression of pluripotency markers Sox2, Nanog, Rex1 and Lin28 compared to H9 cells cultured in the adherent plates (Fig. 2H). HE staining results showed that H9 cells cultured in the 48-well plate give rise to classical teratomas with ectodermal, mesodermal and endodermal features in NOD/SCID mice (Fig. 2I), indicating that H9 cells cultured in the 48-well plate possessed pluripotency. Moreover, H9 cell cultured in the 48-well maintained a normal karyotype as revealed by G-banding karyotyping analysis (Supplementary Fig. 2). These results demonstrated that a small-scale suspension culture system with 48-well plates for hESCs expansion was established.

Optimization of H9 cell suspension culture in 48-well plates. (A) Effect of culture volume on H9 cell cluster morphology when the rotational speed was set as 180 rpm. (B) Cell viability of H9 cell clusters at each cell passage, for which suspensions were cultured in 0.6 mL culture medium and maintained at 180 rpm rotational speed. (C) Fold change in cell number (n = 3). The results are expressed as fold-change relative to the initial cell numbers. (D) Particle size (n = 50) of H9 cell clusters at each passage. (E) Representative results of flow cytometry analyses for SSEA4 and OCT4 of H9 cell clusters at passages P0 and P3. (F) Comparison of the expression ratio of OCT4 and SSEA4 cells at passages P0 and P3 by flow cytometry analysis. (G) Representative images of immunofluorescence staining of SOX2, TRA-1-60, SSEA4 and OCT4 of H9 cells cultured in 48-well plate at passage 3. (H) Comparison of the expression ratio of pluripotency markers (Sox2, Nanog, Rex1 and Lin28) in H9 cells at passages P0 and P3 by qPCR. (I) Representative histology of teratomas formed from H9 cells culture in 48-well plates. The images present histology consistent with endoderm (glandular epithelium), mesoderm (cartilage) or ectoderm (melanin pigmentation)
Effect of Cell Density on the Pancreatic Differentiation Efficiency from H9 Cells in 48-Well Plates
Methods for generating pluripotent stem cell-derived pancreatic β-like cells in vitro have been published, but a method for the derivation of β-like cells in 48-well plates has not been developed. Based on the established suspension culture system in 48-well plates, we intended to develop the method to differentiate hESCs into β-like cells in 48-well plates. The diameter of cell aggregates affects mass transfer efficiency and therefore influences differentiation efficiency [26]. Hence, H9 cells at three different densities were initially adopted in this study, including 9 × 104 cells/mL, 1.8 × 105 cells/mL and 5.4 × 105 cells/mL, respectively. Figure 3A shows the morphologies of cells cultured at various cell densities at the end of stage 0, and the result suggested that the sizes of cell aggregates positively correlated with cell density. The ratio of OCT4 and SSEA4 double positive cells in the three experimental groups was over 98% (Fig. 3B), indicating that the pluripotency of H9 cells was successfully maintained at stage 0; thus, all the cell aggregates of different concentrations were proceeded for further differentiation. At stage 1 of the differentiation process, H9 cells were required to differentiate into definitive endoderm with a ratio of SOX17+ cells greater than 95% [27]. As shown in Fig. 3D, cells cultured at highest density (5.4 × 105 cells/mL) achieved this goal, whereas cells cultured at the density of 9 × 104 cells/mL and 1.8 × 105 cells/mL did not. The reason for the results may be that ESC aggregates that are too small may result in less efficient cell expansion and differentiation, which is consistent with the views reported in the literature [28]. Furthermore, qPCR results revealed that the pluripotency of H9 cells and Sox17 of S1 cells, respectively, was expressed at relatively high levels in H9 cells initially seeded at highest density (Fig. 3E and F).

Optimization of H9 cell differentiation in 48-well plates. (A) Aggregates were formed in 48-well suspension culture plates at various seeding densities while the rotational speed was set as 180 rpm. (B) Representative results of flow cytometry analyses for SSEA4 and OCT4 of H9 cell clusters at various seeding densities. (C) Morphology of various seeding densities of differentiated aggregates at stage 1. (D) Representative results of flow cytometry analyses for SOX17 and FOXA2 of differentiated aggregates at stage 1. (E) Comparison of the expression ratio of pluripotency markers (Sox2, Nanog, Rex1 and Lin28) in H9 cells at various seeding densities by qPCR. (F) Comparison of the gene expression of Sox17 at various seeding densities by qPCR
Establishment of the Optimal Pancreatic Differentiation from H9 Cells in 48-Well Plates
The effect of cell density on the differentiation efficiency of H9 cells in 48-well plates was initially studied, and the results showed that a cell density of 5.4 × 105 cells/mL may be suitable for the differentiation of stem cells into β-like cells. Thus, we chose a cell density of 5.4 × 105 cells/mL to complete the differentiation process of H9 cells in 48-well plates.
Before starting the differentiation experiment, the pluripotency of H9 cells was analysed, and Fig. 4A shows that the batch of H9 cells met the requirement for further derivation (>95% OCT4+ cells, >95%SSEA4+ cells). At the end of each differentiation stage, cells were collected and dissociated into single cells to analyse the key surface markers by flow cytometry. Figure 4B shows that >95% of cells were SOX17 positive, indicating that the H9 cells successfully differentiated into definitive endoderm. The expression level of SOX17 was dramatically reduced in stage 2, which was defined as primitive gut tube [11]. As the protocol demonstrated, on day 2 of stage 3, most cells were PDX1+, indicating that the cells had differentiated into PDX1+ pancreatic progenitors. By day 5 of stage 4, the expression of NKX6.1 had rapidly increased, and the expression of PDX1 was maintained at a high level, demonstrating that the cells had differentiated into PDX1+/NKX6.1+ pancreatic progenitors. As shown in Fig. 4C-D, the expression levels of PDX1 and NKX6.1 in stage 3 and stage 4 remained consistent with the protocol [11], indicating the successful differentiation of the stem cells into cells of pancreatic lineage. By stage 5, the cells had differentiated into EN cells, and by stage 6, into functional beta-like cells expressing the NKX6.1 and C-peptide markers. The population of NKX6.1+/C-peptide+ cells were slightly increased in stage 6 compared with stage 5. Ultimately, the static glucose-stimulated insulin secretion was carried out to test the function of SC-derived β-like cells at stage 6. In addition, we performed immunofluorescent staining of C-peptide with NKX6.1 and C-peptide with GCG on S6 cells to further characterize our 48-well dynamic suspension culture system (see newly added data in Fig. 4E and F). As shown in Fig. 4E, most cells in the clusters showed C-peptide staining in the cytoplasm and were also positive for NKX6.1 in the nucleus, with few cells expressing the α-cell hormone glucagon. These results demonstrate that H9 cells are capable of differentiating to SC-β cells that co-express C-peptide/NKX6–1. We observed that SC-derived β-like cells could display the physiological function in vitro of secreting insulin in response to glucose (Fig. 4G) and their morphologies in Fig. 4H showed preferably homogeneity. These findings indicated that our 48-well dynamic suspension culture system was suitable for pancreatic differentiation from human embryonic stem cells.

Representative results of flow cytometry analyses of H9 cell differentiation in 48-well plates. (A) Summary of flow cytometry analyses for SSEA4 and OCT4 of H9 cell clusters. (B) Summary of flow cytometry analyses for SOX17 and FOXA2 of differentiated aggregates at stage 1 and stage 2. (C) Summary of flow cytometry analyses for PDX1 and NKX6.1 of differentiated aggregates at stage 3 and stage 4. (D) Summary of flow cytometry analyses for PDX1 and NKX6.1 of differentiated aggregates at stage 5 and stage 6. (E) Immunofluorescent staining for C-peptide (green) with NKX6.1 (red) on S6 cells. (F) Immunofluorescent staining for C-peptide (green) with GCG (red) on S6 cells. Scale bar, 50 μm. (G) Insulin secretion in response to glucose and KCl challenges on differentiated aggregates at stage 6. (H) Morphologies of differentiated aggregates at stage 6
Discussion
Pluripotent stem cell-based cell therapy is an extremely promising approach for curing diabetes [29]. Despite significant recent advances in the differentiation of hPSCs into pancreatic cells, the challenge remains to obtain functional human beta cells, which have robust physiologic function [30, 31]. Thus, current protocols need to be optimized to consistent generation of pure populations of fully functional beta cells by screening and identifying novel compounds or cytokines. And, a rapid and efficient screening system for pancreatic differentiation from human embryonic stem cells is urgent. In this study, we present a 48-well culture system of dynamic suspension culture and direct pancreatic differentiation from human embryonic stem cells, which may provide an efficient platform for screening novel compounds or cytokines for pancreatic differentiation. As far as I know, this is the first report of a 48-well suspension culture system for hESC maintenance and pancreatic differentiation.
For 48-well suspension culture system for hESCs, our study reveals that hESCs could be maintained and consistently expanded in 48-well culture system while retaining normal karyotype, appropriate marker expression, and pluripotency. We also find that the 48-well dynamic microplate system was sensitive to rotational speed and culture medium volume (Fig. 2A and Supplementary Fig. 1). As shown, within a certain range, the diameter of cell clusters was negatively correlated with the rotational speed but positively correlated with the culture medium volume, which may fundamentally be attributed to the alteration of shear stress [23, 32, 33]. In addition, cell proliferation analysis and measurement of the average diameter of cell aggregates (Fig. 2C and D), indicated that the culture time at each passage should be flexibly adjusted according to the diameter of cell aggregates, and a range of 200 μm to 300 μm in diameter is preferable [18].
In regard to the optimization of the differentiation system, we find that cells cultured at the lowest cell density failed to reach the target of >95% SOX17+ cells in stage 1 (Fig. 3D). It has been reported that the size of hPSC aggregates has a strong impact on the differentiation efficiency [34]. By choosing the appropriate cell density, we finally obtained stem cell-derived β-like cells expressing NKX6.1+/C-peptide+, which display the physiological function in vitro of secreting insulin in response to glucose. It would be preferable to carry out further functional assessment of stem cell-derived β-like cells in vivo to strengthen the conclusion. However, due to the SC-β cells yields per well, we are facing a lot of difficulties in securing a sufficient number of samples for in vivo assay. Typical yields of current system were approximately (2–3) × 104 cells per well when starting with ~3.24 × 105 H9 cells (5.4 × 105 cells/mL, 0.6 mL), which was consistent with previous reports cultured and differentiated in the spinner flask [21]. For in vivo functional assay, at least 5 mice are required for each group. And, a mouse requires about 5 × 106 transplanted SC-β cells [21, 24]. It has been calculated that this will require 833–1250 wells of SC-β cells to meet the demand. The amount of time for a difficult task seems too overwhelming.
Owing to its mini-system, the dynamic suspension culture and differentiation system in 48-well plates is suitable for screening novel compounds or cytokines for pancreatic differentiation that regulate stem cell fate, which could be applied toward systematically optimizing production strategies for cell replacement therapies to accelerate the pace of their discovery and development toward the clinic. Moreover, in combination with high-content techniques, we could track the process of differentiation of stem cells in microplates, which may help to advance the understanding of corresponding diseases.
Conclusion
We report a controllable 48-well suspension culture system for hESCs maintenance, expansion and pancreatic differentiation, which may provide an efficient platform for high-throughput drug screening using pancreatic β like cells.
Data Availability
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.
Code Availability
Nothing to declare.
References
Yamanaka, S. (2020). Pluripotent stem cell-based cell therapy- promise and challenges. Cell Stem Cell, 27(4), 523–531. https://doi.org/10.1016/j.stem.2020.09.014
Robinton, D. A., & Daley, G. Q. (2012). The promise of induced pluripotent stem cells in research and therapy. Nature, 481(7381), 295–305. https://doi.org/10.1038/nature10761
Shi, Y., Inoue, H., Wu, J. C., & Yamanaka, S. (2017). Induced pluripotent stem cell technology: A decade of progress. Nature Reviews. Drug Discovery, 16(2), 115–130. https://doi.org/10.1038/nrd.2016.245
da Cruz, L., Fynes, K., Georgiadis, O., et al. (2018). Phase 1 clinical study of an embryonic stem cell-derived retinal pigment epithelium patch in age-related macular degeneration. Nature Biotechnology, 36(4), 1-+. https://doi.org/10.1038/nbt.4114
Song, W. K., Park, K. M., Kim, H. J., et al. (2015). Treatment of macular degeneration using embryonic stem cell-derived retinal pigment epithelium: Preliminary results in Asian patients. Stem Cell Reports, 4(5), 860–872. https://doi.org/10.1016/j.stemcr.2015.04.005
Bloor, A. J. C., Patel, A., Griffin, J. E., et al. (2020). Production, safety and efficacy of iPSC-derived mesenchymal stromal cells in acute steroid-resistant graft versus host disease: A phase I, multicenter, open-label, dose-escalation study. Nature Medicine, 26(11), 1720-+. https://doi.org/10.1038/s41591-020-1050-x
Henry, R. R., Pettus, J., Wilensky, J., et al. (2018). Initial clinical evaluation of VC-01TM combination product-a stem cell-derived islet replacement for type 1 diabetes (T1D). Diabetes, 67. https://doi.org/10.2337/db18-138-OR
Ben-David, U., Gan, Q. F., Golan-Lev, T., et al. (2013). Selective elimination of human pluripotent stem cells by an Oleate synthesis inhibitor discovered in a high-throughput screen. Cell Stem Cell, 12(2), 167–179. https://doi.org/10.1016/j.stem.2012.11.015
Sampaziotis, F., de Brito, M. C., Madrigal, P., et al. (2015). Cholangiocytes derived from human induced pluripotent stem cells for disease modeling and drug validation. Nature Biotechnology, 33(8), 845–852. https://doi.org/10.1038/nbt.3275
Scudellari, M. (2016). How iPS cells changed the world. Nature, 534(7607), 310–312. https://doi.org/10.1038/534310a
Sharma, R., Khristov, V., Rising, A., et al. (2019). Clinical-grade stem cell-derived retinal pigment epithelium patch rescues retinal degeneration in rodents and pigs. Science Translational Medicine, 11(475). https://doi.org/10.1126/scitranslmed.aat5580
Avior, Y., Sagi, I., & Benvenisty, N. (2016). Pluripotent stem cells in disease modelling and drug discovery. Nature Reviews. Molecular Cell Biology, 17(3), 170–182. https://doi.org/10.1038/nrm.2015.27
Aigha, I. I., Memon, B., Elsayed, A. K., & Abdelalim, E. M. (2018). Differentiation of human pluripotent stem cells into two distinct NKX6.1 populations of pancreatic progenitors. Stem Cell Research & Therapy, 9. https://doi.org/10.1186/s13287-018-0834-0
Millman, J. R., Xie, C. H., Van Dervort, A., Gurtler, M., Pagliuca, F. W., & Melton, D. A. (2016). Generation of stem cell-derived beta-cells from patients with type 1 diabetes. Nature Communications, 7. https://doi.org/10.1038/ncomms11463
Rigamonti, A., Repetti, G. G., Sun, C. C., et al. (2016). Large-scale production of mature neurons from human pluripotent stem cells in a three-dimensional suspension culture system. Stem Cell Reports, 6(6), 993–1008. https://doi.org/10.1016/j.stemcr.2016.05.010
Borys, B. S., So, T., Colter, J., et al. (2020). Optimized serial expansion of human induced pluripotent stem cells using low-density inoculation to generate clinically relevant quantities in vertical-wheel bioreactors. Stem Cells Translational Medicine, 9(9), 1036–1052. https://doi.org/10.1002/sctm.19-0406
Keller, K. C., Rodrigues, B., & zur Nieden, N. I. (2014). Suspension culture of pluripotent stem cells: Effect of shear on stem cell fate. Critical Reviews in Eukaryotic Gene Expression, 24(1), 1–13.
Chen, V. C., & Couture, L. A. (2015). The suspension culture of undifferentiated human pluripotent stem cells using spinner flasks. Methods in Molecular Biology (Clifton, NJ), 1283, 13–21. https://doi.org/10.1007/7651_2014_118
Tsai, A. C., & Ma, T. (2016). Expansion of human mesenchymal stem cells in a microcarrier bioreactor. Methods in Molecular Biology (Clifton, NJ), 1502, 77–86. https://doi.org/10.1007/7651_2016_338
Li, Y. T., Liu, D. W., Liu, D. M., et al. (2012). Experimental study on the differentiation of human induced pluripotent stem cells into epidermal-like stem cells. Zhonghua shao shang za zhi Zhonghua shaoshang zazhi Chinese Journal of Burns, 28(4), 274–277.
Veres, A., Faust, A. L., Bushnell, H. L., et al. (2019). Charting cellular identity during human in vitro β-cell differentiation. Nature, 569(7756), 368–373. https://doi.org/10.1038/s41586-019-1168-5
Papadopoulos, A., Chalmantzi, V., Mikhaylichenko, O., et al. (2021). Combined transcriptomic and phosphoproteomic analysis of BMP4 signaling in human embryonic stem cells. Stem Cell Research, 50. https://doi.org/10.1016/j.scr.2020.102133
Chen, V. C., Couture, S. M., Ye, J. J., et al. (2012). Scalable GMP compliant suspension culture system for human ES cells. Stem Cell Research, 8(3), 388–402. https://doi.org/10.1016/j.scr.2012.02.001
Pagliuca, F. W., Millman, J. R., Gurtler, M., et al. (2014). Generation of functional human pancreatic beta cells in vitro. Cell, 159(2), 428–439. https://doi.org/10.1016/j.cell.2014.09.040
Rezania, A., Bruin, J. E., Arora, P., et al. (2014). Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nature Biotechnology, 32(11), 1121–1133. https://doi.org/10.1038/nbt.3033
Lee, B., Borys, B. S., Kallos, M. S., Rodrigues, C. A. V., Silva, T. P., & Cabral, J. M. S. (2020). Challenges and solutions for commercial scale manufacturing of allogeneic pluripotent stem cell products. Bioengineering (Basel), 7(2). https://doi.org/10.3390/bioengineering7020031
Pagliuca, F. W., Millman, J. R., Gürtler, M., et al. (2014). Generation of functional human pancreatic β cells in vitro. Cell, 159(2), 428–439. https://doi.org/10.1016/j.cell.2014.09.040
Nogueira, D. E. S., Rodrigues, C. A. V., Hashimura, Y., Jung, S., Lee, B., & Cabral, J. M. S. (2021). Suspension culture of human induced pluripotent stem cells in single-use vertical-wheel™ bioreactors using aggregate and microcarrier culture systems. Methods in Molecular Biology, 2286, 167–178. https://doi.org/10.1007/7651_2020_287
Brusko, T. M., Russ, H. A., & Stabler, C. L. (2021). (2021). Strategies for durable β cell replacement in type 1 diabetes. Science., 373(6554), 516–522. https://doi.org/10.1126/science.abh1657
Jacobson, E. F., & Tzanakakis, E. S. (2017). Human pluripotent stem cell differentiation to functional pancreatic cells for diabetes therapies: Innovations, challenges and future directions. Journal of Biological Engineering, 11, 21. https://doi.org/10.1186/s13036-017-0066-3
Odorico, J., Markmann, J., Melton, D., et al. (2018). Report of the key opinion leaders meeting on stem cell-derived Beta cells. Transplantation, 102(8), 1223–1229. https://doi.org/10.1097/TP.0000000000002217
DiStefano, T., Chen, H. Y., Panebianco, C., et al. (2018). Accelerated and improved differentiation of retinal organoids from pluripotent stem cells in Rotating-Wall vessel bioreactors. Stem Cell Reports, 10(1), 300–313. https://doi.org/10.1016/j.stemcr.2017.11.001
Fridley, K. M., Kinney, M. A., & McDevitt, T. C. (2012). Hydrodynamic modulation of pluripotent stem cells. Stem Cell Research & Therapy, 3(6), 45. https://doi.org/10.1186/scrt136
Bauwens, C. L., Song, H., Thavandiran, N., et al. (2011). Geometric control of cardiomyogenic induction in human pluripotent stem cells. Tissue Engineering. Part A, 17(15–16), 1901–1909. https://doi.org/10.1089/ten.TEA.2010.0563
Acknowledgements
The authors thank Dr. Jiayin Yang (Cell Inspire Biotechnology Co., Ltd., Shenzhen, China) for helping revising the manuscript. The authors also gratefully thank the support from Analysis and Testing Center of Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences.
Funding
This project was supported by the National Key R&D Program 2019YFE0117700 and National Natural Science Foundation of China (31771068).
Author information
Authors and Affiliations
Contributions
Yizhe Song, Jing Liu, Wenjia Li, Yinghua Chen, and Xiuli Wang designed and coordinated the study; Yizhe Song, Xiaoqian Chen, Decan Liang, Jingqiu Li, Zhensheng Ou, Ting-ting Tang, and Qunrui Ye performed the experiments, acquired and analysed data; Yizhe Song, Peiwen Xing, Leilei Guo, and Shidu Zhang interpreted the data; Yizhe Song, Xiaoqian Chen, Yinghua Chen, and Xiuli Wang wrote the manuscript; all authors approved the final version of the article.
Corresponding authors
Ethics declarations
Consent for Publication
The participants have consented to the submission of the report to the journal.
Competing Interests
The authors declare that they have no competing financial interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Yizhe Song, Xiaoqian Chen and Decan Liang are Co-first authors.
Supplementary Information
ESM 1
(DOCX 450 kb)
Rights and permissions
About this article

Cite this article
Song, Y., Chen, X., Liang, D. et al. Development of a 48-Well Dynamic Suspension Culture System for Pancreatic Differentiation from Human Embryonic Stem Cells. Stem Cell Rev and Rep 18, 1423–1433 (2022). https://doi.org/10.1007/s12015-021-10312-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12015-021-10312-w