
Abstract
Cerebral ischemia–reperfusion injury (CIRI) is a significant pathological process in stroke, characterized by neuronal cell death and neurological dysfunction. Metformin, commonly used for diabetes management, has been noted for its neuroprotective properties, though its effects on CIRI and the mechanisms involved remain unclear. This study explored the neuroprotective impact of metformin on CIRI, focusing on its potential to modulate the c-Jun N-terminal kinase (JNK) and p38 MAP kinase (p38) signaling pathways. Using in vitro models of oxygen–glucose deprivation/reperfusion (OGD/R) in neuronal cells and in vivo mouse models of middle cerebral artery occlusion (MCAO), the effects of metformin were assessed. Cell viability was measured with Cell Counting Kit-8 (CCK-8), protein expression via Western Blot (WB), and apoptosis through flow cytometry. The extent of brain injury in mice was evaluated using 2,3,5-triphenyltetrazolium chloride (TTC) staining, while JNK and p38 activation statuses were detected through WB and phospho-JNK (p-JNK) immunofluorescence staining. Results showed that metformin significantly improved the viability of HT22 cells post-OGD/R, reduced apoptosis, and decreased OGD/R-induced phosphorylation of JNK and p38 in vitro. In vivo, metformin treatment notably reduced brain infarct volume in MCAO mice, inhibited p-p38 and p-JNK expression, and enhanced neurological function. These findings suggest that metformin exerts neuroprotective effects against CIRI by modulating the JNK/p38 signaling pathway, highlighting its potential therapeutic value in treating cerebral ischemia–reperfusion injury and paving the way for clinical applications.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cerebral ischemia-reperfusion injury (CIRI) is a common and severe pathological condition following stroke. It occurs when blood flow is restored to previously obstructed areas of the brain [1, 2]. Theoretically, this restoration should be beneficial, but in reality, it triggers a series of complex pathophysiological reactions leading to oxidative stress, inflammatory responses, increased apoptosis, and blood-brain barrier disruption, exacerbating brain tissue damage [3, 4].
Two key stress-activated signaling pathways, c-Jun N-terminal kinase (JNK) and p38 MAP kinase (p38) play crucial roles in CIRI [5]. The activation of these pathways is involved in regulating cell survival and death, with the abnormal activation of JNK and p38 being closely associated with neuronal death in CIRI [6, 7].
Metformin is a drug widely used to treat type 2 diabetes, known for improving insulin sensitivity and reducing blood glucose levels. Recent studies have indicated that, beyond its hypoglycemic effects, metformin also possesses broad anti-inflammatory and neuroprotective effects [8, 9]. It has been found that metformin can act through pathways other than the AMP-activated protein kinase (AMPK) pathway [10,11,12], including the inhibition of JNK and p38 mitogen-activated protein kinase (MAPK) pathways, to reduce inflammation and apoptosis [13,14,15].
Therefore, this study aims to investigate whether metformin can alleviate neurological damage caused by CIRI by regulating the JNK/p38 signaling pathway and to elucidate the potential mechanisms of its neuroprotective effects. The study will use in vitro neuronal cell models induced by oxygen-glucose deprivation/reperfusion (OGD/R), as well as in vivo mouse models of middle cerebral artery occlusion (MCAO), employing various experimental methods, including Cell Counting Kit-8 (CCK-8), Western Blot (WB), flow cytometry, 2,3,5-triphenyltetrazolium chloride (TTC) staining, and p-JNK immunofluorescence staining to assess the neuroprotective effects of metformin.
The outcomes of this study may offer new insights into the clinical treatment of CIRI and pave the way for new applications of metformin.
Methods and Materials
Cell Culture
The induction method for OGD/R was consistent with previously reported procedures [16]. In brief, HT22 cells were first subjected to OGD induction in low-glucose DMEM under an atmosphere of 94% N2, 5% CO2, and 1% O2 for 6 h. Subsequently, the HT22 cells were reoxygenated for 24 h in high-glucose DMEM containing 10% fetal bovine serum under 95% air and 5% CO2. Based on the different treatment protocols, cells were divided into six groups: Sham, OGD/R, Low, Mid, High, and ANI. The Low, Mid, High, and ANI groups were pre-treated with various concentrations of metformin for 24 h before OGD/R treatment (10 µmol/L for the Low group, 20 µmol/L for the Mid group, 40 µmol/L for the High group, and 40 µmol/L for the ANI group). For the ANI group, an additional dose of 20 µmol/L was administered at the end of the OGD induction. All cells mentioned above were passaged for no more than six months following recovery.
CCK-8
Approximately 5000 cells per well were seeded into a 96-well plate. Upon completion of the cell treatment for each well, the original culture medium was discarded and replaced with 100 μL of fresh DMEM, supplemented with ten μL of Cell Counting Kit-8 (CCK-8) solution (Beyotime, China). The cells were then incubated for 2 h. Each well’s absorbance at 450 nm was measured using an enzyme-linked immunosorbent assay (ELISA) reader to facilitate subsequent analysis.
Flow Cytometry Analysis of Cell Apoptosis
The anti-apoptotic effect of metformin was confirmed by Annexin V-FITC staining. HT22 cells were collected from 96-well plates and washed twice with phosphate-buffered saline (PBS). The cells were then resuspended in a binding buffer. After the addition of 5 µL of Annexin V-fluorescein isothiocyanate (FITC) and 5 µL of propidium iodide (PI) (provided by Solarbio Science & Technology Co., Ltd., Beijing, China), the cells were incubated at 18–25 °C for 15 min before analysis. Following staining, the cells were analyzed using a flow cytometer (Muse Cell Analyzer, Merck Millipore, USA).
Animals
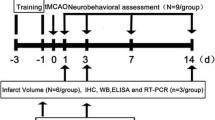
Sixty healthy C57BL/6 mice, aged 4–6 weeks and weighing 20–25 grams, were acquired from Beijing SPF Biotechnology Co., Ltd. (Beijing, China). The animals were housed in a room with controlled temperature (22–25 °C) and lighting (12-hour light-dark cycle), with ad libitum access to water and standard mouse chow. All mice were treated and used in accordance with the Guide for the Care and Use of Laboratory Animals (Federal Register Vol. 76, No. 36, 2011–11490; National Institutes of Health, Bethesda, MD). The experimental protocol was approved by the Ethics Committee of the Second Affiliated Hospital of Qiqihar Medical University (Qiqihar, China) with ethical approval number: 2021-0507. The mice were randomly divided into six groups (n = 10 each): Sham, IRI, Low, Mid, High, and ANI groups.
Construction of the MCAO Model and In Vivo Drug Administration
Anesthesia was induced with 1% sodium pentobarbital (50 mg/kg, intraperitoneal injection), and cerebral ischemia-reperfusion injury was modeled using the transient occlusion of the middle cerebral artery. Briefly, a neck incision was made, the common carotid artery was exposed, and the internal and external carotid arteries were carefully separated. Next, blood flow in the left middle cerebral artery was obstructed using a monofilament nylon suture coated with poly-L-lysine (Cinontech, Beijing, China). The suture was removed after 60 min of ischemia. Reperfusion was initiated seven days before the commencement of the experiment. Metformin was administered via gavage according to experimental requirements (Low group 1 mg/kg, Mid group 2 mg/kg, High group 5 mg/kg; Sigma-Aldrich, USA) along with the compound ANI (Anisomycin, 2 mg/kg, intraperitoneal injection; a JNK activator, APExBIO, USA). The sham-operated group underwent the same surgical procedures without suture insertion and received an equivalent volume of the vehicle at the onset of reperfusion. All mice were euthanized 24 h after reperfusion, and tissues from the peripheral blood, ischemic core, ischemic penumbra, and contralateral cerebral hemisphere were collected and stored at −80 °C for later use. Mice were maintained at a body temperature of 37.1 ± 0.5 °C using a heating pad from the time of anesthesia until euthanasia.
Neurological Assessment
24 and 72 h post-reperfusion, all mice underwent a neurological function deficit assessment using the Zea-Longa five-point scoring system prior to euthanasia (Reversible middle cerebral artery occlusion without craniectomy in rats). Scores were assigned as follows: 0 points, normal; 1 point, failure to extend left forepaw fully; 2 points, circling to the left; 3 points, falling to the left; 4 points, no spontaneous walking with a depressed level of consciousness.
Infarct Area Measurement
Twenty-four hours after reperfusion, mice from each group were euthanized, and their brains were rapidly harvested. The brain samples were quickly frozen, and six coronal brain slices, each 2 mm thick, were prepared. Each slice was initially immersed in a 1% solution of 2,3,5-triphenyltetrazolium chloride (TTC) supplied by Sigma-Aldrich (St. Louis, MO, USA). The staining procedure was carried out at a controlled temperature of 37 °C in the dark for 15 min to ensure adequate staining. Afterwards, the slices were fixed in a 4% solution of paraformaldehyde. The volume of cerebral infarction was measured using the Image-Pro Plus analysis system.
Brain Water Content Determination
To ascertain the water content of mice brain tissue and assess the extent of cerebral edema, we employed the methodology previously established by Wang et al. (MicroRNA-130a Regulates Cerebral Ischemia-Induced Blood-Brain Barrier Permeability by Targeting Homeobox A5). Following the brain extraction and the cerebellum and brainstem removal, the cerebrum’s wet weight (WW) was determined. The brain was then subjected to dehydration in an oven maintained at 100 °C for a period of 24 h to obtain the dry weight (DW). The cerebral water content was calculated using the formula: Percentage of Brain Water Content (%) = [(WW − DW)/WW] × 100%. This procedure facilitates the quantification of cerebral water content, thereby allowing for the evaluation of brain edema.
ELISA
Twenty-four hours after reperfusion, blood samples were collected into 2 ml Eppendorf tubes before euthanasia. The serum was separated by centrifugation at 3000 rpm for 10 min. Subsequently, the concentrations of IL-1β and IL-18 in each sample were measured using ELISA kits (Solarbio, China), strictly following the manufacturer’s instructions.
Western Blot
Total protein from mouse brain tissues and HT22 cells was extracted using RIPA lysis buffer (Beyotime, Shanghai, China). The lysates were centrifuged to separate supernatants. The total protein concentration in each supernatant was determined using a BCA protein assay kit (Beyotime, Shanghai, China) according to the provided instructions. The protein samples were separated via sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Following electrophoresis, the proteins were transferred onto polyvinylidene fluoride (PVDF) membranes. The membranes were blocked with 5% non-fat milk at room temperature for 2 h. Subsequently, the membranes were incubated with primary antibodies (1:2000 dilution) overnight at four °C, followed by a 2-hour incubation with secondary antibodies (1:5000 dilution) at room temperature.
Immunofluorescence Staining for p-JNK and TUNEL
Brain sections were subjected to immunofluorescence staining to detect phosphorylated p-JNK activation and apoptotic cells. Before staining, samples were fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100 in phosphate-buffered saline (PBS). Nonspecific binding sites were blocked using 5% bovine serum albumin (BSA) in PBS for 1 h at room temperature.
For p-JNK detection, the sections and cells were incubated overnight at four °C with a primary antibody specific for p-JNK (1:200 dilution) followed by incubation with a fluorescently labeled secondary antibody (1:500 dilution) for 1 h at room temperature, protected from light. After several washes with PBS, the nuclei were counterstained with DAPI (4’,6-diamidino-2-phenylindole) for 5 min to visualize the cell nuclei. The TUNEL assay followed the manufacturer’s instructions for detecting apoptotic cells. After sealing the slide with cover glass, the images were observed and collected by fluorescence microscope in the dark room.
Statistical Analysis
Unless otherwise specified, all experiments were conducted in triplicate. Data were analyzed using GraphPad Prism 8.0 software and are presented as mean ± standard deviation (X ± s). One-way ANOVA was employed to compare multiple groups, and the Student-Newman-Keuls (SNK) test was used for pairwise comparisons between groups. Results with p-values less than 0.05 were considered statistically significant.
Results
Metformin (Met) Effectively Ameliorates Apoptosis and Promotes Proliferation in HT22 Cells Post-OGD/R Treatment
Annexin V-FITC staining was utilized for quantitative apoptosis analysis. Data revealed that post-OGD/R, the apoptosis rate in the OGD/R group was significantly higher compared to the Sham group (p < 0.01) (Fig. 1A, B). Met pre-treatment enhanced the anti-apoptotic capacity of HT22 cells dose-dependently. However, the addition of the JNK activator ANI reversed Met’s protective effects, suggesting Met pre-treated HT22 cells exhibit resistance to apoptosis. Moreover, a CCK-8 assay (Fig. 1C) indicated that OGD/R markedly reduced HT22 cell viability compared to the control; this was significantly improved post-Met pre-treatment, indicating Met’s substantial protective role against OGD/R-induced HT22 cell damage. These findings provide experimental evidence for Met’s application in treating cerebral ischemia-reperfusion injuries.

The Effect of Metformin on OGD/R-treated Cells. A Flow cytometry results of each group. B Relative apoptosis rate of HT22 cells in each group. C Proliferation curve of HT22 cells in different treatment groups. D Protein expression levels in HT22 cells after OGD/R treatment in each group. E Relative ratio of p-p38 to p38. F Relative ratio of Bcl-2 to Bax. G Relative ratio of p-JNK to JNK. H Relative expression levels of Caspase-3. (* compared to Sham, p < 0.05; # compared to High group, p < 0.05)
Met Effectively Reduces Infarct Volume in MCAO-Injured Mice
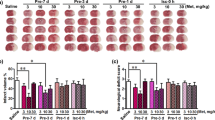
Infarct Volume Assessed by TTC Staining at 1 h Post-Ischemia and 24 h After Reperfusion in Mice. As illustrated in Fig. 2A, C, TTC staining revealed a significant increase in brain infarct area in the MCAO group compared to the Sham group (p < 0.001). TUNEL staining indicated a considerable rise in apoptotic cells within the MCAO group (Fig. 2B, E). With metformin intervention, both the infarct area and the percentage of apoptotic cells were significantly reduced. Furthermore, increased metformin concentration correlated with enhanced anti-apoptotic capability, suggesting that the reduction in infarct volume induced by MCAO under metformin pretreatment is dose-dependent. However, the co-administration of ANI with high-dose metformin attenuated metformin’s protective effect on MCAO mice. (White areas represent infarcted tissue, while red areas indicate normal tissue.)

Metformin Improves CIRI in Mice. A, C Assessment of myocardial infarct area using TTC staining. B, E Evaluation of apoptosis in brain tissues using TUNEL staining.Blue indicates cell nuclei, and green indicates TUNEL-positive cells. D Assessment of brain tissue water content. (* compared to Sham, p < 0.05; # compared to High group, p < 0.05)
Met Ameliorates Brain Edema and Improved Neurological Deficit Scores
Figure 3J, K shows that pretreatment with metformin significantly improved neurological deficit scores in MCAO-treated mice. Over time, the therapeutic effect of Met became more pronounced by the 72-h mark. Notably, a significant effect was only observed after reaching a specific prophylactic dosage of the drug, which may be related to the size of the ischemic brain region. Additionally, weighing the brains of the mice indicated that metformin significantly reduced brain edema (Fig. 2D). The same conclusion was reached, demonstrating the therapeutic efficacy of metformin at a specific dosage.

Protein Expression Levels in Mouse Brain After CIRI. A, G Expression levels of p-JNK in brain tissues of mice in each group. Blue indicates cell nuclei, and red indicates p-JNK-positive cells. B Protein expression levels in brain tissues of mice after CIRI in each group. C Relative ratio of p-p38 to p38. D Relative ratio of Bcl-2 to Bax. E Relative ratio of p-JNK to JNK. F Relative expression levels of Caspase-3. H, I Expression levels of IL-1β and IL-18. J, K Neurological deficit scores assessed using the Zea-Longa five-point scoring system. (* compared to Sham, p < 0.05; # compared to High group, p < 0.05)
Met Reduces Levels of IL-1β and IL-18
Figure 3H, I demonstrates that, compared to the sham-operated group, levels of IL-1β and IL-18 were significantly increased in the peripheral blood of the MCAO group (p < 0.01). With the addition of metformin, a trend toward reduced levels of these inflammatory factors was observed at low dosages, although p > 0.05. However, with increased metformin dosages, there was a significant improvement in these inflammatory factors. The anti-inflammatory effects, as well as the reduction in secretion of these factors, increased with dosage. The addition of ANI negated metformin’s benefits, reinforcing the conclusion that the protective effects of metformin on CIRI are dose-dependent.
Met Protects Brain Cells Through Regulation of JNK and p38 Phosphorylation
In vitro experiments simulated mouse MCAO using the OGD/R approach. Western blot analysis (Fig. 1D–H) revealed that JNK and p38 phosphorylation levels significantly increased in HT22 cells after OGD/R treatment, with a corresponding decrease in anti-apoptotic protein expression and increased pro-apoptotic protein expression. The in vivo experiments yielded similar conclusions (Fig. 3B-F). With the addition of metformin, these results showed marked improvement, and JNK and p38 phosphorylation levels decreased dose-dependently. The addition of the JNK activator ANI reversed the beneficial effects of metformin. This result aligns with conclusions derived from Fig. 3A, G, where a significant reduction in p-JNK positive signals was observed following metformin treatment. Conversely, ANI addition led to a substantial increase in positive signals compared to the high-dose group. This further suggests that metformin can protect against CIRI by mediating p38 by regulating JNK phosphorylation.
Met Improves the Expression of Apoptosis-Related Proteins
In vitro experiments (Fig. 1F, H) revealed that metformin effectively increases the Bcl-2/Bax ratio and decreases Caspase-3 expression, thereby exerting an anti-apoptotic effect. In vivo experiments (Fig. 3D, F) corroborated these findings, replicating the expression above results. The addition of ANI inhibited the anti-apoptotic action of metformin in both scenarios. This further confirms metformin’s protective effect on CIRI mice by regulating the JNK/p38 pathway.
Discussion
Metformin, a drug traditionally prescribed for managing diabetes, has garnered attention for its potential neuroprotective properties [17]. Our investigation delves into its impact on cerebral ischemia, a condition often preceding strokes and causing severe neurological impairments. Our research uncovered compelling evidence indicating that metformin significantly mitigates the detrimental effects of cerebral ischemia.
While the role of Metformin in CIRI has been partially reported, its specific mechanisms of action through the regulation of JNK and p38 MAPK signaling pathways have not been fully explored. Our study focuses on a detailed analysis of how Metformin exerts its protective effects via these pathways and reveals new regulatory mechanisms, differentiating our research from existing studies.
Our research delves into the neuroprotective effects of Metformin on cerebral ischemia, focusing specifically on its regulatory actions through the JNK and p38 MAPK signaling pathways. While previous studies have demonstrated Metformin’s neuroprotective effects, they primarily concentrated on the AMPK pathway [18], whereas we provide a detailed analysis of its impact on JNK and p38 MAPK pathways. Additionally, existing literature has primarily addressed Metformin’s role in diabetes and metabolic syndrome [19], with less attention given to its signaling mechanisms in neurological diseases. Our study reveals that Metformin alleviates CIRI-induced neuronal damage by inhibiting the phosphorylation of JNK and p38 MAPK, a novel mechanism not previously reported. To validate these findings, we employed various experimental models, including in vitro HT22 cells and in vivo MCAO mouse models, enhancing the credibility and applicability of our results.
One of the key findings of our study is the substantial reduction in cerebral edema associated with metformin administration. Cerebral edema, a common outcome of stroke, contributes to increased intracranial pressure and tissue damage [20, 21]. By attenuating cerebral edema, metformin demonstrates its ability to preserve tissue integrity and potentially improve neurological outcomes following ischemic events. Moreover, our research highlights a notable decrease in the volume of cerebral infarcts, directly indicative of the extent of stroke-induced injury and serving as a crucial prognostic marker for recovery.
Central to the neuroprotective effects of metformin are its interactions with signaling pathways involved in CIRI [22, 23], notably the JNK and p38 MAPK pathways [17, 24]. Activation of these pathways exacerbates neuronal damage by promoting inflammatory responses, oxidative stress, and apoptotic cell death. Our study demonstrates that metformin modulates these pathways, particularly the JNK cascade, thereby mitigating ischemia-induced neuronal cell death. This underscores the therapeutic potential of targeting these pathways in stroke management.
Furthermore, our investigation reveals improvements in neurological deficit scores following metformin administration, suggesting enhanced neurofunctional performance. These improvements encompass sensory, motor, and reflex capabilities, indicating the broader beneficial effects of metformin on neurological outcomes beyond histopathological changes. Such enhancements are pivotal indicators of metformin efficacy in promoting post-stroke recovery and enhancing overall quality of life.
A significant aspect of our findings pertains to the modulation of apoptotic pathways and preservation of mitochondrial integrity by metformin. Dysregulation of apoptotic pathways, involving proteins such as Bcl-2, Bax, and Caspase-3, contributes to neuronal cell death during cerebral ischemia-reperfusion injury. Caspase-3, in particular, plays a central role in executing apoptosis by cleaving various cellular substrates, leading to cell death [17, 24]. Metformin’s ability to upregulate Bcl-2 expression while inhibiting Bax activation and Caspase-3 activation underscores its neuroprotective effects by preventing mitochondrial dysfunction and apoptotic cell death, promoting neuronal survival in the ischemic brain [25, 26].
Studies have shown that metformin affects apoptotic pathways through various mechanisms, including activating AMPK and modulation of downstream signaling pathways [27, 28]. Activation of AMPK by metformin leads to inhibition of pro-apoptotic proteins such as Bax and Caspase-3 while simultaneously promoting the expression of anti-apoptotic proteins like Bcl-2 [29]. This dual action helps maintain mitochondrial integrity and cellular homeostasis, ultimately enhancing neuronal survival in the face of ischemic insults.
Furthermore, metformin’s ability to regulate apoptotic pathways extends beyond its direct effects on mitochondrial function. Recent studies have highlighted its role in modulating inflammatory responses and oxidative stress, intricately linked to apoptotic cell death in the ischemic brain [30, 31]. By mitigating inflammation and oxidative stress, metformin provides additional protection against neuronal damage, further enhancing its neuroprotective effects in cerebral ischemia-reperfusion injury [32, 33].
The clinical implications of our discoveries are substantial, positioning metformin as a promising candidate for adjunctive therapy in ischemic stroke management. Its ability to counteract inflammatory responses, oxidative stress, and apoptotic pathways underscores its potential to improve outcomes for stroke patients. Furthermore, the well-established safety profile of metformin in diabetes management facilitates its translation into clinical practice for stroke treatment.
Despite these promising findings, further research is warranted to validate our results in diverse animal models and evaluate the optimal dosing regimens and treatment durations of metformin in stroke therapy. Clinical trials are essential to assess the efficacy and safety of metformin as an adjunctive therapy in stroke patients. Additionally, elucidating the molecular mechanisms underlying metformin’s neuroprotective effects will provide valuable insights and facilitate the development of targeted interventions for stroke management.
Conclusion
In conclusion, our study underscores the significant neuroprotective effects of metformin in the context of cerebral ischemia. Metformin attenuates cerebral edema, reduces infarct volume, and enhances neurological function; metformin shows promise beyond its traditional role in glycemic control. The observed downregulation of the JNK signaling pathway and consequent decrease in neuronal apoptosis position metformin as a potential therapeutic agent in stroke management. These findings advocate for expanding metformin’s clinical applications to include neuroprotection, warranting further investigation into its mechanistic pathways and therapeutic viability in stroke patients.
References
Li, Y., Sandeep, B., & Xiao, Z. (2024). An overview regarding the article ‘clinical outcomes and mortality in patients with atrial fibrillation and recently diagnosed lung cancer in oncology outpatient settings’. Curr. Probl. Cardiol., 49(3), 102414.
Jain, S., Buttar, H. S., & Chintameneni, M., et al. (2018). Prevention of cardiovascular diseases with anti-inflammatory and anti-oxidant nutraceuticals and herbal products: an overview of pre-clinical and clinical studies. Recent Pat. Inflamm. Allergy Drug Discov., 12(2), 145–157.
Yu, L., Jin, Z., & Li, M., et al. (2022). Protective potential of hydroxysafflor yellow A in cerebral ischemia and reperfusion injury: An overview of evidence from experimental studies. Front. Pharm., 13, 1063035.
Maida, C. D., Daidone, M., & Pacinella, G., et al. (2022). Diabetes and ischemic stroke: an old and new relationship an overview of the close interaction between these diseases. Int. J. Mol. Sci., 23(4), 2397.
Song, N. N., Zhao, Y., & Sun, C., et al. (2023). DUSP10 alleviates ischemic stroke-induced neuronal damage by restricting p38/JNK pathway. Behav. Brain Res., 450, 114478.
Xing, Y., Yang, S. D., & Wang, M. M., et al. (2018). Electroacupuncture alleviated neuronal apoptosis following ischemic stroke in rats via midkine and ERK/JNK/p38 signaling pathway. J. Mol. Neurosci., 66(1), 26–36.
Huang, W., Lv, B., & Zeng, H., et al. (2015). Paracrine factors secreted by MSCs promote astrocyte survival associated With GFAP downregulation after ischemic stroke via p38 MAPK and JNK. J. Cell Physiol., 230(10), 2461–2475.
Akhtar, N., Singh, R., & Kamran, S., et al. (2022). Diabetes: chronic metformin treatment and outcome following acute stroke. Front. Neurol., 13, 849607.
Zhang, L., Zhang, J., & Zhu, X., et al. (2023). Metformin enhances neural precursor cells migration and functional recovery after ischemic stroke in mice. Exp. Brain Res., 241(2), 505–515.
Yang, F., Qin, Y., & Wang, Y., et al. (2019). Metformin inhibits the NLRP3 inflammasome via AMPK/mTOR-dependent effects in diabetic cardiomyopathy. Int. J. Biol. Sci., 15(5), 1010–1019.
Zheng, Z., Bian, Y., & Zhang, Y., et al. (2020). Metformin activates AMPK/SIRT1/NF-kappaB pathway and induces mitochondrial dysfunction to drive caspase3/GSDME-mediated cancer cell pyroptosis. Cell Cycle, 19(10), 1089–1104.
Ma, T., Tian, X., & Zhang, B., et al. (2022). Low-dose metformin targets the lysosomal AMPK pathway through PEN2. Nature, 603(7899), 159–165.
Chen, J., Zhang, S., & Pan, G., et al. (2020). Modulatory effect of metformin on cardiotoxicity induced by doxorubicin via the MAPK and AMPK pathways. Life Sci., 249, 117498.
Jin, J., Ma, Y., & Tong, X., et al. (2020). Metformin inhibits testosterone-induced endoplasmic reticulum stress in ovarian granulosa cells via inactivation of p38 MAPK. Hum. Reprod., 35(5), 1145–1158.
Ruiz-Mitjana, A., Vidal-Sabanes, M., & Navaridas, R., et al. (2023). Metformin exhibits antineoplastic effects on Pten-deficient endometrial cancer by interfering with TGF-beta and p38/ERK MAPK signaling. Biomed. Pharmacother., 168, 115817.
Wang, R., Wei, Y., & Deng, W., et al. (2022). Pratensein mitigates oxidative stress and NLRP3 inflammasome activation in OGD/R-injured HT22 Cells by activating Nrf2-anti-oxidant signaling. Neurotox. Res., 40(2), 384–394.
Zhu, J., Zheng, Y., & Zhang, H., et al. (2016). Targeting cancer cell metabolism: the combination of metformin and 2-Deoxyglucose regulates apoptosis in ovarian cancer cells via p38 MAPK/JNK signaling pathway. Am. J. Transl. Res., 8(11), 4812–4821.
Wang, J., Gallagher, D., & Devito, L. M., et al. (2012). Metformin activates an atypical PKC-CBP pathway to promote neurogenesis and enhance spatial memory formation. Cell Stem Cell, 11(1), 23–35.
Rena, G., Hardie, D. G., & Pearson, E. R. (2017). The mechanisms of action of metformin. Diabetologia, 60(9), 1577–1585.
Cappellari, M., Pracucci, G., & Saia, V., et al. (2023). Predictors for hemorrhagic transformation and cerebral edema in stroke patients with first-pass complete recanalization. Int. J. Stroke, 18(10), 1238–1246.
Gu, Y., Zhou, C., & Piao, Z., et al. (2022). Cerebral edema after ischemic stroke: pathophysiology and underlying mechanisms. Front. Neurosci., 16, 988283.
Paudel, Y. N., Angelopoulou, E., & Piperi, C., et al. (2020). Emerging neuroprotective effect of metformin in Parkinson’s disease: a molecular crosstalk. Pharm. Res, 152, 104593.
Karami, F., Jamaati, H., & Coleman-Fuller, N., et al. (2023). Is metformin neuroprotective against diabetes mellitus-induced neurodegeneration? An updated graphical review of molecular basis. Pharm. Rep., 75(3), 511–543.
Liu, C., Zhang, D., & Lu, Z., et al. (2022). Metformin protects against pericyte apoptosis and promotes neurogenesis through suppressing JNK p38 MAPK signalling activation in ischemia/reperfusion injury. Neurosci. Lett., 783, 136708.
El, K. S., Omran, M. M., & Mansour, H. H., et al. (2020). Metformin and/or low dose radiation reduces cardiotoxicity and apoptosis induced by cyclophosphamide through SIRT-1/SOD and BAX/Bcl-2 pathways in rats. Mol. Biol. Rep., 47(7), 5115–5126.
Cui, D., Xu, Z., & Qiu, S., et al. (2022). Nasturtium officinale L. and metformin alleviate the estradiol- induced polycystic ovary syndrome with synergistic effects through modulation of Bax/Bcl-2/p53/caspase-3 signaling pathway and anti-inflammatory and anti-oxidative effects. J. Food Biochem., 46(12), e14462.
Guo, Y., Jiang, H., & Wang, M., et al. (2023). Metformin alleviates cerebral ischemia/reperfusion injury aggravated by hyperglycemia via regulating AMPK/ULK1/PINK1/Parkin pathway-mediated mitophagy and apoptosis. Chem. Biol. Interact., 384, 110723.
Chen, Y. H., Yang, S. F., & Yang, C. K., et al. (2021). Metformin induces apoptosis and inhibits migration by activating the AMPK/p53 axis and suppressing PI3K/AKT signaling in human cervical cancer cells. Mol. Med. Rep., 23(1), 88.
Huang, P., Wu, S. P., & Wang, N., et al. (2021). Hydroxysafflor yellow A alleviates cerebral ischemia reperfusion injury by suppressing apoptosis via mitochondrial permeability transition pore. Phytomedicine, 85, 153532.
Ren, H., Shao, Y., & Wu, C., et al. (2020). Metformin alleviates oxidative stress and enhances autophagy in diabetic kidney disease via AMPK/SIRT1-FoxO1 pathway. Mol. Cell Endocrinol., 500, 110628.
Xu, L., Zhang, X., & Zhao, Y., et al. (2023). Metformin protects trabecular meshwork against oxidative injury via activating integrin/ROCK signals. Elife, 12, e81198.
Qin, Z., Zhou, C., & Xiao, X., et al. (2021). Metformin attenuates sepsis-induced neuronal injury and cognitive impairment. BMC Neurosci., 22(1), 78.
Abdelaziz, D. H., Thapa, S., & Abdulrahman, B., et al. (2020). Metformin reduces prion infection in neuronal cells by enhancing autophagy. Biochem. Biophys. Res. Commun., 523(2), 423–428.
Acknowledgements
Z.M. and L.Z. contributed equally to this study. This study was supported by the Clinical Research Fund Project of Qiqihar Academy of Medical Sciences: QMSI2020L-04.
Author information
Authors and Affiliations
Contributions
Shicun Zhang conceived and designed the study, conducted experiments, analyzed data, and wrote the manuscript. Wei Zou contributed to the study design, performed experiments, and assisted with data analysis and interpretation. Yan Leng participated in data acquisition, analysis, and interpretation, and contributed to the manuscript writing. Zhuang Mu provided critical input on study design, data interpretation, and manuscript revision. Lan Zhan supervised the study, provided resources, and critically revised the manuscript for important intellectual content. All authors have read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhang, S., Zou, W., Leng, Y. et al. Neuroprotective Effects of Metformin on Cerebral Ischemia-Reperfusion Injury: Modulation of JNK and p38 MAP Kinase Signaling Pathways. Cell Biochem Biophys (2024). https://doi.org/10.1007/s12013-024-01373-y
Accepted:
Published:
DOI: https://doi.org/10.1007/s12013-024-01373-y