
Abstract
Umbilical cord blood (CB) transplantation has been used successfully in humans for three decades due to its rapid availability for patients lacking a suitable allogeneic donor, less stringent HLA matching requirements, and low rates of relapse and chronic graft-versus-host disease (GVHD). However, CB transplantation is associated with complications, such as delayed hematopoietic engraftment, graft failure, which increases infection and bleeding and causes longer hospital stays, and transplant-related mortality. The majority of these biological limitations are due to the unforeseeable functional potency of multipotent hematopoietic stem cells (HSCs), which reduce the predictability of successful transplantation; however, several strategies have been developed to increase the number of hematopoietic stem progenitor cells (HSPCs) infused during CB transplantation. This review primarily addresses the methods that promote ex vivo CB expansion within the context of symmetrical and asymmetrical HSC division and those that rely on epigenetic mechanisms, along with the reportedly most successful cytokine combinations. We also review recent clinical research on small molecules (StemRegenin-1, UM171, and nicotinamide) in ex vivo expanded CB and discuss yet unvalidated preclinical strategies. Expanding and transplanting CB graft enriched in HSPCs in a single CB unit is a particularly exciting prospect with the potential to improve the use and availability of CB grafts. Greater knowledge of optimal ex vivo expansion strategies, cell longevity, and graft potency will expand the scope of cellular therapies. Also the development of adequate ex vivo HSPC expansion strategies could bring expanded cord blood grafts to the forefront of transplant therapy and regenerative medicine.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Umbilical cord blood (CB) transplants have been performed successfully since 1989 and provide an alternative graft source in many patients for whom a matched donor is not available [1, 2]. CB grafts offer many advantages, including a less stringent tissue matching requirement, a low rate of graft-versus-host disease (GVHD), and ready availability. The main disadvantage of CB transplants is the protracted time to full blood and immune cell reconstitution, which leaves the recipient vulnerable to infection and bleeding. It is believed that this is a consequence of the relatively low absolute numbers of hematopoietic stem progenitor cells (HSPCs) infused [3]; however, this issue can be overcome by increasing the infused cell dose present in a single CB unit by instead transplanting two CB units. Although this has been relatively successful in reducing the time taken to achieve blood cell reconstitution following double-unit CB transplantation, the cost of transplanting two CB units is prohibitive for many institutions [4].
An alternative strategy is the ex vivo expansion of one CB unit to generate sufficient HSPCs [5, 6], with a retrospective review showing no difference in mortality between one and two CB unit recipients [7]. Initial attempts have included the use of a cocktail containing human recombinant cytokines, such as thrombopoietin (TPO), fetal liver tyrosine 3 ligand (Flt3L), granulocyte colony-stimulating factor (G-CSF), stem cell factor (SCF), interleukin 3 (IL-3), megakaryocyte growth and development factor (MGDF), erythropoietin (EPO), and interleukin 6 (IL-6) to expand CB-derived HSPCs in culture [8], with additional small molecules and feeder cell layers included at a later stage. Several phase I/II clinical trials have been conducted [9] with varying degrees of success (Table 1). However, the latest iterations of CB expansion, including copper chelation tetraethylenepentamine (TEPA) [10], Notch ligand [11], StemRegenin-1 (SR-1) [12], UM171, a pyrimido indole derivative that likely promotes hematopoietic stem cell (HSC) self-renewal in vitro [13], and nicotinamide (NAM; NiCord) [14] have shown promising results and are currently being evaluated in phase I/ II clinical trials (Table 1) [15,16,17].
To date, none of these approaches have been thoroughly evaluated in a randomized trial comparing double CB transplantation versus one expanded and one unexpanded CB unit or the transplantation of a single expanded CB unit alone. A phase I/II clinical trial has been conducted for the copper chelator TEPA (StemEx), which attenuates the differentiation of ex vivo cultured HSPCs resulting in the preferential expansion of early progenitors and hematopoietic engraftment; however, the protocol has not yet been optimized for clinical implementation [10]. Here, we discuss recent progress and encouraging results from the transplantation of expanded CB grafts using small molecules, including SR-1, NiCord, and UM171, which have all shown early clinical success.
Expansion Mechanisms
To date, the discovery of ex vivo HSPC expansion using small molecules termed next-generation molecules (NGMs), including notch ligand, TEPA, SR-1, NAM, and UM171, has been either serendipitous or the result of extensive chemical screenings rather than the product of knowledge regarding the processes that govern the decision between maintaining multipotency and differentiation in hematopoietic progenitor cells (HPC). Several studies have reported that epigenetic modification by chromatin-modifying agents (CMAs) promotes HSC expansion, likely by promoting symmetrical HSC self-renewal divisions [18]. Recently, it has been proposed that relatively mature HPC can be reverted to more primitive HSCs via mitochondrial network reprogramming by epigenetic modifiers, although this requires further validation [19]. Thus, for this review article HSPC is considered as a population of cells consisting of both primitive multipotent HSCs that possess self-renewing ability and relatively mature HPCs that lack self-renewal capacity. The holy grail of CB expansion strategies is to achieve symmetric HSC division in culture as the primary mechanism of expansion (Fig. 1) since asymmetric division will only maintain a multipotent HSC pool under the best-case scenario without rendering true expansion. In contrast, symmetric differentiating division gives rise to two daughter cells that lack HSC properties as they commit to become progenitors and lose their self-renewal ability. As shown in Fig. 1, although asymmetric HSC division only maintains HSC numbers, the newly generated relatively mature progenitors HPCs can serve to repopulate blood cells to shorten the neutropenic period immediately after transplant. Thus the primary goal of CB expansion is twofold, namely to reduce the time taken to achieve blood cell reconstitution following transplantation, and ensuring sustained long-term trilineage hematopoiesis. Preclinical models indicate that the recovery of blood cells immediately after transplantation is repopulated primarily by short term HPCs while sustenance of trilineage hematopoiesis is the contribution of long-term multipotent HSCs that have the ability to self-renew. While long-term HSCs drive sustained lifelong hematopoiesis, these cells are relatively quiescent, hence resulting in a prolonged period of leukopenia. The period immediately after transplantation is primarily repopulated by relatively mature progenitors (HPC). Ex vivo expansion strategies resulting in expansion of HSPCs would, in theory, provide progenitors the capability of giving rise to blood cells immediately after transplantation as well as provide long-term blood cell production for the host. Various strategies have been employed to either augment HSPC numbers or increase the ability of infused HPC progenitors to repopulate the bone marrow (BM) niche via manipulation (Table 1). The foremost ex vivo CB expansion strategies include: (1) HSPC expansion with cytokines including SCF, TPO, and Flt-3 L in liquid culture; (2) mesenchymal stromal cell (MSC) co-culture; (3) chemical molecules such as TEPA (copper chelator), NAM (sirtuin 1 inhibitor), SR1 (aryl hydrocarbon receptor antagonist), immobilized delta-1 (Notch ligand), and bioreactors that allow continuous perfusion culture [9]; and (4) improving BM homing following intravenous graft infusion, intra-bone (i.b.) CB graft infusion, or CB co-infusion with mesenchymal stromal cells [20].

Fate of hematopoietic stem cell (HSC) division: HSC expansion, HSC maintenance, and HSC extinction
Enhanced CB graft homing following transplantation has been attempted more recently using pharmacological strategies such as CD26/Dipeptidylpeptidase-4 (DPP-4) inhibition with sitagliptin [21]. In a phase I clinical trial, DPP-4 inhibition showed only modest benefits in terms of the speed of hematopoietic engraftment (day 21, comparable with the controls at day 19) [21]. CD34 + cells possess complement C3a receptors that allow calcium influx into the cells to modulate SDF-1-dependent chemotaxis. This system has been exploited by using complement C3a priming to expand HSPCs [22], with the transplantation of C3a-primed CB alongside a second unmanipulated CB unit resulting in a median time to neutrophil engraftment of seven days (historical control of D12 at the same institution) [23]. It has also been shown that enhancing prostaglandin E2 (PGE2) synthesis in a zebrafish model increases the number of HSPCs [24], while the transplantation of CB cells briefly exposed to PGE2 in a clinical trial reduced the time to neutrophil engraftment by 3.5 days in one of the cohorts [25]; a phase II randomized trial of PGE2-treated CB cell transplantation is also underway. The homing of transplanted HSCs requires interactions between cell surface molecules, including E and P-selectins. Binding to selectins requires glycoprotein fucosylation to form cell surface glycan determinants such as sialyl Lewis X (sLex) [26]. CD34 + CB cells have been shown to display significantly lower alpha 1–3 fucosylation levels [27], which may explain their reduced ability to populate the BM niche. Several strategies have been used to correct this deficit, including exposing CD34 + CB cells to human fucosyl-transferase VI (FTVI), FTVII, or its substrate GDP-fucose to increase fucosylation levels, with preclinical studies demonstrating improved BM homing following CD34 + cell fucosylation [26,27,28]. An ongoing clinical trial at the M.D. Anderson Cancer Center is exploring this strategy further [Clinical trial.gov NCT01471067] (Table 1).
Another common strategy for improving ex vivo HSPC expansion has been the use of different combinations of cytokines and small molecules (Table 2) [29]. For instance, a preclinical study found that the use of CMA, the hypomethylating drug 5-aza-2’ deoxycytidine (5azaD), and then trichostatin a (TSA) in fetal bovine serum (FBS)-containing culture for nine days in the presence of a cytokine cocktail (SCF, TPO, FL, and IL-3), increased the number of severe combined immune deficiency (SCID) repopulating cells (SRC) by 9.6-fold (Table 2) [30]. Various other studies have found that SR1 with cytokines (SCF, TPO, FL, IL-6) increases the number of SRCs by 17-fold after 21 days of culture [31], UNC0638 with cytokines (SCF, TPO, FL, IL-6) yields a 2-fold increase in 14 days [32], NAM with a cytokine cocktail (SCF, TPO, FL, IL-6, and 10% FBS) increases the number of SRCs by 9-fold in 21 days [33], and the small molecule UM171 with a cytokine cocktail (SCF, TPO, FL) yields a 13-fold increase in 12 days [13]. In our laboratory, we are currently investigating the potential role of stress signals (i.e., pro-inflammatory pathways) in promoting HSC expansion [unpublished data]. The ex vivo recapitulation of physiological events that culminate in the generation of multipotent HSPCs may provide reproducible expansion strategies that can optimize CB grafts, ultimately allowing successful and timely blood cell reconstitution following transplantation. The reliable amplification of HSPC precursors may have significant economic impacts and provide potent CB grafts for an increasing number of patients in need.
Epigenetic Modification As an Initiation Event for Promoting HSC Expansion
The process via which HSCs either maintain a relatively primitive multipotent state or adopt a more differentiated state is thought to be regulated by epigenetic modifications. Therefore, several groups have attempted to use CMAs to induce cultured HSCs to retain a multipotent state and generate two daughter HSCs rather than one primitive multipotent HSC and one relatively committed HPC [18]. By doing so, a small number of HSCs could be amplified by self-renewal divisions to facilitate successful transplantation capable of sustaining long-term hematopoiesis. The main problem with these approaches is that they generate numerous relatively mature HPCs rather than primitive HSCs and may not yield true functional, transplantable HSC expansion in culture. Besides, the sequence in which the agents are added in culture and intrinsic cellular determinants may significantly affect the outcome. Since the initial chromatin state is not known when the modification process starts, cell membrane surface markers are used to identify potential HSCs (i.e., CD34, CD90), and the quality of the starting cell pool is only inferred to be equal. Moreover, it is unclear how many replication cycles HSPCs can undergo before triggering senescence and apoptosis (Hayflick’s limit) or whether HSPCs can bypass these crucial cellular transformation checkpoints.
To date, our group and others have performed preclinical studies on several CMAs, including valproic acid (VPA) and 5azaD/TSA [18, 19, 34,35,36,37,38] (Table 2), intending to induce epigenetic reprogramming to either allow symmetric HSC division or allow committed HPCs to return to HSC status. The ability to promote symmetrical HSC division may potentiate the effects of NGMs and improve their in vivo hematopoietic engraftment ability. For instance, it has been shown that CB grafts expanded with NGMs (NAM, UM171, etc.) become detectable as the engrafted CB unit in approximately 40% of recipients when an unmanipulated CB graft is transplanted simultaneously [12, 14]. Since double-unit CB transplantation was performed, it remains unclear whether these molecules promote symmetrical or asymmetrical HSC division; however, a more recent clinical trial transplanted a UM171-expanded CB graft as a single-unit [39], which may indirectly demonstrate that self-renewing HSC divisions occur in culture.
To determine the mechanisms via which CMA-mediated epigenetic modification promotes the expansion of transplantable HSCs in culture [18], we performed global microarray and pathway analyses. Using expanded enriched CD34 + cells from culture, we demonstrated the expansion and maintenance of transplantable HSCs when cultured with 5azaD/TSA and VPA, respectively, compared to culture lacking these NGMs (control), which lacked the ability to repopulate human CB cells following transplantation in a xenogeneic host. Intriguingly, the sequential addition of a hypomethylating drug (5-azaD) followed by a histone deacetylase (HDAC) inhibitor (TSA) transiently activated inflammatory molecules such as S100A8 and leukotriene B4 (LTB4); however, it remains unclear whether this transient activation promotes the symmetric self-renewing division of transplantable HSCs in culture [unpublished observations, [40]. Mitochondrial activity may also be a crucial determinant of HSC fate [41], since low mitochondrial mass, reactive oxygen species (ROS), and functionally immature mitochondria are all hallmarks of self-renewing HSCs [42]. Upon cellular commitment, HSCs switch to mitochondrial oxidative phosphorylation (OXPHOS) to increase the generation of energy required for differentiation [19]. Indeed, it has been postulated that mitochondrial activity is a critical driver of HSC fate [42] and that interactions with the BM milieu may trigger mitochondrial programs linked to the ultimate destiny of HSCs, such as quiescence, self-renewal, and differentiation [42].
Environmental cues are also thought to play a crucial role in controlling HSPC division, with previous studies indicating that interactions between cell-intrinsic factors are likely influenced by epigenetic regulators and environmental factors such as growth factors and cytokines [37]. For instance, Araki et al. demonstrated that environmental cues could augment epigenetic influences on HSCs in culture and that intricate control over the rate of cell division may be a critical determinant of the maintenance of HSC functional potency during ex vivo expansion. The same study also found that the combination of 5azaD/TSA with an optimal cytokine cocktail in culture increases the expression of the adhesion molecule CD62L and diminishes CD26 expression on HSPCs, likely improving BM homing and ultimately the hematopoietic engraftment of the expanded cells in a xenotransplant model. CD62L also has a crucial role in targeting cells to inflamed tissues and may upregulate inflammatory molecules upon HSPC expansion. Previous work in our laboratory revealed that the demethylation of CpG sites in the promoters of individual inflammatory/stress response genes corresponds to increases in the expression of genes encoding a calcium-binding protein (S100A8) and cytochrome P450 (Cyp11A1) in 5azaD/TSA-expanded culture [18]. S100A8 is a known agonist of toll-like receptor 4 (TLR-4), which plays a role in the maintenance and proliferation of endothelial progenitors [43]. In addition, the transcript levels of inflammatory cytokines such as IL-8, TNFα, and Alox 5, which are involved in the production of the inflammatory molecule leukotriene, were also found to be higher in 5azaD/TSA-expanded cells by quantitative polymerase chain reaction (qPCR) [18]. Consistently, the levels of the inflammatory mediators/cytokines leukotriene B4, TNFα, and IL-8 were found to be significantly higher in the conditioned medium of 5azaD/TSA-expanded cells by ELISA [unpublished data, [40]. Notably, CMA-expanded human CB cells displayed a reduced allostimulatory capacity, as determined by mixed lymphocyte culture (MLC), likely due to the reversible inhibition of STAT3-dependent differentiation in these cells [44]. Automated process development for ex vivo expansion of growth factor-mobilized peripheral blood (mPB)-derived autologous CD34 + cells has also been reported [45]. Previous studies from our laboratory have also shown that human BM [35], CB [30, 37, 38], and mPB [34] grafts promote transplantable HSC expansion. Interestingly, CB-derived CD34 + cells exposed to CMA in culture displayed the highest expansion potential, followed by mPB and BM [35]. Although the underlying mechanisms remain unclear, the apparent correlation between expansion potential and cell ontogeny is intriguing.
It would be interesting to determine whether CMA pretreatment followed by the sequential addition of NGMs (SR-1, UM171, or NAM) in culture promotes symmetrical HSC division, thus improving HSC expansion in culture. Although improved blood cell reconstitution kinetics have begun to emerge following expanded single-unit CB transplantation (i.e., UM171, NiCord), only long-term follow-ups in these patients will allow us to ascertain the benefits of ex vivo expansion reliably. Once established, double-unit CB transplantation may be deemed unnecessary, and many patients who do not currently qualify for HSCT will likely be eligible to receive an expanded CB unit.
Combining Multiple Agents Activating Sequential Pathways
The transcriptional analysis of VPA-expanded CD34 + CB cells has revealed the activation of pathways that were not expected to be involved in cell fate choices; for instance, inflammatory mediators are upregulated as a consequence of CMA treatment [18, 40], unpublished observations]. A cascade of events may, therefore, occur after epigenetic activation, indicating that a sequence of events is needed to promote symmetrical HSC division; however, the cascade of events is currently unknown. Our laboratory is currently studying the possible involvement of stress signaling pathway components [unpublished observations] and attempting to position each cascade event in hierarchical order. Determining the sequence of events may allow them to be recapitulated by sequentially activating each pathway to generate the HSPC graft, while the addition of HSPC amplification agents may further improve the yield and engraftment potential of the expanded cells.
Our group has also proposed, and is currently investigating, a ‘Composite Graft’ approach, that involves high numbers of both short-term (ST) - and long-term (LT)-HSCs expanded in culture with VPA and 5azaD/TSA, respectively (Fig. 2). The rationale behind this approach is to bridge the neutropenic period following CB transplantation and ensure that blood cell production is sustained in the long-term [manuscript under preparation]. Therefore, the integration of the composite CB graft strategy with the NGM-expanded CB graft could serve as a powerful strategy for circumventing cell dose limitations. VPA-expanded grafts do not expand LT-HSCs in culture instead maintains its number, as evidenced by lack of a serial transplantability [18]; however, VPA-expanded CB grafts are primarily enriched in ST-HSCs which could serve as a bridge graft when the hematopoietic function needs to be supplemented transiently prior to the endogenous recovery of blood cells from residual HSPCs (i.e., after myeloablative treatments, graft failure, or ionizing radiation exposure). In contrast, using serum-free culture conditions, Chaurasia et al. suggested that VPA results in expansion of HSCs [36]. Whether VPA is capable of promoting symmetric HSC divisions in culture, or rather reverts more committed HPC to HSC to render HSC expansion, has yet to be fully proven. Although the combination of CMA and NGMs has yet to be studied in CD34 + CB cell culture, it would be interesting to determine whether the symmetrical HSC division promoted by CMA can be improved further by the subsequent addition of another HSC division agonist, such as SR-1 or UM171. Similar to CMAs, the presence of prostaglandin E synthase 3 (PTGES3; an inflammatory mediator) in an inactive cytoplasmic complex with aryl hydrocarbon receptor (AhR) can upregulate inflammatory pathway molecules during HSPC expansion [46]. Further studies of the AhR pathway in conjunction with CMAs may elucidate the role of inflammatory molecules in promoting symmetrical and asymmetrical HSC division.

Composite graft. Expansion of short-term HSCs by VPA and long-term HSCs by 5azaD/TSA
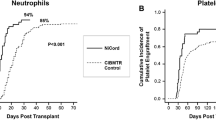
Recent Progress in Ex Vivo CB Expansion
In recent years, significant advancements have been achieved using ex vivo expanded CB grafts [6]. Here, we explore the three main HSPC-promoting agents that have demonstrated benefits in clinical trials.
-
1)
The AhR antagonist SR-1 results in CB expansion
AhR is a ligand-activated basic helix-loop-helix transcription factor that is involved in the biological responses to aromatic (aryl) hydrocarbons and upregulates enzymes (cytochrome P450) involved in xenobiotic detoxification. Non-ligand-bound AhR is retained in the cytoplasm as an inactive protein complex along with a dimer of HSP90, PTGES3, XAP2, AIP, and ARA9 [46], which prevent its inappropriate translocation to the nucleus. A recent phase I/II trial at the University of Minnesota revealed that SR-1 could increase the number of CB CD34 + cells by 330-fold after 15 days in culture, resulting in successful engraftment in 17/17 patients after a median of 15 days for neutrophils and 49 days for platelets [12]. Although these results are encouraging, further follow-up studies are needed to determine the relapse-free survival and long-term utilization of expanded CB grafts, given that early expansion may lead to later exhaustion (12 months follow-up) and graft failure.
A novel product using SR-1, known as MGTA-456 (Magenta Therapeutics, Cambridge, MA), has been developed by combining CD34 + cells expanded by culture with an AhR antagonist (AhRa) for 15 days and the CD34 negative fraction from the same CB unit. An industry-sponsored clinical trial revealed that MGTA-465 promoted faster hematopoietic engraftment than historical controls in 36 patients with hematological malignancies and five patients with non-malignant diseases, with a median follow-up of 2.5 years (range: 0.1-5 years) and 75 days (80–203 days), respectively [47]. The effects of SR-1 in symmetric and asymmetric HSC division or earlier versus later expansion have not yet been evaluated; however, such studies would help to elucidate whether expanded CB cells can sustain long-term hematopoiesis or provide the milieu for the hematopoietic engraftment of a second unmanipulated CB unit.
-
2)
The epigenetic modifier NAM results in CB graft expansion (NiCord)
NAM is a vitamin B3 complex co-factor, also known as niacinamide that acts as a precursor of NAM adenine dinucleotide (NAD+) and potently inhibits enzymes that require NAD for their activities, such as mono-ADP-ribosyltransferases, poly-ADP-ribose polymerases, CD38, and cyclic ADP ribose/NADase. NAM is also a potent inhibitor of the sirtuin family of histone/protein deacetylases and the NAD-dependent class III histone deacetylase (HDAC). SIRT1 catalyzes the deacetylation of acetyl-lysine residues in a reaction that cleaves NAD + and generates NAM, which inhibits SIRT1 in a feedback loop mechanism that blocks NAD + hydrolysis. The genetic deletion of SIRT1 in knock-out mice has been shown to increase HSC proliferation, while the ex vivo treatment of CB CD34 + cells with NAM delays differentiation and enhances migration, homing, and engraftment [48]. The same group also showed that another specific SIRT1 inhibitor mediated the same effects [33]. Based on these preclinical successes, a phase I clinical trial was initiated to test whether a CB-derived cell product consisting of HSPCs expanded with NAM (NiCord) for 21 days and a non-cultured T cell fraction could accelerate hematopoietic recovery and provide long-term hematopoietic engraftment [14]. In another clinical trial, 11 adult patients with hematologic malignancies were recruited from two participating institutions and received a pre-transplant myeloablative-conditioning regimen followed by CB transplantation with NiCord and a second unmanipulated CB unit [14]. NiCord infusion resulted in no detectable adverse events, while complete or partial neutrophil recovery and NiCord-derived T cell engraftment was observed in eight patients and the graft remained stable in all transplanted patients (median follow-up, 21 months). Another clinical trial of 21 patients found that two achieved long-term engraftment with the unmanipulated CB unit while 19 achieved engraftment from the NAM-expanded CB unit [14], in marked contrast to similar previous strategies [11]. Furthermore, patients transplanted with NiCord have been shown to achieve earlier median neutrophil recovery (13 versus 25 days, P < 0.001) than historical controls [14], with one-year overall and progression-free survival rates of 82 and 73%, respectively. In conclusion, rapid blood cell recovery following the NAM-expanded CB transplantation approach is associated with early clinical benefits [48]. Based on these successes, an international multicenter phase I/II clinical trial has been launched utilizing a single NAM-expanded NiCord CB unit [NCT01816230].
-
3)
CB expansion using UM171
A landmark paper demonstrated that the next-generation pyrimidoindole derivative, UM171, which acts independently of AhR and targets cells with a more limited regenerative potential, attenuates cell differentiation and promotes the ex vivo expansion of LT-HSCs [13]. This molecule appears to inhibit transcripts involved in erythroid and megakaryocytic differentiation while most commonly upregulating cell surface molecules. Ex vivo treatment of very small embryonic-like stem cells with UM171 resulted in expansion without compromising their hematopoietic- and endothelial lineage differentiating capacity [49]. An open-label, non-randomized phase I-II clinical trial was conducted in Montreal (Quebec, Canada) to evaluate the use of CB cells expanded with UM171 in ex vivo culture in patients without an available suitable matched donor graft [NCT02668315]. A total of 17 adult patients (out of 25 targeted in the trial NCT02668315) were treated with a myeloablative conditioning regimen prior to transplantation, and CD34 + cells were supplemented with UM171-containing media in a closed culture system until day 0 when the cells were washed and infused. A 36-fold net increase in the absolute number of CD34 + cells was observed alongside relatively rapid neutrophil engraftment (day 10). The clinical trial study (NCT02668315) that was presented as an abstract was recently published showing results from UM171-expanded single CB transplantation trial, demonstrating the feasibility and safety of expanded single-unit CB transplantation [39]. Notably, the patients who received the UM171-expanded CB graft were free of fever much earlier than the historical controls post-transplant (day 7 vs. day 15 p < 0.001), while the length of overall hospitalization was reduced by 11 days and all patients who received UM171-expanded CB grafts achieved full donor chimerism. Since this was a rather small-scale study, its findings should be interpreted with caution and require validation by randomized studies with other CB expansion strategies. Notably, other ex vivo expansion strategies utilizing SR-1 or NAM successfully achieved CD34 + cell expansion, yet the survival benefit observed for UM171-expanded grafts was not apparent in other trials using expanded CB grafts. However, these were small-scale studies and retrospective analyses compared to historical controls, only future randomized trials comparing multiple ex vivo expansion strategies will facilitate the identification of the most potent CB expansion strategy.
Conclusions
In general, mPB and BM grafts provide an optimal number of CD34 + cells as a hematopoietic graft source; however, around one-third of patients are unable to find an appropriately matched allogeneic graft from an adult source. The ex vivo manipulation of human CB cells under appropriate conditions has the potential to expand a limited number of HSPCs in a single-unit of CB to improve the kinetics of hematopoietic engraftment. The epigenetic manipulation of CB HSPCs in culture has resulted in successful preclinical stem cell transplantation and is being actively pursued in clinical trials with some success; however, significant obstacles remain before CB transplantation can be considered a viable alternative source of HSPC grafts [50]. Adding a secondary agent in culture may allow the amplification of functional HSPCs present in a single CB unit; therefore, the identification of non-redundant agonistic pathways may yield a compound effect and thus increase the efficacy of HSPC expansion ex vivo [51].
Given that several NGMs are reportedly effective in promoting HSPC expansion in culture, carefully selected combinatorial approaches may further improve the expansion of HSPCs. Indeed, the identification and validation of a given sequence of compounds that result in enhanced functional HSPC expansion would be an additional advantage in the field of stem cell expansion. To determine the safety of these procedures, long-term follow-ups are needed to demonstrate whether expanded CB cells continue to repopulate the marrow without untoward side effects successfully. An attractive alternative strategy could be to split the CB graft into several components that could be independently manipulated, given that long-term, sustained repopulation appears to be the concerted action of LT-HSCs rather than ST-HSCs. Emerging cell-based therapies under development also include induced pluripotent stem cell (iPSC)-derived tissue-restricted differentiated cells (i.e., HSCs), which also require ex vivo expansion. Besides, genetic modification via the viral delivery of a therapeutic gene to HSPCs for gene therapy or gene editing purposes [52, 53] will likely require brief ex vivo manipulation without triggering HSPC differentiation. The preclinical CB expansion strategies that are currently under investigation may allow the identification of novel sequences of events that are essential for the self-renewing division of HSCs in culture, with the aid of chemical agents that specifically activate symmetric or asymmetric HSC division. Agents that specifically target LT- or ST-HSCs may help to develop tailored HSPC grafts that either facilitate sustained blood cell reconstitution or temporarily improve blood cell counts as a bridge following anti-cancer radiation therapy or chemotherapy. In summary, the use of expanded CB grafts is on the cusp of becoming a clinical reality, and the combination of current ex vivo CB expansion strategies may ultimately result in a better understanding of how to stimulate self-renewing HSC division to engineer an optimal graft for transplantation. Consequently, chemical agonists (alone or in combination) that promote HSC self-renewal could be developed as in vivo drugs to rescue or boost failing HSPC grafts post-transplant and to treat acquired BM failure syndromes.
References
Gluckman, E., Devergié, A., Bourdeau-Esperou, H., Thierry, D., Traineau, R., Auerbach, A., & Broxmeyer, H. E. (1990). Transplantation of umbilical cord blood in Fanconi’s anemia. Nouvelle Revue Francaise d’Hematologie, 32(6), 423–425.
Voelker, R. (2011). FDA grants approval for first cord blood product. JAMA, 306(22), 2442. https://doi.org/10.1001/jama.2011.1759
Sauter, C., & Barker, J. N. (2008). Unrelated donor umbilical cord blood transplantation for the treatment of hematologic malignancies. Current Opinion in Hematology, 15(6), 568–575.
Majhail, N. S., Brunstein, C. G., & Wagner, J. E. (2006). Double umbilical cord blood transplantation. Current Opinion in Immunology, 18(5), 571–575.
Aljitawi, O. S. (2012). Ex vivo expansion of umbilical cord blood: where are we? International Journal of Hematology, 95(4), 371–379.
Maung, K. K., & Horwitz, M. E. (2019). Current and future perspectives on allogeneic transplantation using ex vivo expansion or manipulation of umbilical cord blood cells. International Journal of Hematology, 110, 50–58.
Wagner, J. E. Jr., Eapen, M., Carter, S., Wang, Y., Schultz, K. R., Wall, D. A., Bunin, N., Delaney, C., Haut, P., Margolis, D., Peres, E., Verneris, M. R., Walters, M., Horowitz, M. M., & Kurtzberg, J. (2014). One-unit versus two-unit cord-blood transplantation for hematologic cancers. New England Journal of Medicine, 371(18), 1685–1694.
Levac, K., Karanu, F., & Bhatia, M. (2005). Identification of growth factor conditions that reduce ex vivo cord blood progenitor expansion but do not alter human repopulating cell function in vivo. Haematologica, 90(2), 166–172.
Bari, S., Seah, K. K., Poon, Z., Cheung, A. M., Fan, X., Ong, S. Y., Li, S., Koh, L. P., & Hwang, W. Y. K. (2015). Expansion and homing of umbilical cord blood hematopoietic stem and progenitor cells for clinical transplantation. Biology of Blood and Marrow Transplantation, 21(6), 1008–1019.
de Lima, M., McMannis, J., Gee, A., Komanduri, K., Couriel, D., Andersson, B. S., Hosing, C., Khouri, I., Jones, R., Champlin, R., Karandish, S., Sadeghi, T., Peled, T., Grynspan, F., Daniely, Y., Nagler, A., & Shpall, E. J. (2008). Transplantation of ex vivo expanded cord blood cells using the copper chelator tetraethylenepentamine: a phase I/II clinical trial. Bone Marrow Transplantation, 41(9), 771–778.
Delaney, C., Heimfeld, S., Brashem-Stein, C., Voorhies, H., Manger, R. L., & Bernstein, I. D. (2010). Notch-mediated expansion of human cord blood progenitor cells capable of rapid myeloid reconstitution. Nature Medicine, 16(2), 232–236.
Wagner, J. E. Jr., Brunstein, C. G., Boitano, A. E., DeFor, T. E., McKenna, D., Sumstad, D., Blazar, B. R., Tolar, J., Le, C., Jones, J., Cooke, M. P., & Bleul, C. C. (2016). Phase I/II Trial of StemRegenin-1 Umbilical Cord Blood Hematopoietic Stem Cells Supports Testing as a Stand-Alone Graft. Cell Stem Cell, 18(1), 144–155.
Fares, I., Chagraoui, J., Gareau, Y., Gingras, S., Ruel, R., Mayotte, N., Csaszar, E., Knapp, D. J., Miller, P., Ngom, M., Imren, S., Roy, D. C., Watts, K. L., Kiem, H. P., Herrington, R., Iscove, N. N., Humphries, R. K., Eaves, C. J., Cohen, S., Marinier, A., et al. (2014). Cord blood expansion. Pyrimidoindole derivatives are agonists of human hematopoietic stem cell self-renewal. Science, 345(6203), 1509–1512.
Horwitz, M. E., Chao, N. J., Rizzieri, D. A., Long, G. D., Sullivan, K. M., Gasparetto, C., Chute, J. P., Morris, A., McDonald, C., Waters-Pick, B., Stiff, P., Wease, S., Peled, A., Snyder, D., Cohen, E. G., Shoham, H., Landau, E., Friend, E., Peleg, I., Aschengrau, D., et al. (2014). Umbilical cord blood expansion with nicotinamide provides long-term multilineage engraftment. The Journal of Clinical Investigation, 124(7), 3121–3128.
Dahlberg, A., Delaney, C., & Bernstein, I. D. (2011). Ex vivo expansion of human hematopoietic stem and progenitor cells. Blood, 117(23), 6083–6090.
Hagedorn, E. J., Durand, E. M., Fast, E. M., & Zon, L. I. (2014). Getting more for your marrow: boosting hematopoietic stem cell numbers with PGE2. Experimental Cell Research, 329(2), 220–226.
Robinson, S. N., Ng, J., Niu, T., Yang, H., McMannis, J. D., Karandish, S., Kaur, I., Fu, P., Del Angel, M., Messinger, R., Flagge, F., de Lima, M., Decker, W., Xing, D., Champlin, R., & Shpall, E. J. (2006). Superior ex vivo cord blood expansion following co-culture with bone marrow-derived mesenchymal stem cells. Bone Marrow Transplantation, 37, 359–366.
Mahmud, N., Petro, B., Baluchamy, S., Li, X., Taioli, S., Lavelle, D., Quigley, J. G., Suphangul, M., & Araki, H. (2014). Differential Effects of Epigenetic Modifiers on the Expansion and Maintenance of Human Cord Blood Stem/Progenitor Cells. Biology of Blood and Marrow Transplantation, 20(4), 480–489.
Papa, L., Zimran, E., Djedaini, M., Ge, Y., Ozbek, U., Sebra, R., Sealfon, S. C., & Hoffman, R. (2018). Ex vivo human HSC expansion requires coordination of cellular reprogramming with mitochondrial remodeling and p53 activation. Blood Advances, 2(20), 2766–2779.
Gonzalo-Daganzo, R., Regidor, C., Martín-Donaire, T., Rico, M. A., Bautista, G., Krsnik, I., Forés, R., Ojeda, E., Sanjuán, I., García-Marco, J. A., Navarro, B., Gil, S., Sánchez, R., Panadero, N., Gutiérrez, Y., García-Berciano, M., Pérez, N., Millán, I., Cabrera, R., & Fernández, M. N. (2009). Results of a pilot study on the use of third-party donor mesenchymal stromal cells in cord blood transplantation in adults. Cytotherapy, 11(3), 278–288.
Farag, S. S., Nelson, R., Cairo, M. S., O’Leary, H. A., Zhang, S., Huntley, C., Delgado, D., Schwartz, J., Zaid, M. A., Abonour, R., Robertson, M., & Broxmeyer, H. (2017). High-dose sitagliptin for systemic inhibition of dipeptidylpeptidase-4 to enhance engraftment of single cord umbilical cord blood transplantation. Oncotarget, 8(66), 110350–110357.
Ratajczak, M. Z., Reca, R., Wysoczynski, M., Kucia, M., Baran, J. T., Allendorf, D. J., Ratajczak, J., & Ross, G. D. (2004). Transplantation studies in C3-deficient animals reveal a novel role of the third complement component (C3) in engraftment of bone marrow cells. Leukemia, 18, 1482–1490.
Brunstein, C. G., McKenna, D. H., DeFor, T. E., Sumstad, D., Paul, P., Weisdorf, D. J., Ratajczak, M., Laughlin, M. J., & Wagner, J. E. (2013). Complement fragment 3a priming of umbilical cord blood progenitors: safety profile. Biology of Blood and Marrow Transplantation, 19(10), 1474–1479.
Porter, R. L., Georger, M. A., Bromberg, O., McGrath, K. E., Frisch, B. J., Becker, M. W., & Calvi, L. M. (2013). Prostaglandin E2 increases hematopoietic stem cell survival and accelerates hematopoietic recovery after radiation injury. Stem Cells, 31(2), 372–383.
Cutler, C., Multani, P., Robbins, D., Kim, H. T., Le, T., Hoggatt, J., Pelus, L. M., Desponts, C., Chen, Y., Rezner, B., Armand, P., Koreth, J., Glotzbecker, B., Ho, V. T., Alyea, E., Isom, M., Kao, G., Armant, M., Silberstein, L., Hu, P., et al. (2013). Prostaglandin-modulated umbilical cord blood hematopoietic stem cell transplantation. Blood, 122(17), 3074–3081.
Popat, U., Mehta, R. S., Rezvani, K., Fox, P., Kondo, K., Marin, D., McNiece, I., Oran, B., Hosing, C., Olson, A., Parmar, S., Shah, N., Andreeff, M., Kebriaei, P., Kaur, I., Yvon, E., de Lima, M., Cooper, L. J. N., Tewari, P., Champlin, R. E., et al. (2015). Enforced fucosylation of cord blood hematopoietic cells accelerates neutrophil and platelet engraftment after transplantation. Blood, 125(19), 2885–2892.
Xia, L., McDaniel, J. M., Yago, T., Doeden, A., & McEver, R. P. (2004). Surface fucosylation of human cord blood cells augments binding to P-selectin and E-selectin and enhances engraftment in bone marrow. Blood, 104(10), 3091–3096.
Robinson, S. N., Simmons, P. J., Thomas, M. W., Brouard, N., Javni, J. A., Trilok, S., Shim, J. S., Yang, H., Steiner, D., Decker, W. K., Xing, D., Shultz, L. D., Savoldo, B., Dotti, G., Bollard, C. M., Miller, L., Champlin, R. E., Shpall, E. J., & Zweidler-McKay, P. A. (2012). Ex vivo fucosylation improves human cord blood engraftment in NOD-SCID IL-2Rγ(null) mice. Experimental Hematology, 40(6), 445–456.
Pineault, N., & Abu-Khader, A. (2015). Advances in umbilical cord blood stem cell expansion and clinical translation. Experimental Hematology, 43(7), 498–513.
Araki, H., Mahmud, N., Milhem, M., Nunez, R., Xu, M., Beam, C. A., & Hoffman, R. (2006). Expansion of human umbilical cord blood SCID-repopulating cells using chromatin-modifying agents. Experimental Hematology, 34(2), 140–149.
Boitano, A. E., Wang, J., Romeo, R., Bouchez, L. C., Parker, A. E., Sutton, S. E., Walker, J. R., Flaveny, C. A., Perdew, G. H., Denison, M. S., Schultz, P. G., & Cooke, M. P. (2010). Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science, 329(5997), 1345–1348.
Chen, X., Skutt-Kakaria, K., Davison, J., Ou, Y. L., Choi, E., Malik, P., Loeb, K., Wood, B., Georges, G., Torok-Storb, B., & Paddison, P. J. (2012). G9a/GLP-dependent histone H3K9me2 patterning during human hematopoietic stem cell lineage commitment. Genes and Development, 26(22), 2499–2511.
Peled, T., Shoham, H., Aschengrau, D., Yackoubov, D., Frei, G., Rosenheimer, G. N., Lerrer, B., Cohen, H. Y., Nagler, A., Fibach, E., & Peled, A. (2012). Nicotinamide, a SIRT1 inhibitor, inhibits differentiation and facilitates expansion of hematopoietic progenitor cells with enhanced bone marrow homing and engraftment. Experimental Hematology, 40(4), 342–355.e1.
Saraf, S., Araki, H., Petro, B., Park, Y., Taioli, S., Yoshinaga, K. G., Koca, E., Rondelli, D., & Mahmud, N. (2015). Ex vivo expansion of human mobilized peripheral blood stem cells using epigenetic modifiers. Transfusion, 55(4), 864–874.
Milhem, M., Mahmud, N., Lavelle, D., Araki, H., DeSimone, J., Saunthararajah, Y., & Hoffman, R. (2004). Modification of hematopoietic stem cell fate by 5aza 2’deoxycytidine and trichostatin A. Blood, 103(11), 4102–4110.
Chaurasia, P., Gajzer, D. C., Schaniel, C., D’Souza, S., & Hoffman, R. (2014). Epigenetic reprogramming induces the expansion of cord blood stem cells. Journal of Clinical Investigation, 124(6), 2378–2395.
Araki, H., Baluchamy, S., Yoshinaga, K., Petro, B., Petiwala, S., Parajuli, R., Milhema, M., Lavellea, D., DeSimonea, J., & Mahmudab, N. (2009). Cord blood stem cell expansion is permissive to epigenetic regulation and environmental cues. Experimental Hematology, 37(9), 1084–1095.
Araki, H., Yoshinaga, K., Boccuni, P., Zhao, Y., Hoffman, R., & Mahmud, N. (2007). Chromatin-modifying agents permit human hematopoietic stem cells to undergo multiple cell divisions while retaining their repopulating potential. Blood, 109(8), 3570–3578.
Cohen, S., Roy, J., Lachance, S., Delisle, J. S., Marinier, A., Busque, L., Roy, D. C., Barabé, F., Ahmad, I., Bambace, N., Bernard, L., Kiss, T., Bouchard, P., Caudrelier, P., Landais, S., Larochelle, F., Chagraoui, J., Lehnertz, B., Corneau, S., Tomellini, E., et al. (2020). Hematopoietic stem cell transplantation using single UM171-expanded cord blood: a single-arm, phase 1–2 safety and feasibility study. The Lancet Haematology, 7(2), e134–e145.
Mahmud, N., Sidani, A., Koca, E., Kim, A., & Petro, B. (2014). Epigenetic Modifiers Promote Expansion of Transplantable Human Cord Blood Stem/Progenitor Cells Likely Through Activation of Inflammatory/Stress Response Signaling Pathways. Vancouver: ISSCR. (Abstract).
Papa, L., Djedaini, M., & Hoffman, R. (2019). Ex vivo HSC expansion challenges the paradigm of unidirectional human hematopoiesis. Annals of the New York Academy of Sciences. https://doi.org/10.1111/nyas.14133
Vannini, N., Girotra, M., Naveiras, O., Nikitin, G., Campos, V., Giger, S., Roch, A., Auwerx, J., & Lutolf, M. P. (2016). Specification of haematopoietic stem cell fate via modulation of mitochondrial activity. Nature Communications, 7, 13125.
He, J., Xiao, Z., Chen, X., Chen, M., Fang, L., Yang, M., Lv, Q., Li, Y., Li, G., Hu, J., & Xie, X. (2010). The expression of functional Toll-like receptor 4 is associated with proliferation and maintenance of stem cell phenotype in endothelial progenitor cells (EPCs). Journal of Cellular Biochemistry, 111(1), 179–186.
Petro, B., Mahmud, D., Taioli, S., Ganapathy, A., Senyuk, V., Yoshinaga, K. G., Suphangul, M., Rondelli, D., & Mahmud, N. (2019). Chromatin-Modifying Agent-Expanded Human Cord Blood Cells Display Reduced Allostimulatory Capacity. Journal of Immunology, 202(8), 2493–2501.
Saucourt, C., Vogt, S., Merlin, A., Valat, C., Criquet, A., Harmand, L., Birebent, B., Rouard, H., Himmelspach, C., Jeandidier, É, Chartois-Leauté, A. G., Derenne, S., Koehl, L., Salem, J. E., Hulot, J. S., Tancredi, C., Aries, A., Judé, S., Martel, E., Richard, S., Douay, L., & Hénon, P. (2019). Design and Validation of an Automated Process for the Expansion of Peripheral Blood-Derived CD34(+) Cells for Clinical Use After Myocardial Infarction. Stem Cells Translational Medicine, 8(8), 822–832.
Esser, C., & Rannug, A. (2015). The aryl hydrocarbon receptor in barrier organ physiology, immunology, and toxicology. Pharmacological Reviews, 67(2), 259–279.
Boitano, A. E., Goncalves, K. A., Sumstad, D., Wagner, J. E., & Cooke, M. P. (2019). Mgta-456 contains large numbers of CD34 + CD90 + hematopoietic stem cells (HSC) which contain the NSG engraftment activity and correlate with time to neutrophil recovery following transplant into patients with hematologic malignancy. Biology of Blood and Marrow Transplantation, 25(3), S2–S3.
Anand, S., Thomas, S., Hyslop, T., Adcock, J., Corbet, K., Gasparetto, C., Lopez, R., Long, G. D., Morris, A. K., Rizzieri, D. A., Sullivan, K. M., Sung, A. D., Sarantopoulos, S., Chao, N. J., & Horwitz, M. E. (2017). Transplantation of Ex Vivo Expanded Umbilical Cord Blood (NiCord) Decreases Early Infection and Hospitalization. Biology of Blood and Marrow Transplantation, 23(7), 1151–1157.
Lahlil, R., Scrofani, M., Barbet, R., Tancredi, C., Aries, A., & Hénon, P. (2018). VSELs Maintain their Pluripotency and Competence to Differentiate after Enhanced Ex Vivo Expansion. Stem Cell Reviews and Reports, 14(4), 510–524.
Stanevsky, A., Shimoni, A., Yerushalmi, R., & Nagler, A. (2011). Cord blood stem cells for hematopoietic transplantation. Stem Cell Reviews and Reports, 7(2), 425–433.
Bueno, C., Montes, R., & Menendez, P. (2010). The ROCK inhibitor Y-27632 negatively affects the expansion/survival of both fresh and cryopreserved cord blood-derived CD34 + hematopoietic progenitor cells: Y-27632 negatively affects the expansion/survival of CD34 + HSPCs. Stem Cell Reviews and Reports, 6(2), 215–223.
Lee, B., & Davidson, B. L. (2011). Gene therapy grows into young adulthood: special review issue. Human Molecular Genetics, 20(R1), R1. https://doi.org/10.1093/hmg/ddr188.
Horn, P. A., Morris, J. C., Bukovsky, A. A., Andrews, R. G., Naldini, L., Kurre, P., & Kiem, H.-P. (2002). Lentivirus-mediated gene transfers into hematopoietic repopulating cells in baboons. Gene Therapy, 9, 1464–1471.
Acknowledgements
We would like to thank Editage (www.editage.com) for English language editing. We would also like to thank James Zacny, PhD, for his critical review of the manuscript and his helpful suggestions. We also acknowledge contributions of Amudha Ganapathy, PhD and Benjamin Petro for their asistance with literature search and technical review of the draft manuscript.
Funding
The studies reported in this review from the Mahmud laboratory were supported in part by the Leukemia Lymphoma Society Translational Research Program, the University of Illinois at Chicago Chancellor’s Innovation Award, and Office of Vice Chancellor for Research Areas of Excellence Award to Dr. Nadim Mahmud. The funders had no role in study design, data collection and analysis, or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical Statement
This manuscript has not been published or presented elsewhere in part or in entirety and is not under consideration by another journal. The authors of this manuscript have read and understood this journal’s policies, and believe that neither the manuscript nor the unpublished studies from the authors laboratory discussed here violates any of these.
Conflicts of Interest
There are no conflicts of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Sica, R.A., Terzioglu, M.K., Mahmud, D. et al. Mechanistic Basis of ex Vivo Umbilical Cord Blood Stem Progenitor Cell Expansion. Stem Cell Rev and Rep 16, 628–638 (2020). https://doi.org/10.1007/s12015-020-09981-w
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12015-020-09981-w