
Abstract
This study aimed to evaluate the effects of electroacupuncture (EA) intervention administered at rats of middle cerebral artery occlusion (MCAO)/reperfusion. Fifty-four male Sprague-Dawley rats were divided into three groups, consisting of sham group, MCAO/R group, and EA group. EA treatment at Quchi and Zusanli acupoints was applied in rats of EA group at 24 h after MCAO once per day for 3 days. Our results indicated that EA treatment reduced infarct volumes and neurological deficits, as well alleviated the apoptotic cells in peri-infarct cortex, indicating that EA exerted neuroprotective effect in cerebral ischemic rats. Moreover, EA treatment may effectively reverse the upregulation of caspase-3 and Bim and alleviate the inhibition of Bcl-2 following 72-h ischemic stroke. EA may significantly reverse the promoted relative density level of p-ERK1/2, p-JNK, and p-p38 in the EA group compared with the MCAO/R group. In addition, the growth factor midkine (MK) was upregulated at 72 h after MCAO/R, and EA treatment may significantly prompt expression of MK. Our study demonstrated that EA exerted neuroprotective effect against neuronal apoptosis and the mechanism might involve in upregulation of MK and mediation of ERK/JNK/p38 signal pathway.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ischemic stroke, as one of the most serious illnesses, is related to high mortality and morbidity and may affect human health as well as result in death worldwide (Go et al. 2014). Complicated pathophysiology of cerebral ischemic injury is regulated by diverse processes, for example, oxidative, apoptotic, excitotoxicity, and inflammatory mechanisms (Park et al. 2017). Cerebral I/R injury triggers several cellular events via production of considerable reactive oxygen species, which contributes to apoptosis and necrosis (Gao et al. 2005).
\EA intervention as a traditional therapeutic method may alleviate cerebral damage induced by I/R injury (Wu et al. 2015). EA intervention shows beneficial effect for reduction of neurological deficit and for recovery of injured cerebral cells in rats with cerebral ischemic stroke via prompting differentiation and proliferation of neural stem cells (Wu et al. 2015). Moreover, EA may simultaneously increase the expression of Bcl-2 at the mRNA level and the expression of p-Bad and Bcl-2 and at the protein level, suppress the expression of cleaved caspase-3 and Bax (Xue et al. 2014), whereas the concrete mechanisms of EA intervention for alleviating neuronal apoptosis are complicated and remain unclear.
Mitogen-activated protein kinase (MAPK) is consisting of extracellular signal-regulated kinase 1/2 (ERK1/2), p38 MAPK, and c-Jun N-terminal kinase (JNK), which transmit signals from extracellular to intracellular targets to modulate cellular activities via diverse signaling pathways (Wang et al. 2015). MAPK signaling pathway is indispensable for the modulation of apoptosis and inflammation during ischemic stroke (Zhen et al. 2016). Several evidences show that ERK1/2 activation may exert neuroprotection against cell apoptosis (Sawe et al. 2008; Wang et al. 2003), but sustained activation of JNK and/or p38 always lead to neuronal death (Liu et al. 2017). It was reported that ERK activation could result in apoptosis and ERK inhibition could induce neuroprotection (Satoh et al. 2000). Tournier et al. (2000) indicate that JNK may inactivate and phosphorylate anti-apoptotic protein Bcl-2. Bayatmakoo et al. (2017) show that JNK causes neuronal apoptosis via directly phosphorylating BimL and BimEL, and phosphorylated JNK results in inactivation of the anti-apoptotic Bcl-2.
Midkine (MK), a heparin-binding growth factor, is characterized by a responsive gene to retinoic acid. As an endogenous neurite outgrowth factor, MK is related to development of the central nervous system (Muramatsu et al. 1993). Neurotrophic factors may activate diverse signal pathways after ischemic stroke, which provides neuroprotective effect against caspase-3-dependent apoptosis (Cheng et al. 2014). Ishikawa et al. demonstrate that MK may lead to neuronal regeneration and MK gene transfer exerts neuroprotection until the subacute stage of cerebral infarction (Ishikawa et al. 2009). Otsuka et al. indicate that MK may be upregulated by exercise pretreatment after ischemic brain injury, along with improvement of motor function, reduction of infarct volume, and alleviation of neuronal apoptosis (Otsuka et al. 2016). Thereby, we speculate that the MK expression may be increased via EA intervention, of which mechanism is no one understand.
In the present study, we speculate that EA intervention at Quchi (LI11) and Zusanli (ST36) acupoints provides neuroprotection by regulating the expression level of MK and inhibiting neuronal apoptosis following ischemic stroke via ERK/JNK/p38MAPK pathway.
Material and Methods
Rat MCAO Model
The rat model of focal cerebral stroke was induced by left middle cerebral artery occlusion (MCAO). Rats were fasted for 10 h and then anesthetized with 10% chloral hydrate (300 mg/kg) by intraperitoneal injection. A surgical nylon suture (diameter, 0.26 mm; Beijing Shadong Biotech Co., Ltd., Beijing, China) was used to occlude the left middle cerebral artery. After 2-h MCAO, reperfusion (R) was induced via withdrawing the nylon suture slowly. The sham group underwent surgical procedures as same as MCAO/R and EA groups without occluding the middle cerebral artery. During the whole surgical procedure, a heating pad was used to keep the rectal temperature of rats at 37 °C.
Experimental Animals and Groups
Male Sprague-Dawley (SD) rats (280–320 g) from Hebei Province Laboratory Animal Center were housed in a controlled condition with a 12-h light/dark cycle at 22 ± 2 °C and 60–70% humidity. Water and food were available ad libitum. All experimental protocols for animals were permitted by the Animal Care and Use Committee of Hebei Medical University.
In the light of a random number table method, 54 rats were randomly divided into three groups (n = 18/group) as follows: (i) sham-operated group (sham group); (ii) MCAO/R group; and (iii) the MCAO/R + EA intervention group (EA group).
Assessment of Neurological Deficit Scores
Neurological function of all rats was evaluated for 72 h after MCAO/R surgery in a blinded method. The standard scoring system was as follows: score 0, no apparent neurologic symptoms; score 1, not able to completely extend the right front jaw; score 2, circling to contralateral when walking; score 3, falling to contralateral when walking; score 4, unable to walk; and score 5, died. Rats with score 1–3 points demonstrated successful establishment of MCAO model.
EA Intervention
EA intervention was administered by using an EA apparatus (Model G6805-2A; Shanghai Huayi Co., Shanghai, China) at 24 h following MCAO (22 h after MCAO/reperfusion) once per day for 3 days. Briefly, rats in the EA group were anesthetized with 10% chloral hydrate and inserted two acupuncture needles (0.3 mm in diameter) 2–3 mm deep at the Quchi (LI11) and Zusanli (ST36) on the paralyzed limb for 30 min once daily. The intervention parameters were set as dense-disperse waves of 4/20 Hz (adjusted to the muscle twitch threshold), intensity of 1 mA, and peak voltage of 6 V. However, rats in the sham and MCAO/R groups were anesthetized without EA intervention. Finally, rats were evaluated by neurological deficit scores and then sacrificed at 72 h following MCAO/R for the next step experiment.
TTC Staining
To detect infarct volume, rats were euthanized using 10% chloral hydrate at 72 h following I/R injury. Brain tissues were removed and coronally cut into six slices and the thickness of each slice was 2 mm. These slices were rapidly placed in 2% TTC (2,3,5-triphenyltetrazolium chloride) solution (37 °C) for 30 min and subsequently fixed in 4% paraformaldehyde buffer (Bederson et al. 1986). The images were measured using imaging software (Adobe Photoshop 7.0, Adobe Systems, Mountain View, CA) and the area of infarct was calculated as previously reported. Following 24-h fixation, the sections were photographed with a digital camera (Kodak DC240; Eastman Kodak Co., Rochester, NY).
Western Blot Analysis
Total protein of the tissues from the cerebral cortex in peri-infarct region and the same region in the sham-operated rats were extracted by lysis buffer. The samples (30 μg/lane) were separated by 12% SDS-PAGE and transferred onto a polyvinylidene difluoride membrane (Roche, Mannheim, Germany) in transfer buffer containing 0.1% SDS. Following that, the membranes were blocked with 5% bovine serum albumin solution and incubated with the primary antibodies against midkine (1:1000, #ab36038, abcam), SAPK/JNK (1:1000, #9252, Cell Signaling Technology), p38 MAPK (1:1000, #8690, Cell Signaling Technology), Erk1/2(1:1000, #4695, Cell Signaling Technology), p-SAPK/JNK(1:1000, #4668, Cell Signaling Technology), p-p38 MAPK (1:1000, #AF4001, Affinity Biosciences), p-Erk1/2(1:1000, #4370, Cell Signaling Technology), caspase-3 (1:2000, #ab184787, abcam), and Bcl-2(1:1000, #ARG55188, arigo) Following washing with TBST for three times, peroxidase-conjugated goat anti-rabbit IgG (1:5000) was used as secondary antibodies for 1 h at room temperature. Finally, Protein bands were visualized using the chemiluminescent HRP substrate. Intensity of bands was quantified using image J software and normalized to GAPDH (1:5000, #10494-1-AP, proteintech).
Immunohistochemistry Analysis
Paraffin-embedded tissues were produced by standard method. The brains of all rats were removed and fixed in 4% paraformaldehyde for 24 h. Following that, cerebral tissues were washed with PBS so as to remove fixative. Then, tissues were dehydrated in graded alcohol and infiltrated in molten paraffin wax followed by embedding in blocks. The paraffin-embedded sections were dewaxed, hydrated, and incubated with 3% H2O2 for 30 min for quenching of endogenous peroxidase. Subsequently, sections were rinsed and exposed to solution consisting of MK (#11009-1-AP, proteintech), caspase-3 (#ab184787, abcam), Bcl-2 (#ab196495, abcam), and Bim (#2933, Cell Signaling Technology) overnight at 4 °C. After washing with TBST, sections were visualized with 3,3′-diaminobenzidine chromogen following incubation with goat anti-rabbit IgG (1:1000) for 60 min at room temperature. Finally, positive staining was performed under a light microscope followed by counterstain with hematoxylin. The positive cells per square millimeter were calculated through an investigator who was ignorant of grouping methods.
TUNEL Staining
The amount of apoptotic cells was detected using an In Situ Cell Death Detection Kit (Roche, Mannheim, Germany) after 72-h reperfusion. The brain (n = 6/group) was fixed with 4% formaldehyde for 24 h at 4 °C and then embedded in paraffin. Following that step, the paraffin was sectioned at a thickness of 2 mm. Brain slices were incubated with proteinase K for 15 min, followed by quenching with 3% H2O2 for 10 min at room temperature. After rinsing three times with PBS, the sections were incubated in TUNEL reaction mixture for 60 min at 37 °C in the dark. A light microscope (Olympus Corporation, Tokyo, Japan) was used to observe and photograph brain slices, and five high-power fields of the cerebral cortex surrounding ischemia were randomly selected. The ImageJ software (National Institute of Health) was used to count the number of apoptotic cells by a pathologist blinded to the treatment. The apoptosis index (AI) = the amount of TUNEL-positive cells / the amount of total cells.
Statistical Analysis
Data were demonstrated as mean ± SEM. Statistical comparisons of results from the three groups were performed through one-way analysis of variance (ANOVA). Data of neurological deficits among the three groups were analyzed by non-parametric test. p < 0.05 was considered as statistical significance.
Results
EA Alleviates Neurological Behavioral Scores and Infarct Volumes in Cerebral I/R Injury Rats
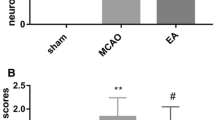
The rats were administered neurological behavioral assessment and infarct area evaluation at 72 h after reperfusion. As shown in Figs. 1 and 2, the rats of the EA group and MCAO/R group demonstrated significantly neurological behavioral symptoms and cerebral infarction; however, the rats of the sham group showed no neurological symptoms and cerebral infarct at 72 h after reperfusion. There is obvious difference of neurological behavioral score and infarct volumes between the EA group and MCAO/R group at 72 h after reperfusion (p < 0.05).The neurological behavioral score and infarct volumes of rats at the EA group were less than those at the MCAO/R group.

Neurological behavioral scores. Neurological behavioral assessment at 72 h after I/R injury. **p < 0.01 vs. sham group; #p < 0.05, vs. MCAO/R group. I/R, ischemia/reperfusion; MCAO/R, middle cerebral artery occlusion/reperfusion

Effect of EA intervention on infarct volume. a TTC staining was performed at 72 h after reperfusion. b Bar graph showed the percentage of cerebral infarct volume among the three groups. #p < 0.05, vs. the MCAO/R group. EA, electroacupuncture; MCAO/R, middle cerebral artery occlusion/reperfusion; TTC, 2,3,5-triphenyltetrazolium chloride
Treatment of EA Alleviates Apoptosis in Cerebral I/R Injury Rats
To further investigate the effect of EA intervention against apoptosis, we performed TUNEL staining to evaluate the percentage of apoptotic cells. As shown in Fig. 3, the percentage of apoptotic cells in the MCAO/R group was significantly raised compared to that in the sham group (p < 0.01) at 72 h following reperfusion, whereas EA intervention decreased the percentage of apoptotic cells compared to the MCAO/R group (p < 0.01).

Effect of EA intervention against apoptosis. a TUNEL staining in infarct area of the cerebral cortex of the sham, MCAO/R, and EA groups (× 400). b Bar graph indicates the percentage of apoptosis-positive cells. **p < 0.01 vs. sham group; ##p < 0.01, vs. MCAO/R group. EA, electroacupuncture; MCAO/R, middle cerebral artery occlusion/reperfusion
Treatment of EA Regulates Apoptosis-Related Proteins
The mechanism of EA neuroprotection was investigated by IHC staining for caspase-3, Bcl-2, and Bim and western blot for caspase-3 and Bcl-2. As shown in Fig. 4, ischemic stroke increased the positive cell percentage of caspase-3 and Bim and reduced the percentage of Bcl-2-positive cells in the ischemic cortex of the MCAO/R group than the sham group (p < 0.01), whereas EA intervention at Zusanli and Quchi acupoints significantly alleviated neuronal apoptosis via reversing the downregulation of Bcl-2 and upregulation of caspase-3 and Bim (p < 0.01). As shown in Fig. 5, EA intervention significantly reduced the enhancement of caspase-3 (p < 0.01) and Bcl-2 (p < 0.05) after 72-h reperfusion by western blot analysis.

EA intervention affects the expression of apoptosis-related proteins in the peri-infarct cortex. Immunohistochemical staining of (a) caspase-3, (b) Bcl-2, and (c) Bim (× 200); d the ratios of apoptosis-positive cells in the penumbra of the cortex were shown by bar graph. **p < 0.01 vs. sham group; ##p < 0.01, vs. MCAO/R group. EA, electroacupuncture; MCAO/R, middle cerebral artery occlusion/reperfusion

EA intervention affects the expression of apoptosis-related proteins. a Western blot analysis was performed to investigate the expression of caspase-3 and Bcl-2; b bar graph shows quantitative analysis of the ratios of apoptosis-related proteins/GAPDH. **p < 0.01 vs. sham group; #p < 0.05, vs. MCAO/R group, ##p < 0.01, vs. MCAO/R group. EA, electroacupuncture; MCAO/R, middle cerebral artery occlusion/reperfusion
Treatment of EA Regulates ERK/JNK/p38 Pathway
As shown in Fig. 6, the relative density level of p-ERK1/2, p-JNK, and p-p38 of MCAO/R group was significantly increased compared with the sham group (p < 0.01). However, EA may significantly reverse the promoted relative density level of p-ERK1/2 (p < 0.01), p-JNK (p < 0.05), and p-p38 (p < 0.05) in the EA group compared with the MCAO/R group.

EA intervention affects the ERK/JNK/p38MAPK pathway. a The western blotting analysis for the levels of ERK1/2, SAPK/JNK, p38, p-ERK1/2, p-SAPK/JNK, and p-p38 in peri-infarct penumbra of the cerebral cortex. b Bar graph shows quantitative analysis of the ratios of phosphorylated protein levels/total proteins levels. #p < 0.05, vs. the MCAO/R group; **p < 0.01 vs. sham group; ##p < 0.01, vs. the MCAO/R group. EA, electroacupuncture; MCAO/R, middle cerebral artery occlusion/reperfusion; ERK1/2, extracellular signal-regulated kinase 1/2; JNK, c-Jun N-terminal kinase
Treatment of EA Regulates the Expression of MK
As demonstrated in Figs. 7 and 8, there is a significant difference in MK levels among the three groups by western blot and immunohistochemistry analysis. The expression of MK in the MCAO/R group (p < 0.01) and EA group (p < 0.01) may significantly increase compared to that in the sham group. Furthermore, The MK levels of rats in the EA group were obvious higher than those in the MCAO/R group (p < 0.01).

EA intervention affects the expression of MK. a Western blotting analysis for the levels of MK in peri-infarct penumbra of the cerebral cortex. b Bar graph shows relative quantitative analysis . **p < 0.01 vs. sham group; ##p < 0.01, vs. the MCAO/R group; &&p < 0.01 vs. EA group. EA, electroacupuncture; MCAO/R, middle cerebral artery occlusion/reperfusion; MK, midkine

Immunohistochemical staining of MK. a Sham, b MCAO/R, and c EA groups (× 200). d Amplification of image in c (× 400); e bar graph shows MK activity. *p < 0.05 vs. sham group; #p < 0.05, vs. the MCAO/R group; &&p < 0.01 vs. EA group. EA, electroacupuncture; MCAO/R, middle cerebral artery occlusion/reperfusion; MK, midkine
Discussion
Ischemic stroke is characterized by deprivation of cerebral blood circulation caused by occluding the cerebral artery and is a widespread neurological illness (Park et al. 2017). Evidence indicates that EA might exert neuroprotective effect against ischemic brain damage via activing diverse survival signaling pathways (Feng et al. 2013; Lan et al. 2013). Liu et al. show that EA may promote recovery of the motor dysfunction and the possible mechanism may be involved in suppression of microglia-mediated neuroinflammation in the ischemic sensorimotor cortex of rats after ischemia (Liu et al. 2016). EA therapy may suppress the activation of apoptotic signal pathways, which resulted in improvement of injury in the ischemic penumbra (Kim et al. 2013). Wu et al. indicate that EA may improve neurological deficits and alleviate cortical neuronal apoptosis in rats following ischemic stroke(Wu et al. 2017).On the basis of these studies, we also demonstrated that EA intervention may significantly decrease neurological deficits and reduce cerebral infarct volumes, as well as alleviate the neuronal apoptosis.
Apoptosis is an important characteristic in mild cerebral ischemic stroke and exerts a key role in the pathological development of late infarction (Cheng et al. 2014). Activated caspase-3, a critical apoptotic executor, was upregulated obviously 24 to 72 h following ischemia/reperfusion and resulted in cells to suffer DNA fragmentation and nuclear condensation(Lee et al. 2002). As a key molecule of the intrinsic apoptosis pathway, Bcl-2 protein may regulate the apoptotic changes in vascular cells (Bayatmakoo et al. 2017). The Bcl-2 protein family is comprised of anti-apoptosis proteins Bcl-2 and pro-apoptotic proteins Bim, exerting important effects in the apoptotic mitochondrial pathway (Kim et al. 2006). It was reported that high expression level of Bcl-2 may suppress the cell apoptosis (Kim et al. 2006; Wang et al. 2011). Richard et al. show that Bim is indispensable for neuronal apoptosis caused by deprivation of growth factor in various cell types, involving in lymphocytes, epithelial cells, endothelial cells, mast cells, osteoclasts, and neurons (Youle and Strasser 2008). In the current study, EA intervention also alleviated apoptosis via increasing the expression of Bcl-2 and reversing the enhancement of Bim and caspase-3 after ischemic stroke.
ERK pathway as a key member of MAPK signal family is chiefly related to apoptosis, cell proliferation, and differentiation (Shen et al. 2004). ERK signaling pathway is involved in sequential promotion of Raf, ERK, p38MAPK, and other related proteins (Johnson and Lapadat 2002). Yang et al. suggest that EA might increase the activation of ERK pathways and affect the hippocampal microenvironment, which may exert benefits in depressive-like behaviors and NSC proliferation (Yang et al. 2013). It was reported that ERK1/2 is overexpressed in the ischemic brain (Wang et al. 2003) and suppression of the ERK1/2 pathway may decrease focal infarct volume and brain injury in mice with ischemic stroke (Namura et al. 2001). Suppression of the enhanced phospho-ERK1/2 following ischemic stroke may provide neuroprotection against cerebral ischemic damage (Zhang et al. 2010), which is consistent with our findings.
The p38 MAPK pathway possibly exerts significant effect during the process of cerebral ischemic stroke (Wang et al. 2014). It was reported that continuously activated p38 MAPK may promote inflammation resulting in aggravation of ischemic damage in the acute phase following transient MCAO (Cheng et al. 2016). Several studies indicated that upregulation of p38 MAPK may prompt neural cell death following ischemic stroke (Irving et al. 2000; Wu et al. 2000). On the contrary, Nishimura et al. suggest that enduring activation of p38 may result in ischemic tolerance in the hippocampus CA1 and that members of the p38 signal pathway play an important role in modification neuronal survival following ischemia (Nishimura et al. 2003). Cheng et al. also indicate that phosphorylated p38 MAPK plays a key role for anti-apoptotic effect in the area of penumbral cortex during the subacute and acute stages of ischemic stroke (Cheng et al. 2016). We observed that phosphorylated p38 may be upregulated after ischemic stroke and reversed after EA intervention, demonstrating that EA may exert neuroprotection via decreasing phosphorylated p38.
The JNK family is related to various pathological and physiological reactions in the central nerve system, for example neuroinflammatory reaction, neuronal apoptosis, neural plasticity, neuronal degeneration, and regeneration (Antoniou and Borsello 2012). c-Jun N-terminal kinase (JNK) as an crucial stress-responsive kinase may be activated via diverse types of brain insults (Gao et al. 2005). Zheng et al. demonstrate that activated JNK signaling pathway may contribute to neuronal apoptosis and inflammation after the MCAO (Zheng et al. 2018). Gao et al. show that JNK as a key cell death mediator in cerebral ischemic damage may activate the mitochondrial apoptotic pathway by prompting Bim and Bax translocation (Gao et al. 2005). However, Murata et al. show that delayed suppression of JNK pathways at 7 days following ischemia may deteriorate ischemic brain injury with increased infarction volumes, and the result may be associated with JNK-regulated endogenous repair mechanisms of neurovascular plasticity (Murata et al. 2012). The studies demonstrate that JNK pathway exerts different functions in early and late phases of ischemic stroke. In this study, we demonstrated that EA intervention following 72-h ischemic injury may decrease phosphorylated SAPK/JNK levels so as to exert neuroprotective effect.
MK as a neurotrophic factor may keep normal adult brain (Yoshida et al. 2008) and is expressed in the astrocytic cytoplasm during the early stage following cerebral ischemia (Wang et al. 1998). According to previous study, MK is expressed in the surrounding tissue of lesion area and may serve as a reparative neurotrophic factor in the early stage following cerebral ischemia (Yoshida et al. 1995). Kadomatsu et al. show that MK exerts beneficial effects following heart ischemia/reperfusion injury via alleviating infarct size and inhibiting cardiac remodeling in a long run (Kadomatsu et al. 2014). Furthermore, Matsuda et al. demonstrate that expression of MK may be upregulated in the early phase of ischemia, which was observed from cerebral ischemic acute to subacute stage (Matsuda et al. 2011). In the present study, results suggest that growth factor MK exerts neuroprotection in neuronal survival and plays an important role in EA intervention at 72 h after ischemic brain injury.
Conclusion
In conclusion, our findings indicated that EA at the Zusanli and Quchi acupoints decreased neurological deficits and alleviated infarct volumes and neuronal apoptosis following ischemic stroke. In addition, the anti-apoptotic mechanism of EA after ischemic stroke might be associated with upregulation of MK and mediation of ERK/JNK/p38MAPK pathway.
Abbreviations
- MK:
-
Midkine
- MAPKs:
-
Mitogen-activated protein kinases
- ERK1/2:
-
Extracellular signal-regulated kinase 1/2
- JNK:
-
c-Jun N-terminal kinase
References
Antoniou X, Borsello T (2012) The JNK signalling transduction pathway in the brain. Front Biosci (Elite Ed) 4:2110–2120
Bayatmakoo R, Rashtchizadeh N, Yaghmaei P, Farhoudi M, Karimi P (2017) Atorvastatin inhibits cholesterol-induced caspase-3 cleavage through down-regulation of p38 and up-regulation of Bcl-2 in the rat carotid artery. Cardiovasc J Afr 28:298–303. https://doi.org/10.5830/CVJA-2017-005
Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H (1986) Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke 17:472–476
Cheng CY, Lin JG, Su SY, Tang NY, Kao ST, Hsieh CL (2014) Electroacupuncture-like stimulation at Baihui and Dazhui acupoints exerts neuroprotective effects through activation of the brain-derived neurotrophic factor-mediated MEK1/2/ERK1/2/p90RSK/bad signaling pathway in mild transient focal cerebral ischemia in rats BMC. Complement Altern Med 14:92. https://doi.org/10.1186/1472-6882-14-92
Cheng CY, Tang NY, Kao ST, Hsieh CL (2016) Ferulic acid administered at various time points protects against cerebral infarction by activating p38 MAPK/p90RSK/CREB/Bcl-2 anti-apoptotic signaling in the subacute phase of cerebral ischemia-reperfusion injury in rats. PLoS One 11:e0155748. https://doi.org/10.1371/journal.pone.0155748
Feng X et al (2013) Electroacupuncture ameliorates cognitive impairment through inhibition of NF-kappaB-mediated neuronal cell apoptosis in cerebral ischemia-reperfusion injured rats. Mol Med Rep 7:1516–1522. https://doi.org/10.3892/mmr.2013.1392
Gao Y et al (2005) Neuroprotection against focal ischemic brain injury by inhibition of c-Jun N-terminal kinase and attenuation of the mitochondrial apoptosis-signaling pathway. J Cereb Blood Flow Metab 25:694–712. https://doi.org/10.1038/sj.jcbfm.9600062
Go AS et al (2014) Heart disease and stroke statistics--2014 update: a report from the American Heart Association. Circulation 129:e28–e292. https://doi.org/10.1161/01.cir.0000441139.02102.80
Irving EA, Barone FC, Reith AD, Hadingham SJ, Parsons AA (2000) Differential activation of MAPK/ERK and p38/SAPK in neurones and glia following focal cerebral ischaemia in the rat. Brain Res Mol Brain Res 77:65–75
Ishikawa E, Ooboshi H, Kumai Y, Takada J, Nakamura K, Ago T, Sugimori H, Kamouchi M, Kitazono T, Ibayashi S, Iida M (2009) Midkine gene transfer protects against focal brain ischemia and augments neurogenesis. J Neurol Sci 285:78–84. https://doi.org/10.1016/j.jns.2009.05.026
Johnson GL, Lapadat R (2002) Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 298:1911–1912. https://doi.org/10.1126/science.1072682
Kadomatsu K, Bencsik P, Gorbe A, Csonka C, Sakamoto K, Kishida S, Ferdinandy P (2014) Therapeutic potential of midkine in cardiovascular disease. Br J Pharmacol 171:936–944. https://doi.org/10.1111/bph.12537
Kim H, Rafiuddin-Shah M, Tu HC, Jeffers JR, Zambetti GP, Hsieh JJ, Cheng EH (2006) Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell Biol 8:1348–1358. https://doi.org/10.1038/ncb1499
Kim YR et al (2013) Effects of electroacupuncture on apoptotic pathways in a rat model of focal cerebral ischemia. Int J Mol Med 32:1303–1310. https://doi.org/10.3892/ijmm.2013.1511
Lan L et al (2013) Electroacupuncture exerts anti-inflammatory effects in cerebral ischemia-reperfusion injured rats via suppression of the TLR4/NF-kappaB pathway. Int J Mol Med 31:75–80. https://doi.org/10.3892/ijmm.2012.1184
Lee SH, Kim M, Kim YJ, Kim YA, Chi JG, Roh JK, Yoon BW (2002) Ischemic intensity influences the distribution of delayed infarction and apoptotic cell death following transient focal cerebral ischemia in rats. Brain Res 956:14–23
Liu W et al (2016) Electroacupunctre improves motor impairment via inhibition of microglia-mediated neuroinflammation in the sensorimotor cortex after ischemic stroke. Life Sci 151:313–322. https://doi.org/10.1016/j.lfs.2016.01.045
Liu Y, Wang Z, Xie W, Gu Z, Xu Q, Su L (2017) Oxidative stress regulates mitogen-activated protein kinases and cJun activation involved in heat stress and lipopolysaccharideinduced intestinal epithelial cell apoptosis. Mol Med Rep 16:2579–2587. https://doi.org/10.3892/mmr.2017.6859
Matsuda F, Sakakima H, Yoshida Y (2011) The effects of early exercise on brain damage and recovery after focal cerebral infarction in rats. Acta Physiol (Oxf) 201:275–287. https://doi.org/10.1111/j.1748-1708.2010.02174.x
Muramatsu H, Shirahama H, Yonezawa S, Maruta H, Muramatsu T (1993) Midkine, a retinoic acid-inducible growth/differentiation factor: immunochemical evidence for the function and distribution. Dev Biol 159:392–402. https://doi.org/10.1006/dbio.1993.1250
Murata Y et al (2012) Delayed inhibition of c-Jun N-terminal kinase worsens outcomes after focal cerebral ischemia. J Neurosci Off J Soc Neurosci 32:8112–8115. https://doi.org/10.1523/JNEUROSCI.0219-12.2012
Namura S et al (2001) Intravenous administration of MEK inhibitor U0126 affords brain protection against forebrain ischemia and focal cerebral ischemia. Proc Natl Acad Sci U S A 98:11569–11574. https://doi.org/10.1073/pnas.181213498
Nishimura M et al (2003) Activation of p38 kinase in the gerbil hippocampus showing ischemic tolerance. J Cereb Blood Flow Metab 23:1052–1059. https://doi.org/10.1097/01.WCB.0000084251.20114.65
Otsuka S, Sakakima H, Sumizono M, Takada S, Terashi T, Yoshida Y (2016) The neuroprotective effects of preconditioning exercise on brain damage and neurotrophic factors after focal brain ischemia in rats. Behav Brain Res 303:9–18. https://doi.org/10.1016/j.bbr.2016.01.049
Park HR, Lee H, Lee JJ, Yim NH, Gu MJ, Ma JY (2017) Protective effects of Spatholobi Caulis extract on neuronal damage and focal ischemic stroke/reperfusion injury. Mol Neurobiol. https://doi.org/10.1007/s12035-017-0652-x
Satoh T, Nakatsuka D, Watanabe Y, Nagata I, Kikuchi H, Namura S (2000) Neuroprotection by MAPK/ERK kinase inhibition with U0126 against oxidative stress in a mouse neuronal cell line and rat primary cultured cortical neurons. Neurosci Lett 288:163–166
Sawe N, Steinberg G, Zhao H (2008) Dual roles of the MAPK/ERK1/2 cell signaling pathway after stroke. J Neurosci Res 86:1659–1669. https://doi.org/10.1002/jnr.21604
Shen CP, Tsimberg Y, Salvadore C, Meller E (2004) Activation of Erk and JNK MAPK pathways by acute swim stress in rat brain regions. BMC Neurosci 5:36. https://doi.org/10.1186/1471-2202-5-36
Tournier C et al (2000) Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 288:870–874
Wang S, Yoshida Y, Goto M, Moritoyo T, Tsutsui JI, Izumo S, Sato E, Muramatsu T, Osame M (1998) Midkine exists in astrocytes in the early stage of cerebral infarction. Brain Res Dev Brain Res 106:205–209
Wang Z, Chen X, Zhou L, Wu D, Che X, Yang G (2003) Effects of extracellular signal-regulated kinase (ERK) on focal cerebral ischemia. Chin Med J (Engl) 116:1497–1503
Wang Y, Sun ZY, Zhang KM, Xu GQ, Li G (2011) Bcl-2 in suppressing neuronal apoptosis after spinal cord injury. World J Emerg Med 2:38–44
Wang L et al (2014) Protective effect of shikonin in experimental ischemic stroke: attenuated TLR4, p-p38MAPK, NF-kappaB, TNF-alpha and MMP-9 expression, up-regulated claudin-5 expression, ameliorated BBB permeability. Neurochem Res 39:97–106. https://doi.org/10.1007/s11064-013-1194-x
Wang JY, Chen SP, Gao YH, Qiao LN, Zhang JL, Liu JL (2015) Effect of repeated electroacupuncture intervention on hippocampal ERK and p38MAPK signaling in neuropathic pain rats. Evid Based Complement Alternat Med 2015:641286. https://doi.org/10.1155/2015/641286
Wu DC, Ye W, Che XM, Yang GY (2000) Activation of mitogen-activated protein kinases after permanent cerebral artery occlusion in mouse brain. J Cereb Blood Flow Metab 20:1320–1330. https://doi.org/10.1097/00004647-200009000-00007
Wu C, Wang J, Li C, Zhou G, Xu X, Zhang X, Lan X (2015) Effect of electroacupuncture on cell apoptosis and ERK signal pathway in the hippocampus of adult rats with cerebral ischemia-reperfusion. Evid Based Complement Alternat Med 2015:414965. https://doi.org/10.1155/2015/414965
Wu C et al (2017) Effects of electroacupuncture on the cortical extracellular signal regulated kinase pathway in rats with cerebral ischaemia/reperfusion. Acupunct Med 35:430–436. https://doi.org/10.1136/acupmed-2016-011121
Xue X et al (2014) Electro-acupuncture at points of Zusanli and Quchi exerts anti-apoptotic effect through the modulation of PI3K/Akt signaling pathway. Neurosci Lett 558:14–19. https://doi.org/10.1016/j.neulet.2013.10.029
Yang L, Yue N, Zhu X, Han Q, Liu Q, Yu J, Wu G (2013) Electroacupuncture upregulates ERK signaling pathways and promotes adult hippocampal neural progenitors proliferation in a rat model of depression. BMC Complement Altern Med 13:288. https://doi.org/10.1186/1472-6882-13-288
Yoshida Y, Goto M, Tsutsui J, Ozawa M, Sato E, Osame M, Muramatsu T (1995) Midkine is present in the early stage of cerebral infarct. Brain Res Dev Brain Res 85:25–30
Yoshida Y et al (2008) Expression of the heparin-binding growth factor midkine in the cerebrospinal fluid of patients with neurological disorders. Intern Med 47:83–89
Youle RJ, Strasser A (2008) The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 9:47–59. https://doi.org/10.1038/nrm2308
Zhang F, Wu Y, Jia J, Hu YS (2010) Pre-ischemic treadmill training induces tolerance to brain ischemia: involvement of glutamate and ERK1/2. Molecules 15:5246–5257. https://doi.org/10.3390/molecules15085246
Zhen Y, Ding C, Sun J, Wang Y, Li S, Dong L (2016) Activation of the calcium-sensing receptor promotes apoptosis by modulating the JNK/p38 MAPK pathway in focal cerebral ischemia-reperfusion in mice. Am J Transl Res 8:911–921
Zheng M et al (2018) Netrin-1 promotes synaptic formation and axonal regeneration via JNK1/c-Jun pathway after the middle cerebral artery occlusion front cell. Neuroscience 12:13. https://doi.org/10.3389/fncel.2018.00013
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The study was approved by the Animal Care and Use Committee of Hebei Medical University.
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Xing, Y., Yang, SD., Wang, MM. et al. Electroacupuncture Alleviated Neuronal Apoptosis Following Ischemic Stroke in Rats via Midkine and ERK/JNK/p38 Signaling Pathway. J Mol Neurosci 66, 26–36 (2018). https://doi.org/10.1007/s12031-018-1142-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-018-1142-y