
Abstract
Ellagic acid (EA) is a phenolic constituent in certain fruits and nuts with wide range of biological activities, including potent antioxidant, antidiabetic, anti-inflammatory, anticancer and antimutagen properties. The aim of this study was to evaluate the effect of EA on sodium arsenic (SA)-induced cardio- and hematotoxicity in rats. Animals were divided into five groups. The first group was used as control. Group 2 was orally treated with sodium arsenite (SA, 10 mg/kg) for 21 days. Group 3 was orally treated with EA (30 mg/kg) for 14 days. Groups 4 and 5 were orally treated with SA for 7 days prior to EA (10 and 30 mg/kg, respectively) treatment and continued up to 21 days simultaneous with SA administration. Various biochemical, histological and molecular biomarkers were assessed in blood and heart. The results indicate that SA-intoxicated rats display significantly higher levels of plasma cardiac markers (AST, CK-MB, LDH and cTnI) than normal control animals. Moreover, an increase in MDA and NO with depletion of GSH and activities of CAT, SOD and GPx occurred in the heart of rats treated with SA. Furthermore, SA-treated rats showed significantly lower WBC, RBC, HGB, HCT and PLT and significantly higher MCV and MCH. Administration of EA (30 mg/kg) resulted in a significant reversal of hematological and cardiac markers in arsenic-intoxicated rats. These biochemical disturbances were supported by histopathological observations of the heart. In conclusion, the results of this study suggest that EA treatment exerts a significant protective effect on SA-induced cardio- and hematotoxicity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
It is estimated that 200 million people around the world are affected by arsenic exposure which has become a serious health problem across the world especially in poor countries [1]. The major routes of exposure to arsenic are through drinking water and industrial pollution [2]. It has been shown that sodium arsenite (SA, NaAsO2) is the most toxic chemical among various arsenic compounds [1,2,3]. Exposure to arsenic is known to induce a wide range of toxic effects in different tissues such as blood and heart [4]. Broad consensus has not been reached regarding the precise mechanisms of arsenic toxicity and these are still being studied intensively [5]. Arsenic induces the over production of reactive oxygen species (ROS), enhances lipid peroxidation and increases the production of nitric oxide (NO) [3]. On the other hand, it is well established that arsenic disturbs antioxidant/pro-oxidant ratio via deleterious effects on the antioxidants enzymes such as superoxide dismutase (SOD), glutathione peroxidase (GPx), catalase (CAT) and glutathione (GSH) [6]. It seems that antioxidant compounds such as linolenic acid, resveratrol and N-acetylcysteine have protective effects on arsenic toxicity through increasing the antioxidant capacity [7,8,9].
Ellagic acid (EA; 2,3,7,8-tetrahydroxy-chromeno[5,4,3-cde]chromene-5,10-dione) is a phenolic constituent found in certain fruits and nuts, such as raspberry, strawberry, walnut and pomegranate [10]. EA has a variety of biological activities, including potent antioxidant, antidiabetic, anti-inflammatory, anticancer and antimutagen properties [11]. EA is a safe substance and Doyle et al. [12] have found have that feeding rats with EA (50 mg/day up to 45 days) does not cause any sign of systemic toxicity. The exact mechanism of EA is not known, but its potent scavenging action against ROS such as hydroxyl and superoxide radicals are well documented [13]. Also, EA exerts its antioxidant effect via activating or inducing cellular antioxidant enzyme systems [14]. Moreover, EA induces its anti-inflammatory effects via inhibiting the cyclooxygenase (COX) enzyme and reducing the expression of this enzyme [15]. It has demonstrated that EA has protective effect on oxidative damage in lung, liver and kidney [16,17,18].
Due to the above-mentioned EA effects, we investigated whether that it has protective effect on SA-induced cardio- and hematotoxicity in rats.
Materials and Methods
Chemicals
Sodium arsenite (SA; Catalog Number: 7784-46-5) and Ellagic acid (EA; Catalog Number: 476-66-4) were purchased from Sigma-Aldrich Chemical Company (St. Louis, MO, USA). All other chemicals and reagents used were analytical grade and prepared from Merck Company (Darmstadt, Germany).
Animals
Thirty-five adult male Wistar rats (180–200 g) were obtained from animal house of Ahvaz Jundishapur University of Medical Sciences. Animals were housed seven per cage and kept on commercial diet and tap water provided ad libitum. Rats were maintained at a controlled condition of temperature (20 ± 2 °C) with a 12 h light: 12 h dark cycle [19]. The experiments were performed according to the Animal Ethics Committee Guidelines for the use of experimental animals (Ethic code: IR.AJUMS.REC.1395.641).
Experimental Protocol
Animals were randomly divided into five groups each consisted of seven rats. Group 1 received normal saline (2 ml/kg) for 21 days. Group 2 received SA 10 mg/kg, p.o., daily for 21 days) [20]. Group 3 received EA (30 mg/kg, p.o.) for 14 consecutive days [21]. Groups 4 and 5 received SA for 7 days prior to EA (in doses of 10 and 30 mg/kg, respectively) treatment and continued up to 21 days in parallel with SA administration (14 days). All drugs were administered through oral gavage.
Sample Collection
The control and SA-treated rats were euthanized 22 days after the first dose of SA, and the rats in EA group were euthanized 16 days after the first dose of EA. The animals were anaesthetized with combination of ketamine/xylazine (60/6 mg/kg, i.p.), and two blood samples were collected through cardiac puncture from the left ventricle. One sample was used for serum separation (centrifugation for 10 min at 3000 rpm) and stored at − 20 °C until analysis. The second sample was used for complete blood count (inverted several times in an EDTA-coated tube to prevent coagulation).
Then animals were sacrificed by rapid decapitation. Then heart tissues were isolated and washed with saline quickly. For histological studies, a part of these samples was fixed in 10% phosphate buffered formalin. For biochemical estimations, the second part of this tissue was homogenized (1/10 w/v) in ice-cold Tris–HCl buffer (0.1 M, pH 7.4).
Blood Parameters
Hematological Analyses
The complete blood count was used for hematological analyses. The WBC indices, RBC indices and platelets were analyzed immediately by an ADVIA 2120 hematology system (Siemens, Munich, Germany), according to the manufacturer’s instructions.
Biochemical Analyses
Specific markers related to cardiac dysfunction were measured in plasma samples. Aspartate Aminotransferase (AST) activities were assayed according to Reitman and Frankel [22] using commercially available kits. Creatine kinase-MB (CK-MB) activity was estimated in serum by commercially available CK-MB assay kit (BioAssay Systems, USA; Catalog Number: ECPK-100) adopting the method of Bishop [23]. Lactate dehydrogenase (LDH) activity was estimated in serum by commercially available LDH kit (Linear Chemicals, S.L., UK; Catalog Number: REF KR10282) according to the method of Whitaker [24]. Cardiac Troponin I (cTnI) was estimated in serum by Rat ELISA Kit (MyBioSource, San Diego, USA; Catalog Number: MBS727624).
Assay for Redox Status in Tissue Homogenates
Activities of antioxidant enzymes (SOD, CAT, GSH, GPx) in the heart tissue were determined following the method as described by Ghosh et al. [25]. Cardiac lipid peroxidation was determined by the method described by Buege and Aust [26], which estimates the malondialdehyde (MDA) formation. Cardiac NO levels were measured by the Griess diazotization reaction after conversion of nitrate to nitrite by nitrate reductase in supernatant [27].
Statistical Analysis
Results were expressed as mean ± SD and all comparisons were made by one-way ANOVA test followed by Tukey’s post hoc analysis and p value less than 0.05 was considered statistically significant.
Results
SA-Induced Alterations in Hematological Parameters are Attenuated by EA
As depicted in Table 1, the SA-treated rats showed significantly lower WBC, RBC, HGB, HCT and PLT and significantly higher MCV and MCH than their control counterparts (all p < 0.05). EA (30 mg/kg) mitigated the effect of SA treatment on the above-mentioned variables (all p < 0.05) reflecting the amelioration of hematotoxicity in SA-intoxicated rats. Moreover, EA administration (30 mg/kg) in normal rats did not affect the hematological parameters compared to the control group.
SA-Induced Alterations in Biochemical Parameters are Attenuated by EA
AST, CK-MB, LDH and cTnI are markers of cardiac dysfunction. As shown in Fig. 1, levels of AST, CK-MB, LDH and cTnI were significantly increased in the SA group compared to the control group (all p < 0.001). Treatment with EA (10 mg/kg) significantly prevented SA-induced increase in AST, LDH and cTnI levels (p < 0.01, p < 0.05 and p < 0.05, respectively). Moreover, treatment with EA (30 mg/kg) significantly prevented SA induced increase in AST, CK-MB, LDH and cTnI levels (p < 0.001, p < 0.01, p < 0.001 and p < 0.001, respectively). In addition, administration of EA at the dose of 30 mg/kg did not change these biochemical parameters in naïve rats compared to control group.

Effect of treatment with EA on AST, CK-MB, cTnI and LDH levels in SA-induced cardiotoxicity. Values are mean ± SD (n = 7). Data were analyzed by one-way ANOVA followed by Tukey’s post hoc test for multiple comparisons. *Significant difference in comparison with the control group (***p < 0.001). #Significant difference in comparison with the SA group (#p < 0.05, ##p < 0.01 and ###p < 0.001)
SA-Induced Alterations in Heart Oxidative Stress Parameters are Attenuated by EA
The effects of EA on the heart oxidative stress parameters are shown in Fig. 2. The MDA and NO levels of heart were significantly increased in the SA group compared to the control rats (all p < 0.001). Treatment with EA at the dose of 10 mg/kg significantly decreased the NO level in heart tissue (p < 0.01) (Fig. 1a). Moreover, treatment with EA at the dose of 30 mg/kg significantly decreased both the MDA and NO levels in heart (all p < 0.001) (Fig. 1b). In addition, administration of EA at the dose of 30 mg/kg to naïve rats did not change these oxidative stress parameters compared to rats in control group.

Effect of treatment with EA on MDA and NO levels in SA-induced cardiotoxicity. Values are mean ± SD (n = 7). Data were analyzed by one-way ANOVA followed by Tukey’s post hoc test for multiple comparisons. *Significant difference in comparison with the control group (***p < 0.001). #Significant difference in comparison with the SA group (##p < 0.01, ###p < 0.001)
SA-Induced Alterations in Antioxidant Enzymes are Attenuated by EA
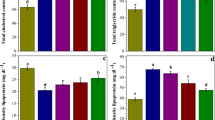
The effects of EA on activity of antioxidant enzymes CAT, SOD and GPx as well as the GSH content are shown in Fig. 3.

Effect of treatment with EA on GSH content and GPx, SOD and CAT activity in SA-induced cardiotoxicity. Values are mean ± SD (n = 7). Data were analyzed by one-way ANOVA followed by Tukey’s post hoc test for multiple comparisons. *Significant difference in comparison with the control group (***p < 0.001). #Significant difference in comparison with the SA group (#p < 0.05, ##p < 0.01)
As shown in Fig. 3a, GSH content was significantly decreased in SA group compared to the control group (p < 0.001). Also, treatment of EA at the dose of 30 mg/kg significantly (p < 0.01) increased GSH content in heart tissue of SA-treated rats.
The activity of GPx was significantly (p < 0.001) decreased in the SA group compared to the control group (Fig. 3b). Treatment with EA at the dose 30 mg/kg significantly (p < 0.05) increased GPx activity compared to the SA group.
The activity of SOD was significantly (p < 0.001) reduced in the SA group compared to the control group (Fig. 3c). Treatment with EA at the dose 30 mg/kg significantly (p < 0.01) increased SOD activity compared to the SA group.
As shown in Fig. 3d, the activity of CAT was significantly (p < 0.001) decreased in the SA group compared to the control group. In contrast, Treatment with EA at the doses of 30 mg/kg significantly (p < 0.05) increased CAT activity compared to the SA group.
In addition, administration of EA at the dose of 30 mg/kg to naïve rats did not change these antioxidant enzymes compared to rats in control group.
SA-Induced Alterations in Histopathology of Heart are Attenuated by EA
The results of microscopic studies in cardiac tissues of different experimental groups are shown in Fig. 4. Our evaluation showed that no pathological changes occur in groups receiving normal saline or EA (30 mg/kg) and cardiac muscle bundles were normal as well. In contrast, rats treated with SA exhibited degenerative changes in cardiomyocytes including; condensed nuclei and increased eosinophilic cytoplasm which indicates necrosis. We observed moderate myocardial necrosis in rats receiving of EA (10 mg/kg). Treatment with EA (30 mg/kg) showed a dramatic improvement in cardiomyocytes structure and nuclei. Also, cytoplasm structure was found normal in these observations.

Histopathological observations (stained with Hematoxylin & Eosin, magnification ×150) showing effects of EA on SA-induced toxicity changes in heart. a Control group; b SA group; c EA 30 group; d EA10 + SA group; EA30 + SA group. Arrow: necrosis
Discussion
Chronic exposure to SA promotes ROS formation and cellular oxidative damage in different tissues such as heart and blood [4, 28]. Also, SA exposure has been shown to disturb the balance between generation of ROS and antioxidant capacity [29]. These events ultimately lead to hematological disturbance and cardiac cell damage as well as apoptosis [4, 28]. Natural antioxidant agents acting as effective ROS scavengers have important role in defense against SA-induced generation of free radicals and the ensuing cardio- and hematotoxicity [7,8,9].
In the present study, we investigated the effect of EA treatment on SA-induced cardio- and hematotoxicity in rats. Our results indicated that oral administration of SA at the dose of 10 mg/kg results in significant cardio- and hematotoxicity as evidenced by increased serum levels of AST, CK-MB, LDH and cTnI. In addition, consistent with previous reports [4, 28], MDA and NO levels were increased; however, CAT, SOD and GPx activity as well as GSH level were decreased in heart tissue. We also observed that the mentioned changes in biochemical parameters are correlated with the heart histological results and hematological tests. Moreover, oral treatment of rats with EA at the dose of 10 and 30 mg/kg (more potently at dose 30 mg/kg) for 21 days could prevent SA-induced cardio- and hematotoxicity by regulation of CAT, SOD and GPx activity as well as MDA, NO, GSH, AST, CK-MB, LDH and cTnI levels. Also, EA treatment attenuated the histological changes of the heart tissue induced by SA. Moreover, we demonstrated that oral use of EA (30 mg/kg/day) for 14 days affects neither the morphology nor the function of heart in naïve rats. The results of our study, for the first time, suggest that EA treatment exerts a significant protective effect on SA-induced cardio- and hematotoxicity.
Hematological parameters have been utilized to analyze the toxicity of SA. Our CBC results indicated that SA decreases the WBC, RBC, HBG, HCT and PLT, but increases the MCH and MCV. Our findings are in partial agreement with previous reports. Recently, it has been reported that arsenic exposure decreases the HCT, MCV, and MCH, but increases the MCHC [4]. In another study, it has been demonstrated that most of the hematological parameters are not affected by SA, except WBC and PLT count [30]. We also found that EA (30 mg/kg) attenuates the effect of the above-mentioned variables, thus reducing hematotoxicity in arsenic-intoxicated rats. These results might be attributed to the antioxidant activity of EA.
Chronic exposure to arsenic triggers the pathological conditions in heart which are characterized by elevated serum levels of AST, CK-MB, LDH and cTnI [28]. The results of our study demonstrated that oral administration of SA increases the markers of cardiac dysfunction. Administration of EA significantly suppressed the SA-induced enhancement of AST, CK-MB, LDH and TnI serum concentration. Our data suggested that EA (more potently at dose 30 mg/kg) restricts the extent of heart injuries by reducing the release of these cardiac markers from the myocardium. Antioxidant agents such as biochanin A suppress these changes in SA-induced cardiotoxicity [28]. EA induces its antioxidant effects directly and/or indirectly by scavenging the free radicals and increasing the activity and expression of antioxidant enzymes [14, 15]. Hence, it seems that treatment with EA may cause cardioprotection at least partially via its antioxidant properties.
It has been shown that SA potentiates the oxidative stress parameters such as MDA and NO levels in different organs [29]. Over production of NO causes oxidative stress through generating the peroxynitrite radical (a potent cytotoxic agent) [31]. On the other hand, MDA is a highly reactive lipid peroxidation end-product and the level of MDA is used as an indirect biomarker of oxidative stress in tissues [32]. It is well established that elevation of oxidative stress causes hematological disturbance and morphological and functional alterations in the heart tissue [4, 28]. In agreement with previous studies, we observed that oral administration of SA elevates MDA and NO levels in heart tissue. Our results also indicate that treatment with EA (more potently at dose of 30 mg/kg) significantly decreases these oxidative stress indices in heart tissue of SA-treated rats. It has been reported that EA decreases lipid peroxidation during oxidative stress in various organs such as liver, kidney and heart [18, 33]. Also, recently Ding et al. [34], reported that dietary EA has protective effects against atherosclerosis and endothelial oxidative stress. They found that EA exerts these protective effects via improving the nitric oxide bioavailability. Hence, our results showed that EA may exert its protective effect against arsenic cardio- and hematotoxicity by decreasing lipid peroxidation and NO synthesis.
In this study, we measured the levels of SOD, CAT and GPx activity as well as GSH content to evaluate the capacity of antioxidant system in heart tissue. These antioxidant enzymes eliminate free radicals produced in SA-treated animals [35]. On the other hand, our results are consistent with previous findings indicating that the over production of free radicals during arsenic exposure is associated with depletion of antioxidant enzymes such as CAT, SOD, GPx and GSH [7, 8, 36]. Our findings also demonstrated that oral administration of EA enhances the activity and level of these enzymes. Several evidences support the idea that EA effectively increases the activity and level of antioxidant enzymes such as CAT, SOD, GPx and GSH in different tissues such as heart, kidney and liver. Hemmati et al. [37] have reported that EA has beneficial cardioprotective effects against arsenic trioxide-induced toxicity in rat via antioxidant properties. Administration of EA has been shown to decrease the MDA levels and increase the level of GSH, GPx and CAT in cisplatin-induced oxidative stress when assessed in rat’s liver and heart tissues [17]. EA has also been found to induce renoprotective effects against diabetic nephropathy via increasing the levels of SOD, GSH, CAT and GPx in kidney tissue [38]. Thus, our results revealed that arsenic toxicity targets antioxidant enzymes and EA treatment could exert cardioprotection, at least in part, via increasing the antioxidant activity.
Consistent with the results of biochemical assessment, our histopathological observations demonstrated structural changes in heart tissue of SA-treated rats. SA administration (10 mg/kg for 21 days) causes histopathological lesions in tissue such as degenerative changes in cardiomyocytes. These results indicate the beneficial effects of EA on SA-induced necrosis of cardiomyocytes. It was also demonstrated that pretreatment with EA before arsenic trioxide administration effectively prevents histopathological alterations in heart tissue [37].
In summary, results of the present study demonstrated that EA treatment is effective in alleviating SA-induced cardio- and hematotoxicity. EA could reduce oxidative stress via decreasing the levels of MDA and NO. On the other hand, EA enhances the activity of endogenous antioxidant enzymes such as CAT, SOD, GPx and thereby attenuates SA-induced cardio- and hematotoxicity. Collectively, our results suggest that EA provides a safe and natural option for prevention of SA-induced toxicity in humans. However, further investigations are required to precisely understand the underlying cellular mechanisms.
References
Jiang, J. Q., Ashekuzzaman, S. M., Jiang, A., Sharifuzzaman, S. M., & Chowdhury, S. R. (2012). Arsenic contaminated groundwater and its treatment options in Bangladesh. International Journal of Environmental Research and Public Health, 10, 18–46.
Anetor, J. I., Wanibuchi, H., & Fukushima, S. (2007). Arsenic exposure and its health effects and risk of cancer in developing countries: Micronutrients as host defence. Asian Pacific Journal of Cancer Prevention: APJCP, 8, 13–23.
Hughes, M. F. (2002). Arsenic toxicity and potential mechanisms of action. Toxicology Letters, 133, 1–16.
Jalaludeen, A. M., Ha, W. T., Lee, R., Kim, J. H., Do, J. T., Park, C., et al. (2016). Biochanin a ameliorates arsenic-induced hepato- and hematotoxicity in rats. Molecules, 21, 69.
Shi, H., Shi, X., & Liu, K. J. (2004). Oxidative mechanism of arsenic toxicity and carcinogenesis. Molecular and Cellular Biochemistry, 255, 67–78.
Pachauri, V., & Flora, S. (2013). Effect of nicotine pretreatment on arsenic-induced oxidative stress in male Wistar rats. Human and Experimental Toxicology, 32, 972–982.
Chen, C., Jiang, X., Hu, Y., & Zhang, Z. (2013). The protective role of resveratrol in the sodium arsenite-induced oxidative damage via modulation of intracellular GSH homeostasis. Biological Trace Element Research, 155, 119–131.
Abu El-Saad, A. M., Al-Kahtani, M. A., & Abdel-Moneim, A. M. (2016). N-acetylcysteine and meso-2,3-dimercaptosuccinic acid alleviate oxidative stress and hepatic dysfunction induced by sodium arsenite in male rats. Drug Design, Development and Therapy, 10, 3425–3434.
Saha, S. S., & Ghosh, M. (2010). Ameliorative role of conjugated linolenic acid isomers against oxidative DNA damage induced by sodium arsenite in rat model. Food and Chemical Toxicology, 48, 3398–3405.
Soong, Y.-Y., & Barlow, P. J. (2006). Quantification of gallic acid and ellagic acid from longan (Dimocarpus longan Lour.) seed and mango (Mangifera indica L.) kernel and their effects on antioxidant activity. Food Chemistry, 97, 524–530.
Garcia-Nino, W. R., & Zazueta, C. (2015). Ellagic acid: Pharmacological activities and molecular mechanisms involved in liver protection. Pharmacological Research, 97, 84–103.
Doyle, B., & Griffiths, L. A. (1980). The metabolism of ellagic acid in the rat. Xenobiotica, 10, 247–256.
Priyadarsini, K. I., Khopde, S. M., Kumar, S. S., & Mohan, H. (2002). Free radical studies of ellagic acid, a natural phenolic antioxidant. Journal of Agricultural and Food Chemistry, 50, 2200–2206.
Pari, L., & Sivasankari, R. (2008). Effect of ellagic acid on cyclosporine A-induced oxidative damage in the liver of rats. Fundamental & Clinical Pharmacology, 22, 395–401.
El-Shitany, N. A., El-Bastawissy, E. A., & El-desoky, K. (2014). Ellagic acid protects against carrageenan-induced acute inflammation through inhibition of nuclear factor kappa B, inducible cyclooxygenase and proinflammatory cytokines and enhancement of interleukin-10 via an antioxidant mechanism. International Immunopharmacology, 19, 290–299.
Al-Kharusi, N., Babiker, H. A., Al-Salam, S., Waly, M. I., Nemmar, A., Al-Lawati, I., et al. (2013). Ellagic acid protects against cisplatin-induced nephrotoxicity in rats: A dose-dependent study. European Review for Medical and Pharmacological Sciences, 17, 299–310.
Yuce, A., Atessahin, A., Ceribasi, A. O., & Aksakal, M. (2007). Ellagic acid prevents cisplatin-induced oxidative stress in liver and heart tissue of rats. Basic & Clinical Pharmacology & Toxicology, 101, 345–349.
Gul, M., Aliosmanoglu, I., Uslukaya, O., Firat, U., Yuksel, H., Gumus, M., et al. (2013). The protective effect of ellagic acid on lung damage caused by experimental obstructive jaundice model. Acta Chirurgica Belgica, 113, 285–289.
Al-Hasan, A. K. J. (2017). Effects of low-and high-level pulsed Nd: YAG laser irradiation on red blood cells and platelets indices of albino rats in vitro. Iraq Medical Journal, 1, 10–19.
Saha, S. S., & Ghosh, M. (2009). Comparative study of antioxidant activity of alpha-eleostearic acid and punicic acid against oxidative stress generated by sodium arsenite. Food and Chemical Toxicology, 47, 2551–2556.
Celik, G., Semiz, A., Karakurt, S., Arslan, S., Adali, O., & Sen, A. (2013). A comparative study for the evaluation of two doses of ellagic acid on hepatic drug metabolizing and antioxidant enzymes in the rat. BioMed Research International, 2013, 358945.
Reitman, S., & Frankel, S. (1957). In vitro determination of transaminase activity in serum. American Journal of Clinical Pathology, 28, 56–63.
Bishop, C., Chu, T., & Shihabi, Z. (1971). Single stable reagent for creatine kinase assay. Clinical Chemistry, 17, 548–550.
Whitaker, J. (1969). A general colorimetric procedure for the estimation of enzymes which are linked to the NADH/NAD + system. Clinica Chimica Acta, 24, 23–37.
Ghosh, A., & Sil, P. C. (2008). A protein from Cajanus indicus Spreng protects liver and kidney against mercuric chloride-induced oxidative stress. Biological and Pharmaceutical Bulletin, 31, 1651–1658.
Buege, J. A., & Aust, S. D. (1978). Microsomal lipid peroxidation. Methods Enzymology, 52, 302–310.
Tracey, W. R., Linden, J., Peach, M. J., & Johns, R. A. (1990). Comparison of spectrophotometric and biological assays for nitric oxide (NO) and endothelium-derived relaxing factor (EDRF): Nonspecificity of the diazotization reaction for NO and failure to detect EDRF. Journal of Pharmacology and Experimental Therapeutics, 252, 922–928.
Jalaludeen, A. M., Lee, W. Y., Kim, J. H., Jeong, H. Y., Ki, K. S., Kwon, E. G., et al. (2015). Therapeutic efficacy of biochanin a against arsenic-induced renal and cardiac damage in rats. Environmental Toxicology and Pharmacology, 39, 1221–1231.
Adil, M., Kandhare, A. D., Visnagri, A., & Bodhankar, S. L. (2015). Naringin ameliorates sodium arsenite-induced renal and hepatic toxicity in rats: Decisive role of KIM-1, Caspase-3, TGF-β, and TNF-α. Renal Failure, 37, 1396–1407.
Dwivedi, N., Flora, G., Kushwaha, P., & Flora, S. J. (2014). Alpha-lipoic acid protects oxidative stress, changes in cholinergic system and tissue histopathology during co-exposure to arsenic-dichlorvos in rats. Environmental Toxicology and Pharmacology, 37, 7–23.
Archer, S. (1993). Measurement of nitric oxide in biological models. The FASEB Journal, 7, 349–360.
Priyamvada, S., Priyadarshini, M., Arivarasu, N., Farooq, N., Khan, S., Khan, S. A., et al. (2008). Studies on the protective effect of dietary fish oil on gentamicin-induced nephrotoxicity and oxidative damage in rat kidney. Prostaglandins Leukotrienes and Essential Fatty Acids, 78, 369–381.
Singh, K., Khanna, A., Visen, P., & Chander, R. (1999). Protective effect of ellagic acid on t-butyl hydroperoxide induced lipid peroxidation in isolated rat hepatocytes. Indian Journal of Experimental Biology, 37, 939–940.
Ding, Y., Zhang, B., Zhou, K., Chen, M., Wang, M., Jia, Y., et al. (2014). Dietary ellagic acid improves oxidant-induced endothelial dysfunction and atherosclerosis: Role of Nrf2 activation. International Journal of Cardiology, 175, 508–514.
Song, L.-L., Tu, Y.-Y., Xia, L., Wang, W.-W., Wei, W., Ma, C.-M., et al. (2014). Targeting catalase but not peroxiredoxins enhances arsenic trioxide-induced apoptosis in k562 cells. PLoS ONE, 9, e104985.
Modi, M., Kaul, R. K., Kannan, G. M., & Flora, S. J. (2006). Co-administration of zinc and n-acetylcysteine prevents arsenic-induced tissue oxidative stress in male rats. Journal of Trace Elements in Medicine and Biology, 20, 197–204.
Hemmati, A. A., Olapour, S., Varzi, H. N., Khodayar, M. J., Dianat, M., Mohammadian, B., and Yaghooti, H. (2017). Ellagic acid protects against arsenic trioxide-induced cardiotoxicity in rat. Human & Experimental Toxicology 960327117701986.
Ahad, A., Ganai, A. A., Mujeeb, M., & Siddiqui, W. A. (2014). Ellagic acid, an NF-κB inhibitor, ameliorates renal function in experimental diabetic nephropathy. Chemico-Biological Interactions, 219, 64–75.
Acknowledgements
This study was funded by Deputy of Research of Ahvaz Jundishapur University of Medical Sciences, Ahvaz, Iran (Grant Number 95s35).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Authors declare no conflict of interest related to this study.
Rights and permissions
About this article

Cite this article
Goudarzi, M., Fatemi, I., Siahpoosh, A. et al. Protective Effect of Ellagic Acid Against Sodium Arsenite-Induced Cardio- and Hematotoxicity in Rats. Cardiovasc Toxicol 18, 337–345 (2018). https://doi.org/10.1007/s12012-018-9446-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12012-018-9446-2