
Abstract
Since an antipsychotic drug haloperidol has been clinically reported to induce QT interval prolongation and torsade de pointes, in this study its risk stratification for the onset of torsade de pointes was performed by using the chronic atrioventricular block canine model with a Holter electrocardiogram. Haloperidol in a dose of 3 mg kg−1 p.o. prolonged the QT interval, but it did not induce torsade de pointes during the observation period of 21 h (n = 4), indicating that the dose would be safe. Meanwhile, haloperidol in a dose of 30 mg kg−1 p.o. significantly increased the short-term variability in beat-to-beat analysis of QT interval (n = 4), and it induced torsade de pointes in 4 animals out of 4, showing that the dose could be torsadogenic. Since 3 mg kg−1 p.o. of haloperidol in this study can be estimated to provide about 8 times higher plasma concentrations than its therapeutic level, haloperidol may be used safely for most of the patients, as long as its plasma drug concentration is kept within the therapeutic range.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Haloperidol is one of the most widely used antipsychotics and is the prototype of the butyrophenone class [1]. Parenteral forms of haloperidol including intravenous, intramuscular and depot injectable preparations in addition to oral one have been used in the management of acute and chronic psychotic disorders as well as delirium-associated agitation [2], because of its effectiveness, low rate of anticholinergic side effects, familiarity with the dosage, and minimal respiratory or sedative properties. In an analysis of total number of 70 cases of intravenous haloperidol-associated QT interval prolongation and/or torsade de pointes, patients who experienced torsade de pointes received total doses of 5–645 mg, whereas those with QT interval prolongation alone took ones of 2–1540 mg [2].
Haloperidol has been shown to block human ether-á-go–go-related gene (hERG) K+ channels [3], which is considered to provide rationale for QT interval prolongation in experimental animals and patients [4–10]. In the torsadogenic propensity categories in the Association of the British Pharmaceutical Industry (ABPI) survey, haloperidol belongs to category 3, which means that the drug has “measurable incidence/numerous reports of torsade de pointes” [2, 6]. However, experimental evidence showing the causal link between the administration of haloperidol and the onset of torsade de pointes is lacking except for the one in vitro study with the rabbit Langendorff heart model [11].
In this study, we clarified the causal link and, moreover, quantified proarrhythmic potential of haloperidol by estimating its safety margin for the onset of torsade de pointes in vivo. For this purpose, we adopted the chronic atrioventricular block canine model, which has common pathophysiological changes in the heart to patients who are the most sensitive to the drug-induced long QT syndrome, and has been widely used for quantifying the torsadogenic potential of various drugs [12].
Materials and Methods
All experiments in this study were approved by Toho University Animal Care and User Committee (No. 15-55-152) and performed in accordance with the Guidelines for the Care and Use of Laboratory Animals of Toho University and Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines for reporting experiments involving animals [13, 14]. Experiments were performed with 4 female beagle dogs weighing approximately 9 kg. Animals were obtained through Kitayama Labes Co., Ltd. (Nagano, Japan). The dogs were kept in individual cages on a 12-h light (6:00–18:00)–12-h dark (18:00–6:00) cycle. The ventilation provided a total air exchange rate of 10–15 times per hour. The room temperature was maintained at 23 ± 2 °C, and relative humidity was 50 ± 30 %. Each dog was fed 200 g day−1 of standard diet (CD-5M, CLEA Japan, Tokyo, Japan) and was allowed free access to tap water.
Production of Complete Atrioventricular Block
The catheter ablation technique for producing atrioventricular block was used as previously described [15]. The dogs were anesthetized with thiopental sodium (30 mg kg−1, i.v.). After intubation with a cuffed endotracheal tube, 100 % oxygen was inhaled with a volume-limited ventilator (SN-480-3; Shinano Manufacturing, Tokyo, Japan). Tidal volume and respiratory rate were set at 20 mL kg−1 and 15 strokes min−1, respectively. To prevent blood clotting, heparin calcium (100 IU kg−1, i.v.) was administered. The surface lead II electrocardiogram was continuously monitored with a polygraph system (RM-6000; Nihon Kohden, Co., Tokyo, Japan). A quadpolar electrodes catheter with a large tip of 4 mm (D7-DL-252; Cordis-Webster, Baldwin Park, CA, USA) was inserted through the right femoral vein by using a standard percutaneous technique under a sterile condition and positioned around the tricuspid valve, watching the bipolar electrograms from the distal electrodes pair. The optimal site for “fast pathway” ablation of atrioventricular node was judged based on the intracardiac electrogram, of which a very small His deflection was recorded and atrial/ventricular voltage ratio was >2 [16, 17]. The site was usually found at 1–2 cm proximal from the position where the largest His bundle electrogram was recorded. The power source for atrioventricular node ablation was obtained from an electrosurgical generator (MS-1500; Mera Senko Medical Instrument Mfg. Co., Ltd., Tokyo, Japan), which delivers continuous unmodulated radiofrequency energy at a frequency of 500 kHz. After proper positioning, the radiofrequency energy of 20 W was delivered for 10 s from the tip electrode to an indifferent patch electrode positioned on the animal’s back, which continued for 30 s if junctional rhythm was induced. The end point of this procedure was the development of the complete atrioventricular block with an onset of stable idioventricular escaped rhythm.
Holter Electrocardiogram Recording
A Holter recording and analysis system (QR2100 and HS1000, Fukuda M-E Kogyo Co., Ltd., Tokyo, Japan) was used to record and analyze electrocardiogram over 24 h. The effects of haloperidol on the ventricular rate, QT interval and corrected QT calculated with the Fridericia’s formula (QTcF): QTcF = QT/(RR/1000)1/3 [18] in addition to their proarrhythmic actions were assessed without anesthesia. The ventricular rate, QT interval and QTcF were expressed as the mean of ten consecutive complexes. In this study, torsade de pointes was defined as a polymorphic ventricular tachycardia associated with QT interval prolongation, consisting of 5 beats or more twisting QRS complexes around the baseline [19].
Experimental Protocol
At least 4 weeks after the induction of complete atrioventricular block, experiments were performed. We have assessed proarrhythmic effects of various drugs in two escalating doses with the group size of 4–6, which has been confirmed to achieve enough sensitivity and reliability to demonstrate the potential for the onset of drug-induced torsade de pointes [12]. At the first day, about 2 h after the start of Holter electrocardiogram recording, 3 mg kg−1 of haloperidol, which is several-fold higher dose than the therapeutic one, was orally administered with gelatin capsules (J.P No.00, Kobayashi capsule, Hyogo, Japan). Ventricular rate and QT interval were assessed at 1, 2, 3, 4, 6, 8, 12 and 21 h after the start of the administration. At the second day, 30 mg kg−1 of the drug, which is several tenfold higher dose than the therapeutic one, was orally administered, and the variables were assessed in the same manner. The oral administration protocol has been used in order to maintain the plasma drug concentration for more than several hours, which would increase the chance to detect the events induced by a parent compound as well as its metabolites [12]. The electrocardiogram parameters at 0.5–1 h before the drug administration were defined as the control, and the electrocardiogram was recorded for total of 24 h when lethal arrhythmia was not induced. Safety margin for the onset of torsade de pointes was defined as a ratio of the torsadogenic doses/concentrations to the therapeutic ones; namely, when torsade de pointes was induced by 1–3 times of the therapeutic doses/concentrations, the safety margin was 1–3 [12].
Beat-to-Beat Analysis
Electrocardiogram of 51 of consecutive beats under the stable idioventricular automaticity without ectopic activity was adopted before and about 2 h after the administration of haloperidol in a dose of 30 mg kg−1. When the torsade de pointes was observed, 51 of consecutive beats were obtained just before the onset of the arrhythmia. When the QT interval was obscured by P wave, we estimated the end of T wave by canceling the component of the P wave from the electrocardiogram waveform on screen. Poincaré plots with QT n versus QT n+1 were prepared for each of the two analysis time points. The mean orthogonal distance from the diagonal to the points of the Poincaré plot was determined as short-term variability (=Σ|QT n+1–QT n |/[50√2]). On the other hand, the mean distance to the mean of the parameter parallel to the diagonal of the Poincaré plot was determined as long-term variability (=Σ|QT n+1 + QT n –2QTmean|/[50√2]). These nomenclatures have been adopted from investigations of heart rate variability in humans [20], which have been applied to the QT interval analysis of normal dogs and chronic atrioventricular block dogs [21].
Drugs
The following drugs were purchased: haloperidol (Sigma-Aldrich, St Louis, MO, USA), thiopental sodium (Ravonal® 0.5 g for Injection, Mitsubishi Tanabe Pharma, Osaka, Japan) and heparin calcium (Caprocin®, Sawai Pharmaceutical Co., Ltd., Osaka, Japan).
Statistics
Data are presented as the mean ± SE. The statistical analysis was performed with software GraphPad Prism 6 (version 6.03, GraphPad software, Inc., La Jolla, CA, USA). The statistical significances within a parameter were evaluated with one-way repeated measures analysis of variance (ANOVA) followed by a post hoc test for mean values comparison. A p value <0.05 was considered to be statistically significant.
Results
Effects on the Electrocardiogram
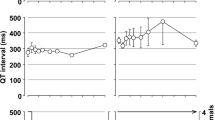
The time courses of the changes in the electrocardiogram variables and the number of surviving animals after the administrations of haloperidol are summarized in Fig. 1 (n = 4). The pre-drug control values (C) of the ventricular rate, QT interval and QTcF were 27 ± 3 beats min−1, 344 ± 20 ms and 257 ± 6 for the low dose of 3 mg kg−1, whereas those were 27 ± 2 beats min−1, 354 ± 10 ms and 269 ± 7 for the high dose of 30 mg kg−1, respectively. The low dose prolonged the QT interval for 3–21 h and QTcF for 4–12 h, whereas no significant change was detected in the ventricular rate. The high dose tended to increase the QT interval and QTcF, which did not achieve statistical significance because of the reduction in the number of surviving animals. The ventricular rate was hardly altered.

Time courses of changes in the ventricular rate (bpm, top), QT interval (ms, middle) and QTcF (bottom) before and after the oral administration of haloperidol at 3 (left) and 30 (right) mg kg−1 (n = 4, each). Data are presented as the mean ± SE. Closed symbols represent significant changes from the respective pre-drug control values (C) by p value <0.05
The low dose did not induce torsade de pointes, whereas the high dose did it in 4 animals out of 4. Typical tracings of the electrocardiogram showing the torsadogenic actions of the high dose are depicted in Fig. 2. It should be noted that the morphology and axis of the QRS complex were changed after the administration of the high dose in each animal, as depicted in Fig. 2. The high dose triggered the initial episode of torsade de pointes at 1.3–3.5 h. The total number of the episodes ranged 1–9. The latest event degenerated into the ventricular fibrillation (#1 and #2) or cardiac arrest (#3), leading to animal’s death as shown in Fig. 1, whereas in the remaining 1 animal (#4), two episodes of torsade de pointes were observed at 17.8 and 17.9 h, which were spontaneously terminated. This animal was found to be dead soon after the cessation of the Holter electrocardiogram recording.

Representative electrocardiogram before (pre-drug control) and 2 h after the oral administration of 30 mg kg−1 of haloperidol (2 h after 30 mg kg−1, p.o.). Hash key (#) represents the animal number. TdP torsade de pointes
Beat-to-Beat Analysis
Beat-to-beat analysis was performed for each animal to quantify the extent of torsadogenic potential of the drug as depicted in Poincaré plots in Fig. 3. The results are summarized in Table 1. The QT interval of electrocardiogram of 51 consecutive beats under stable idioventricular rhythm was measured in each animal before and 1.4–2.6 h after the high dose. The short-term variability of repolarization increased in each animal irrespective of the QT interval changes, whereas no significant change was detected in the long-term variability of it.

Effects of 30 mg kg−1 p.o. of haloperidol on the Poincaré plots of the QT interval in the canine chronic atrioventricular block dogs. Fifty-one beats were plotted for each of the two analysis time points, before (gray) and 1.4–2.6 h after (black) the administration. The short-term variability (STV) and long-term variability (LTV) in each animal (#1, #2, #3 and #4) are depicted in their respective panels. Hash key (#) represents the animal number
Discussion
Using the chronic atrioventricular block canine model [12], we performed the risk stratification of haloperidol for the onset of torsade de pointes. Haloperidol in a dose of 3 mg kg−1 p.o. prolonged the QT interval, but it did not induce torsade de pointes, indicating that the dose would be safe, which supports a previous knowledge [2] that the risk of torsade de point is low if the cumulative dose was less than 2 mg. Meanwhile, haloperidol in a dose of 30 mg kg−1 p.o. significantly increased the short-term variability in beat-to-beat analysis of QT interval, resulting in the onset of torsade de pointes in 4 animals out of 4, which suggests the dose could be torsadogenic.
The Rationale for the Drug Doses
In a previous clinical study in psychotic patients (n = 39), administration of a single oral dose of 0.2 mg kg−1 was reported to achieve C max of 10.5 ng mL−1 (27.9 nmol L−1) at a mean of 4.5 h (T max), and the range of elimination half-life was shown to be between 8.5 and 66.6 h with a mean of 19.5 h [1]. In another study with psychotic patients treated with 15 mg day−1 of haloperidol for 12 days (n = 27), peak plasma/serum concentration (C max) was reported to be 16.1 ng mL−1 (43 nmol L−1) in average [22], which could reflect its therapeutic drug level. Meanwhile, in previous experimental studies with dogs, intravenous administration of 0.3 and 3 mg kg−1 of haloperidol provided C max of 140 ± 6 ng mL−1 (359 nmol L−1) and 1371 ± 170 ng mL−1 (3642 nmol L−1), respectively [4], whereas oral administration of 3 mg kg−1 of it attained C max of 362 ± 101 nmol L−1 with T max of 2.8 ± 1.1 h [23], suggesting that 3 and 30 mg kg−1 p.o. dose of haloperidol in this study could have provided similar peak levels of the plasma drug concentrations after the administration of 0.3 and 3 mg kg−1 i.v., respectively, and that episodes of torsade de pointes leading to animals’ death might have occurred around T max. Thus, the doses assessed in this study can be considered to provide ≥8 times higher concentrations than the therapeutic level.
QT Interval Prolongation
IC50 values of haloperidol have been reported to be 7.0 µmol L−1 for Na+ channels [24], 500 µmol L−1 for L-type Ca2+ channels [25], 1.6 µmol L−1 for ATP-sensitive K+ channels [26] and 26 nmol L−1 for hERG K+ channels [3]. The protein-binding ratio of haloperidol has been reported to be 92 % [27, 28], suggesting that 3 and 30 mg kg−1 p.o. dose in this study could have provided free plasma concentrations of 29–290 nmol L−1, respectively. Based on this previously obtained knowledge, the electrophysiological events observed in this study are considered to largely depend on hERG K+ channel inhibition by haloperidol.
Proarrhythmic Effect
Since the chronic atrioventricular block canine model was found to simulate a patient who would be the most sensitive to inhibitors of rapidly activating delayed rectifier K+ current (I Kr), we have proposed the following risk stratification of proarrhythmia [12]. When the torsade de pointes was induced by 1–3 times of the therapeutic dose/concentration, the drug can be considered to have high risk of proarrhythmia. If it was not observed by 1–3 times of the therapeutic dose/concentration, but induced by its 10–30 times, the drug was judged to have low risk [12]. Thus, torsadogenic risk of haloperidol could be classified to be low by our risk stratification of proarrhythmia [12].
Utility of previously described surrogate markers for proarrhythmia deserves comments. Using beat-to-beat analysis of QT interval, we showed that oral administration of 30 mg kg−1 of haloperidol increased the short-term variability to >8.5 ms in each animal irrespective of the extent of the QT interval changes (Fig. 3). These results confirm that the short-term variability may be a highly sensitive and reliable marker for predicting the onset of haloperidol-induced torsade de pointes [20, 21]. On the other hand, in our previous study using the halothane-anesthetized dogs, haloperidol increased the terminal repolarization period [4], which can reflect the increase in electrical vulnerability during phase 3 repolarization [12]. The current results also confirm that terminal repolarization period can be efficacious for estimating the proarrhythmic potential of haloperidol.
The high dose of haloperidol changed the morphology and axis of the QRS complex from those at control, which also needs comments. Since haloperidol potently inhibits hERG K+ channel together with modest Na+ channel inhibition [3, 24], it may cause intraventricular conduction delay including fascicular block at high levels [4, 5], therefore explaining QRS widening and axis shift.
Limitation
In this study, we could not measure plasma concentrations of haloperidol to prevent the impact of the blood collection procedure on the onset rate of proarrhythmic events. Although there has been no report describing pharmacokinetic profile of oral administration of 30 mg kg−1 of haloperidol in dogs, that of 3 mg kg−1 was reported previously [23]. Also, pharmacokinetic information of intravenous administration of 0.3 and 3 mg kg−1 of haloperidol in dogs was available [4]. Using these previous results [4, 23] together with our experiences with this canine model [12], we estimated the pharmacokinetic profile of oral administration of 3 and 30 mg kg−1 of haloperidol in this study.
We could not assess the electrocardiograms just before the onset of unexpected death in one animal (#4), since it occurred after the experimental periods of 24 h. However, we believe that there may be causal relationship between the haloperidol administration and the sudden death in this case, since the animal had been alive for >1 month after the induction of complete atrioventricular block without any symptom of heart failure including appetite loss and general edema.
Currently used chronic atrioventricular block dog is an animal model for assessing proarrhythmic effect of a drug. However, it may not necessarily reflect the clinically observed drug-induced torsade de pointes, since the clinical QT-prolonging effect as well as proarrhythmic potential of a drug is dependent on many factors including age, gender, presence of structural heart disease, genetic predisposition, individual pharmacodynamics and pharmacokinetic sensitivity.
Conclusion
Haloperidol in a dose of 3 mg kg−1, p.o., which would have provide approximately 8 times higher concentration than the therapeutic level, delayed the ventricular repolarization, whereas its 10 times higher dose induced torsade de pointes in the chronic atrioventricular block dogs, which could be largely explained by its hERG K+ channel inhibitory profile, indicating that the safety margin of haloperidol for the onset of torsade de pointes might be at least >8 times of the therapeutic concentration. Thus, haloperidol can be used safely for most of the patients, as long as its plasma drug concentration is kept within the therapeutic range.
References
Khot, V., DeVane, C. L., Korpi, E. R., Venable, D., Bigelow, L. B., Wyatt, R. J., et al. (1993). The assessment and clinical implications of haloperidol acute-dose, steady-state, and withdrawal pharmacokinetics. Journal of Clinical Psychopharmacology, 13, 120–127.
Meyer-Massetti, C., Cheng, C. M., Sharpe, B. A., Meier, C. R., & Guglielmo, B. J. (2010). The FDA extended warning for intravenous haloperidol and torsades de pointes: How should institutions respond? Journal of Hospital Medicine, 5, E8–E16.
Polak, S., Wiśniowska, B., & Brandys, J. (2009). Collation, assessment and analysis of literature in vitro data on hERG receptor blocking potency for subsequent modeling of drugs’ cardiotoxic properties. Journal of Applied Toxicology, 29, 183–206.
Sugiyama, A., Satoh, Y., & Hashimoto, K. (2001). In vivo canine model comparison of cardiohemodynamic and electrophysiological effects of a new antipsychotic drug aripiprazole (OPC-14597) to haloperidol. Toxicology and Applied Pharmacology, 173, 120–128.
Mörtl, D., Agneter, E., Krivanek, P., Koppatz, K., & Todt, H. (2003). Dual rate-dependent cardiac electrophysiologic effects of haloperidol: Slowing of intraventricular conduction and lengthening of repolarization. Journal of Cardiovascular Pharmacology, 41, 870–879.
Redfern, W. S., Carlsson, L., Davis, A. S., Lynch, W. G., MacKenzie, I., Palethorpe, S., et al. (2003). Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: Evidence for a provisional safety margin in drug development. Cardiovascular Research, 58, 32–45.
Kijtawornrat, A., Ozkanlar, Y., Keene, B. W., Roche, B. M., Hamlin, D. M., & Hamlin, R. L. (2006). Assessment of drug-induced QT interval prolongation in conscious rabbits. Journal of Pharmacological and Toxicological Methods, 53, 168–173.
Ando, K., Hombo, T., Kanno, A., Ikeda, H., Imaizumi, M., Shimizu, N., et al. (2005). QT PRODACT: In vivo QT assay with a conscious monkey for assessment of the potential for drug-induced QT interval prolongation. Journal of Pharmacological Sciences, 99, 487–500.
Kano, M., Toyoshi, T., Iwasaki, S., Kato, M., Shimizu, M., & Ota, T. (2005). QT PRODACT: Usability of miniature pigs in safety pharmacology studies: Assessment for drug-induced QT interval prolongation. Journal of Pharmacological Sciences, 99, 501–511.
Testai, L., Breschi, M. C., Martinotti, E., & Calderone, V. (2007). QT prolongation in guinea pigs for preliminary screening of torsadogenicity of drugs and drug-candidates. II. Journal of Applied Toxicology, 27, 270–275.
Hondeghem, L. M., Lu, H. R., van Rossem, K., & De Clerck, F. (2003). Detection of proarrhythmia in the female rabbit heart: Blinded validation. Journal of Cardiovascular Electrophysiology, 14, 287–294.
Sugiyama, A. (2008). Sensitive and reliable proarrhythmia in vivo animal models for predicting drug-induced torsades de pointes in patients with remodelled hearts. British Journal of Pharmacology, 154, 1528–1537.
Kilkenny, C., Browne, W., Cuthill, I. C., Emerson, M., & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579.
McGrath, J., Drummond, G., McLachlan, E., Kilkenny, C., & Wainwright, C. (2010). Guidelines for reporting experiments involving animals: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1573–1576.
Sugiyama, A., Ishida, Y., Satoh, Y., Aoki, S., Hori, M., Akie, Y., et al. (2002). Electrophysiological, anatomical and histological remodeling of the heart to AV block enhances susceptibility to arrhythmogenic effects of QT-prolonging drugs. Japanese Journal of Pharmacology, 88, 341–350.
Sugiyama, A., Satoh, Y., Ishida, Y., Yoneyama, M., Yoshida, H., & Hashimoto, K. (2002). Pharmacological and electrophysiological characterization of junctional rhythm during radiofrequency catheter ablation of the atrioventricular node: Possible involvement of neurotransmitters from autonomic nervous system. Circulation Journal, 66, 696–701.
Rubart, M., & Zipes, D. P. (2008). Arrhythmias, sudden death, and syncope. In P. Libby, R. O. Bonow, D. L. Mmann, D. P. Zipes, & E. Braunwald (Eds.), Braunwald’s heart disease (8th ed., pp. 763–778). Philadelphia: Saunders.
Fridericia, L. S. (1920). Die systolendauer in elektrokardiogramm bei normalen menschen und bei herzkranken. Acta Medica Scandinavica, 53, 469–486.
Satoh, T., & Zipes, D. P. (1996). Rapid rates during bradycardia prolong ventricular refractoriness and facilitate ventricular tachycardia induction with cesium in dogs. Circulation, 94, 217–227.
Brennan, M., Palaniswami, M., & Kamen, P. (2001). Do existing measures of Poincaré plot geometry reflect nonlinear features of heart rate variability? IEEE Trans BioMedical Engineering, 48, 1342–1347.
Thomsen, M. B., Verduyn, S. C., Stengl, M., Beekman, J. D., de Pater, G., van Opstal, J., et al. (2004). Increased short-term variability of repolarization predicts d-sotalol-induced torsades de pointes in dogs. Circulation, 110, 2453–2459.
Harrigan, E. P., Miceli, J. J., Anziano, R., Watsky, E., Reeves, K. R., Cutler, N. R., et al. (2004). A randomized evaluation of the effects of six antipsychotic agents on QTc, in the absence and presence of metabolic inhibition. Journal of Clinical Psychopharmacology, 24, 62–69.
Chaves, A. A., Zingaro, G. J., Yordy, M. A., Bustard, K. A., O’Sullivan, S., Galijatovic-Idrizbegovic, A., et al. (2007). A highly sensitive canine telemetry model for detection of QT interval prolongation: Studies with moxifloxacin, haloperidol and MK-499. Journal of Pharmacological and Toxicological Methods, 56, 103–114.
Ogata, N., & Narahashi, T. (1989). Block of sodium channels by psychotropic drugs in single guinea-pig cardiac myocytes. British Journal of Pharmacology, 97, 905–913.
Tarabová, B., Nováková, M., & Lacinová, L. (2009). Haloperidol moderately inhibits cardiovascular L-type calcium current. General Physiology and Biophysics, 28, 249–259.
Yang, S. B., Proks, P., Ashcroft, F. M., & Rupnik, M. (2004). Inhibition of ATP-sensitive potassium channels by haloperidol. British Journal of Pharmacology, 143, 960–967.
Wishart, D. S., Knox, C., Guo, A. C., Shrivastava, S., Hassanali, M., Stothard, P., et al. (2006). DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Research, 34(Database issue): D668–D672.
DrugBank Version 4.3. http://www.drugbank.ca/drugs/DB00502 (haloperidol). Accessed August 18, 2015.
Acknowledgments
This study was funded in part by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology in Japan JSPS KAKENHI (16K08559); the Research Promotion Grant from Toho University Graduate School of Medicine (No. 16-02 to AS); and Toho University Joint Research Fund (No. 16-05 to AS). We thank Ms. Misako Nakatani and Mrs. Yuri Ichikawa for their technical assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Izumi-Nakaseko, H., Nakamura, Y., Cao, X. et al. Assessment of Safety Margin of an Antipsychotic Drug Haloperidol for Torsade de Pointes Using the Chronic Atrioventricular Block Dogs. Cardiovasc Toxicol 17, 319–325 (2017). https://doi.org/10.1007/s12012-016-9388-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12012-016-9388-5