
Abstract
Notch3 and TGF-β1 signaling play a key role in the pathogenesis and progression of chronic cardiovascular disease. However, whether Notch3 protects against myocardial infarction (MI) and the underlying mechanisms remains unknown. C57BL/6 mice were randomized to be treated with Notch3 siRNA (siNotch3) or lentivirus carrying Notch3 cDNA (Notch3) before coronary artery ligation. Four weeks after constructing MI model, cardiac function and fibrosis were compared between groups. The cardiac fibroblast cells (CFs) were isolated from newborn C57BL/6 mice (1–3 days old) and transfected with lentivirus carrying Notch3 cDNA. TGF-β1 (5 ng/ml), a well-known pro-fibrotic factor, was administered 72 h after Notch3 cDNA administration in CFs. The related proteins of fibrosis such as a-smooth muscle actin (a-SMA), Type I collagen, metalloprotease (MMP)-9 and the tissue inhibitor of metalloproteinases (TIMP)-2 were examined by western blot analysis. Notch3 cDNA treatment attenuated cardiac damage and inhibited fibrosis in mice with MI. Meanwhile, Notch3 siRNA administration aggravated cardiac function damage and markedly enhanced cardiac fibrosis in mice with MI. Overexpression of Notch3 inhibited TGF-β1-induced fibroblast–myofibroblast transition of mouse cardiac fibroblast cells, as evidenced by down-regulating a-SMA and Type I collagen expression. Notch3 cDNA treatment also increased MMP-9 expression and decreased TIMP-2 expression in the TGF-β1-stimulated cells. This study indicates that Notch3 is an important protective factor for cardiac fibrosis in a MI model, and the protective effect of Notch3 is attributable to its action on TGF-β1/Smad3 signaling.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cardiac fibrosis which is characterized by excessive deposition of scar tissue significantly reduces cardiac function after myocardial infarction (MI). Thus, anti-fibrotic therapies are likely to be promising in curbing fibrosis and enhance cardiac performance after MI. However, there is no effective anti-fibrotic therapy for MI, largely because the underlying mechanism of fibrosis is unclear [1]. During post-MI tissue repair, α-smooth muscle actin (α-SMA)-expressing myofibroblast migrates into the infarcted myocardium and synthesizes newly created extracellular matrix. The reason why the infarcted myocardium adopts fibrosis as its primary repair process remains unknown. One of the key regulators of fibrosis is transforming growth factor β1 (TGF-β1)/Smad3 signaling [2]. Strategies inhibiting TGF-β1/Smad3 signaling may serve as a potential therapeutic target for anti-fibrotic treatments [2–5].
Notch pathway is an evolutionarily conserved signaling system which has been shown to mediate cellular physiology process in developmental and differentiated tissues [6, 7]. In mammals, four members of Notch receptors (Notch-1 to 4) are known [8]. Upon ligand binding, the Notch intracellular domain (NICD) is released by proteolytic cleavage, which then migrates into the nucleus and regulates Notch-dependent gene expression [9, 10]. Moreover, Hes-1 is a canonical Notch transcriptional target gene. It has also been shown that activating Notch signaling pathway has a beneficial effect in metabolic diseases and cardiac hypertrophy [11, 12]. Interestingly, Notch signaling has been recently shown to be involved in tissue fibrosis. Notch activation, primarily via its actions on the fibroblast population, prevents pro-fibrotic factor production and limits cardiac fibrosis in response to pressure overload. It can positively influence the switching of fibroblasts into myofibroblasts [12]. Cross talk between Notch and TGF-β pathways is crucial during the development of the heart and has been described to regulate fibrosis in several organs in the adult [13, 14].
Several in vitro studies have reported that Notch1 inhibits TGF-β-induced fibroblast–myofibroblast transition and stimulates the expression and release of ECM-degrading enzymes [3, 15]. More recently, Djudjaj and colleagues demonstrated Notch3 as a regulator of fibrosis in kidney injury. On the basis of these observations, the aims of the present study were to examine the role of Notch3 in myocardial fibrosis after MI and the underlying mechanisms.
Materials and Methods
Animal Models
Male C57BL/6 mice (6–8 weeks) were purchased from the Fourth Military Medical University Medical Animal Center. All experiments were performed in adherence with the National Institutes of Health Guidelines on the use of laboratory animals and approved by the Fourth Military Medical University Committee on Animal Care (ID:20140321). Mice were anesthetized with 2 % isoflurane, and a left thoracotomy incision was made between the fourth and fifth ribs to expose the heart. Notch3 siRNA (20 μg) or scrambled siRNA was dissolved in 30 μl of vivo-jetPEI™ (Invitrogen, USA) and 10 % glucose mixture. Intramyocardial injections (5 μl per injection) were made at six separate sites of the free anterior wall of the left ventricle (LV) with a total volume of 30 μl solution containing Notch3 siRNA or scrambled siRNA using a 30-gauge needle [16]. Notch3 cDNA lentivirus was injected into the free anterior wall of the left ventricle (LV) at six different sites. Three days after infection of Notch3 siRNA or Notch3 cDNA lentivirus, cardiac MI injury model was constructed as previously described [17]. The incision was opened under anesthetic condition once again. A 6–0 silk suture slipknot was placed at the proximal one-third of the left anterior descending (LAD) artery. The ligation was accomplished at the same segment of LAD, and then, the anterior wall of left ventricle (LV) turned pale. The same surgical protocols were performed in the Sham group except that the suture placed under the left coronary artery was not tied. Mice were randomly allocated into the following groups: (1) Sham group (n = 20); (2) MI group (n = 20); (3) MI + siControl group (n = 20); (4) MI + siNotch3 group (n = 20); (5) MI + Control vector group (n = 20); (6) MI + Notch3 group (n = 20). Four weeks after MI, survival rate was recorded.
Cell Culture and Treatments
Primary cultures of mouse cardiac fibroblast cells were obtained from newborn C57BL/6 mice (1–3 days old). Briefly, myocardium specimen was cut into small pieces and washed, followed by digestion steps with collagenase type 2. The tissue lysate was filtered through a 70-mm cell strainer (Millipore, Billerica, MA, USA) and pre-plated for 1 h in a humidified cell culture incubator (21 % O2, 5 % CO2, 37 °C). The cells adherent to the culture plate, mainly composed of cardiac fibroblast cells (CFs), were cultured in DMEM supplemented with 10 % FBS, penicillin (100 U/ml) and streptomycin (100 U/ml) (Sigma) at 37 °C in a humidified atmosphere of 5 % CO2. After 48 h, Notch3 cDNA lentivirus was transfected into CFs. After 72-h transfection, the CFs were treated with TGF-β1 (5 ng/ml, 24 h), a well-known pro-fibrotic factor. Cells were randomly divided into the following groups: (1) Control (CON); (2) TGF-β1; (3) TGF-β1 +Control vector; (4) TGF-β1 + Notch3.
Determination of Cardiac Function
Echocardiography was conducted at 4 weeks after MI. Sedated mice (2 % isoflurane) were studied by using an echocardiography system (Sequoia Acuson, Siemens, 15-MHz linear transducer, Erlangen, Germany). Cardiac dimensions and function were assessed by M-mode echocardiography. Left ventricular end-diastolic diameter (LVEDD) and left ventricular end-systolic diameter (LVESD) were measured on the parasternal left ventricular long-axis view. All measurements represented the mean of five consecutive cardiac cycles. Left ventricular end-systolic volume (LVESV), left ventricular end-diastolic volume (LVEDV) and left ventricular ejection fraction (LVEF) were calculated by the use of computer algorithms. All of these measurements were conducted under the same conditions in a blinded manner. A Millar Mikro-tip catheter transducer that was inserted into the left ventricular cavity through the left carotid artery was used to measure the left ventricular pressure (LVP). The left ventricular systolic pressure (LVSP), left ventricular end-diastolic pressure (LVEDP), first derivative of the left ventricular pressure (±LV dp/dt max) and heart rate were obtained by computer algorithms and an interactive videographics program (Po-Ne-Mah Physiology Platform P3 Plus, Gould Instrument Systems, Valley View, Ohio).
Notch3 siRNA or Lentivirus Carrying Notch3 cDNA Construction
Notch3 siRNA oligonucleotides were purchased from GenePharma Company (Shanghai, China). The RNAi sequence targeting Notch3 against murine is 50-GGCCAGUUUACUUGCAUCUTT-30 (sense) and 50-AGAUGCAAGUAAACGGCCTT-30 (antisense). Lentivirus carrying Notch3 cDNA was purchased from GeneChem Company (Shanghai, China)
Immunofluorescence Staining
Immunofluorescence staining was conducted with standard protocols. Cells slides were fixed with 4 % paraformaldehyde for 15 min and then incubated with 10 % normal goat serum containing 0.1 % Triton X-100 (Sigma) for 1 h. The α-SMA was detected by incubating with rabbit anti-α-SMA (diluted 1:150; Abcam, Cambridge, MA, UK) overnight at 4 °C. The slides were incubated for 2 h with a fluorescein isothiocyanate (FITC)-conjugated anti-rabbit (Sigma) secondary antibodies, and 4,6-diamidino-2-phenylindole (DAPI) (Sigma) stained all cell nuclei.
Masson’s Trichrome Staining
Four weeks after MI operation, mice myocardial tissues were fixed with 4 % paraformaldehyde, dehydrated through graded solutions of ethanol and embedded in paraffin. Heart sections were cut (4–5 μm thickness) and stained with Masson’s trichrome (Sigma-Aldrich; St. Louis, MO). The extent of fibrosis was expressed as the Masson’s trichrome-stained area/total LV area ratio.
Western Blot Evaluation
Cardiac tissues and CFs proteins were harvested with RIPA lysis buffer. After blocking with 5 % milk for 1 h in 1 × TBS-Tween-20 buffer, membranes were incubated overnight at 4 °C with primary antibody. The primary antibodies used were anti-GAPDH antibody (Santa Cruz, CA, USA), anti-Notch3 (Santa Cruz, CA, USA), anti-Hes1, anti-Smad3, anti-Phospho-Smad3 (Ser423/425) (Cell Signaling Technology, Beverly, MA, USA), anti-α-SMA, anti-Type I collagen, anti-MMP-9 and anti-TIMP-2 (Abcam, Cambridge, MA, UK). After three washes with TBST, the membrane was incubated with the corresponding secondary antibodies (Abcam, Cambridge, MA, UK) for 1 h at room temperature. Protein concentrations were quantified using the Bradford assay (Bio-Rad Laboratories, Hercules, CA, USA). The bands were detected using a chemiluminescence system (Amersham Bioscience, Buchinghamshire, UK). Band intensities were quantified by densitometry and normalized to GAPDH.
Statistical Analysis
Continuous variables that approximated the normal distribution were expressed as mean ± standard deviation (SD). Comparison between groups was subjected to ANOVA followed by Bonferroni correction for post hoc t test. A Chi-square test was used to assess the data expressed as proportions. Two-sided tests have been used throughout, and p values <0.05 were considered statistically significant. SPSS software package version 15.0 (SPSS, Chicago, IL) was used for data analysis.
Results
Examination of Notch3 Protein Level in Heart Post-MI Operation
Western blot analysis demonstrated that the expression of Notch3 and Hes1 increased rapidly after 24 h of MI and declined to baseline level until 2 weeks after MI (Fig. 1a–c). The expression of α-SMA and Type I collagen increased at 1 week after MI and further increased at 4 weeks after MI (Fig. 1a, d, e). The survival rate of mice decreased after MI (100 vs. 50 %, p < 0.05) (Fig. 1f). Notch3 cDNA treatment significantly enhanced (67 vs 50 %, p < 0.05), while Notch3 siRNA administration decreased (25 vs. 50 %, p < 0.05) the survival rate of mice as compared with the MI group (Fig. 1f).

Changes of Notch3 expression in mice heart post-MI injury. a–e Analysis of Notch3, Hes-1, a-SMA and Type I collagen protein levels for indicated time periods from injured areas of mouse heart after MI injury using western blot. f Kaplan–Meier plots of survival rates. The columns and errors bars represent means and SD. *p < 0.05 versus Sham, # p < 0.05 versus MI (24 h), § p < 0.05 versus MI (1 week), $ p < 0.05 versus MI (2 weeks)
Notch3 Attenuates Cardiac Damage in Mice with Myocardial Infarction
Echocardiography evaluated at 4 weeks after MI injury revealed that Notch3 cDNA treatment significantly enhanced, while Notch3 siRNA administration decreased LVEF as compared to the MI group (Fig. 2a, b). Notch3 cDNA treatment significantly decreased, while Notch3 siRNA administration increased LVESV and LVEDV as compared to the MI group (Fig. 2a, c, d). Hemodynamic measurements performed at 4 weeks after MI injury indicated that the ±LV dp/dt max was increased in the MI + Notch3 group as compared to the MI group (Fig. 2e, f). The ±LV dp/dt max was decreased in the MI + siNotch3 group as compared to the MI group (Fig. 2e, f). There was no statistical difference in heart rate (HR) between groups (Fig. 2g).

Notch3 attenuates cardiac damage in the mice with myocardial infarction. a Representative images of echocardiography at 4 weeks after MI injury. b–d LVEF, LVESV, LVEDV measured by echocardiography. e, f The ± LV dp/dt max obtained by hemodynamic evaluation at 4 weeks after MI injury. g Heart rates. The columns and errors bars represent means and SD. *p < 0.05 versus Sham, # p < 0.05 versus MI, § p < 0.05 versus MI + siControl, $ p < 0.05 versus MI + siNotch3, & p < 0.05 versus MI + Control vector
Notch3 Attenuates Cardiac Fibrosis in Mice with Myocardial Infarction
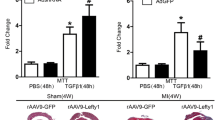
To analyze whether Notch3 modifies fibrosis in the infarcted myocardium, we performed Masson trichrome staining on day 28 post LAD ligation. Masson trichrome staining showed that fibrosis was markedly alleviated in the MI + Notch3 group compared with the MI group (Fig. 3a, b). Notch3 siRNA administration further enhanced MI-induced increase in cardiac fibrosis (Fig. 3a, b). The α-SMA and Type I collagen expression in the heart 4 weeks after MI were significantly higher in the MI group than the sham group (Fig. 3c, i, g). Notch3 cDNA treatment significantly reduced α-SMA and Type I collagen expression (Fig. 3c, i, g). Notch3 siRNA administration significantly increased α-SMA and Type I collagen expression (Fig. 3c, i, g).

Overexpression of Notch3 reduces significant the extent of cardiac fibrosis in the mice with myocardial infarction. a Representative images of Masson’s trichrome staining of each group 4 weeks after MI to assess the degree of cardiac fibrosis. The positively stained area (blue) represents fibrosis; b Quantitative analysis of Masson’s trichrome staining. Fibrosis level was presented as the ratio of fibrotic area (blue)/LV; c–j Western blot analysis of Notch3, Hes-1, p-Smad3, Smad3, TIMP-2, MMP-9, a-SMA and Type I collagen expression. The columns and errors bars represent means and SD. *p < 0.05 versus Sham, # p < 0.05 versus MI, § p < 0.05 versus MI + siControl, $ p < 0.05 versus MI + siNotch3, & p < 0.05 versus MI + Control vector
Notch3 Inhibits the TGF-β1/Smad3 Signaling Pathway in Mice with Myocardial Infarction
Notch3 cDNA treatment significantly reduced p-Smad3 and TIMP-2 expression and increased MMP-9, Notch3 and Hes1 expression (Fig. 3c–h). Notch3 siRNA administration significantly increased p-Smad3 and TIMP-2 expression and decreased MMP-9, Notch3 and Hes1 expression (Fig. 3c–h).
Notch3 Inhibits the TGF-β1/Smad3 Signaling Pathway in CFs
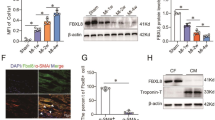
Immunofluorescence staining demonstrated that TGF-β1 increased a-SMA expression in CFs cells (Fig. 4a). Meanwhile, overexpression of Notch3 inhibited TGF-β1-induced fibroblast–myofibroblast transition, as evidenced by its ability to down-regulate a-SMA(Fig. 4a). In cultured CFs, TGF-β1 (5 ng/ml, 24 h) significantly increased p-Smad3 level (Fig. 4b, e), a typical downstream event of TGF-β1 receptor activation. TGF-β1-enhanced α-SMA and Type I collagen expression levels (Fig. 4b, h, i), decreased Notch3 and Hes-1 expression levels (Fig. 4b–d). Notch3 cDNA administration to TGF-β1-treated cardiac fibroblasts caused a marked decrease in p-Smad3, α-SMA and Type I collagen expression (Fig. 4b, e, h, i) and increased Notch3 and Hes-1 expression (Fig. 4b–d). Notch3 cDNA treatment inhibited TGF-β1-induced fibroblast–myofibroblast transition, as evidenced by its ability to down-regulate α-SMA and Type I collagen expression by western blot analysis. We also found that Notch3 cDNA administration up-regulated MMP-9 expression and down-regulated TIMP-2 expression in TGF-β1-stimulated cells (Fig. 4b, f, g).

Overexpression of Notch3 inhibits cardiac fibroblast–myofibroblast transition on primary neonatal cardiac fibroblasts by interfering with TGF-β1/Smad3 signaling pathway. a a-SMA level in CFs after different treatments evaluated by immunofluorescence. Representative immunofluorescence images, a-SMA (green). Nuclei are marked in blue with DAPI. b–i Western blot analysis of Notch3, Hes-1, p-Smad3, Smad3, TIMP-2, MMP-9, a-SMA and Type I collagen expression levels. The columns and errors bars represent means and SD. *p < 0.05 versus CON, # p < 0.05 versus TGF-β1, § p < 0.05 versus TGF-β1 + Control vector (Color figure online)
Discussion
Myofibroblasts regulate the balance between collagen synthesis and degradation after tissue injury [3, 15, 18]. When undergoing ischemia, autocrine and paracrine factors (such as TGF-β1 and pro-inflammatory cytokines), cardiac fibroblasts increase synthesis of ECM proteins which repair the necrotic myocardium with a scar [19]. Although myofibroblasts stabilize the infarcted area and promote scar tissue contraction, they can lead to abnormal myocardial stiffness and impairment of cardiac function [20]. Thus, we could find therapeutic strategies to inhibit excessive left ventricular stiffness after myocardial infarction by identifying the factors which are capable of preventing myofibroblast generation and defining their molecular targets.
In the developing heart, Notch regulates cardiomyocyte proliferation and valve formation. Mutations in components of the Notch pathway lead to cardiac structural abnormalities and dysfunction [21]. Augmentation of Notch activation exerts protective effects in alcoholic cardiomyopathy, myocardial infarction and cardiac hypertrophy [16, 22]. In our study, the activation of Notch signaling in response to cardiac injury was assessed in a mouse model of MI. The expression levels of Notch3 and the Notch targets Hes1 were increased at several time points within 2 weeks following MI. The levels of Notch3 and Hes1 reverted to normal values (sham) at 2 weeks after MI. Therefore, activation of Notch signaling was transient following MI. In the present study, augmentation of endogenous Notch reactivation in the mice with myocardial infarction, by intramyocardial injections of Notch3 cDNA lentivirus, increases mice survival rate, improves cardiac function and minimizes MI-induced increase in cardiac fibrosis. Moreover, Notch3 siRNA administration decreases survival rate, exacerbates cardiac function and increases cardiac fibrosis in the mice with myocardial infarction. This new study confirms and extends the knowledge on the anti-fibrotic effect of Notch and on its beneficial actions in the cardiovascular diseases [23, 24].
ECM homeostasis is regulated by a coordination between proteolytic enzymes and their specific tissue inhibitors (TIMPs) [25, 26]. MMPs can degrade collagen and other ECM proteins. High TIMP-2 level has a potent inhibitory effect on MMPs activation [27]. Our results have shown that Notch3 cDNA administration down-regulated multiple processes involved in adverse cardiac remodeling, reduced TIMP-2 and increased MMP-9 expression levels in the mice with myocardial infarction. In cultured CFs, our data showed that Notch3 cDNA treatment inhibited TGF-β1-induced fibroblast–myofibroblast transition by up-regulating MMP9 expression and reducing the expression of TIMP-2.
Previous studies have showed that TGF-β1/Smad3 axis takes part in tissue fibrosis [28, 29]. TGF-β1activates Smad3, which then translocates into the nucleus and induces the expression of pro-fibrotic target genes [30, 31]. The a-SMA and Type I collagen are the typical markers of myocardium fibrosis. Our results have shown that Notch3 treatment reduced p-Smad3, a-SMA and Type I collagen expression levels in mice with myocardial infarction. In vitro studies performed with cultured CFs also demonstrated that Notch3 treatment reduced p-Smad3, a-SMA and Type I collagen expression levels. Therefore, Notch3 overexpression may inhibit cardiac fibroblast–myofibroblast transition in vitro and in vivo by interfering with TGF-β1/Smad3 signaling.
Previous in vitro observations have shown that down-regulation of Notch pathway in neonatal rat cardiac fibroblasts is necessary for their differentiation into a-SMA-positive myofibroblasts [15]. A recent study also revealed that Notch regulates the balance between fibrotic and regenerative repair in the adult heart and represents a unique therapeutic target for inducing regeneration via mobilization of cardiac precursor cells in the adult heart [32]. The results of the present study demonstrated that Notch3 inhibited the phenotypic switch of cardiac fibroblasts to myofibroblasts in the mice with myocardial infarction and in cultured CFs with TGF-β1 through TGF-β1/Smad3 axis. It is conceivable that Notch3 may link with other important pathways to inhibit cardiac fibrosis in the mice with myocardial infarction.
Conclusions
The present study offers new evidence that Notch3 antagonizes the effects of TGF-β1 on cardiac fibroblast–myofibroblast transition. The salient finding of the present study provides important experimental evidence supporting Notch3 as a potential therapeutic target for the chronic ischemic heart disease by inhibiting TGF-β1/Smad3 signaling pathway to reduce cardiac fibrosis.
References
Leask, A. (2010). Potential therapeutic targets for cardiac fibrosis: TGF beta, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circulation Research, 106, 1675–1680.
Flanders, K. C. (2004). Smad3 as a mediator of the fibrotic response. International Journal of Experimental Pathology, 85, 47–64.
Sassoli, C., Chellini, F., Pini, A., Tani, A., Nistri, S., Nosi, D., et al. (2013). Relaxin prevents cardiac fibroblast-myofibroblast transition via notch-1-mediated inhibition of TGF-b/Smad3 signaling. PLoS ONE, 8, e63896.
Docherty, N. G., Murphy, M., Martin, F., Brennan, E. P., & Godson, C. (2014). Targeting cellular drivers and counter-regulators of hyperglycaemia and TGF-β1 associated pro-fibrotic responses in diabetic kidney disease. Experimental Physiology, 99, 1154–1162.
Vivar, R., Humeres, C., Ayala, P., Olmedo, I., Catalán, M., García, L., et al. (2013). TGF-β1 prevents simulated ischemia/reperfusion-induced cardiac fibroblast apoptosis by activation of both canonical and non-canonical signaling pathways. Biochimica et Biophysica Acta, 1832, 754–762.
Lassaletta, A. D., Elmadhun, N. Y., Burgess, T. A., Bianchi, C., Sabe, A. A., Robich, M. P., et al. (2014). Microvascular Notch signaling is upregulated in response to vascular endothelial growth factor and chronic myocardial ischemia. Circulation Journal, 78, 743–751.
Zhou, X. L., & Liu, J. C. (2014). Role of Notch signaling in the mammalian heart. Brazilian Journal of Medical and Biological Research, 47, 1–10.
Artavanis-Tsakonas, S., Rand, M. D., & Lake, R. J. (1999). Notch signaling: Cell fate control and signal integration in development. Science, 284, 770–776.
Bray, S. J. (2006). Notch signalling: A simple pathway becomes complex. Nature Reviews Molecular Cell Biology, 7, 678–689.
Mazzone, M., Selfors, L. M., Albeck, J., Overholtzer, M., Sale, S., Carroll, D. L., et al. (2010). Dose-dependent induction of distinct phenotypic responses to Notch pathway activation in mammary epithelial cells. Proceedings of the National Academy of Sciences of the United States of America, 107, 5012–5017.
Wickremasinghe, R. G., Prentice, A. G., & Steele, A. J. (2011). p53 and Notch signaling in chronic lymphocytic leukemia: Clues to identifying novel therapeutic strategies. Leukemia, 25, 1400–1407.
Kennard, S., Liu, H., & Lilly, B. (2008). Transforming growth factor-beta (TGF-1) down-regulates Notch-3 in fibroblasts to promote smooth muscle gene expression. Journal of Biological Chemistry, 283, 1324–1333.
MacGrogan, D., Nus, M., & de la Pompa, J. L. (2010). Notch signaling in cardiac development and disease. Current Topics in Developmental Biology, 92, 333–365.
Hurlbut, G. D., Kankel, M. W., Lake, R. J., & Artavanis-Tsakonas, S. (2007). Crossing paths with Notch in the hyper-network. Current Opinion in Cell Biology, 19, 166–175.
Fan, Y. H., Dong, H., Pan, Q., Cao, Y. J., Li, H., & Wang, H. C. (2011). Notch signaling may negatively regulate neonatal rat cardiac fibroblast-myofibroblast transformation. Physiological Research, 60, 739–748.
Gude, N. A., Emmanuel, G., Wu, W., Cottage, C. T., Fischer, K., Quijada, P., et al. (2008). Activation of Notch-mediated protective signaling in the myocardium. Circulation Research, 102, 1025–1035.
Gao, E., Lei, Y. H., Shang, X., Huang, Z. M., Zuo, L., Boucher, M., et al. (2010). A novel and efficient model of coronary artery ligation and myocardial infarction in the mouse. Circulation Research, 107, 1445–1453.
Li, P., Wang, D., Lucas, J., Oparil, S., Xing, D., Cao, X., et al. (2008). A trial natriuretic peptide inhibits transforming growth factor beta-induced Smad signaling and myofibroblast transformation in mouse cardiac fibroblasts. Circulation Research, 102, 185–192.
Li, Y., Hiroi, Y., Ngoy, S., Okamoto, R., Noma, K., Wang, C. Y., et al. (2011). Notch1 in bone marrow derived cells mediates cardiac repair after myocardial infarction. Circulation, 123, 866–876.
Zakharova, L., Nural-Guvener, H., Nimlos, J., Popovic, S., & Gaballa, M. A. (2013). Myocardial infarction is associated with transforming growth factor beta-dependent yield and functional decline in atrial explant-derived c-Kit + cells. Journal of the American Heart Association, 2, e000317.
Kratsios, P., Catela, C., Salimova, E., Huth, M., Berno, V., Rosenthal, N., et al. (2010). Distinct roles for cell-autonomous notch signaling in cardiomyocytes of the embryonic and adult heart. Circulation Research, 106, 559–572.
Ge, W., & Ren, J. (2012). mTOR-STAT3-notch signalling contributes to ALDH2-induced protection against cardiac contractile dysfunction and autophagy under alcoholism. Journal of Cellular and Molecular Medicine, 16, 616–626.
Russell, J. L., Goetsch, S. C., Gaiano, N. R., Hill, J. A., Olson, E. N., & Schneider, J. W. (2011). A dynamic Notch injury response activates epicardium and contributes to fibrosis-repair. Circulation Research, 108, 51–59.
Li, Y., Hiroi, Y., & Liao, J. K. (2010). Notch signaling as an important mediator of cardiac repair and regeneration after myocardial infarction. Trends in Cardiovascular Medicine, 20, 228–231.
Visse, R., & Nagase, H. (2003). Matrix metalloproteinases and tissue inhibitors of metalloproteinases: Structure, function, and biochemistry. Circulation Research, 92, 827–839.
Barnes, B. R., Szelenyi, E. R., Warren, G. L., & Urso, M. L. (2009). Alterations in mRNA and protein levels of metalloproteinases-2, -9, and -14 and tissue inhibitor of metalloproteinase-2 responses to traumatic skeletal muscle injury. American Journal of Physiology. Cell Physiology, 297, C1501–C1508.
Lu, K. V., Jong, K. A., Rajasekaran, A. K., Cloughesy, T. F., & Mischel, P. S. (2004). Upregulation of tissue inhibitor of metalloproteinases (TIMP)-2 promotes matrix metalloproteinase (MMP)-2 activation and cell invasion in a human glioblastoma cell line. Laboratory Investigation, 84, 8–20.
Yang, J., Zheng, J., Wu, L., Shi, M., Zhang, H., Wang, X., et al. (2011). NDRG2 ameliorates hepatic fibrosis by inhibiting the TGF-b1/Smad pathway and Altering the MMP2/TIMP2 ratio in rats. PLoS ONE, 6, e27710.
Huang, X. Z., Wen, D., Zhang, M., Xie, Q., Ma, L., Guan, Y., et al. (2014). Sirt1 activation ameliorates renal fibrosis by inhibiting the TGF-b/Smad3 pathway. Journal of Cellular Biochemistry, 115, 996–1005.
Feng, X. H., & Derynck, R. (2005). Specificity and versatility in TGF-beta signaling through Smads. Annual Review of Cell and Developmental Biology, 21, 659–693.
Samarakoon, R., Overstreet, J. M., & Higgins, P. J. (2013). TGF-b signaling in tissue fibrosis: Redox controls, target genes and therapeutic opportunities. Cellular Signalling, 25, 264–268.
Nemir, M., Metrich, M., Plaisance, I., Lepore, M., Cruchet, S., Berthonneche, C., et al. (2014). The Notch pathway controls fibrotic and regenerative repair in the adult heart. European Heart Journal, 35, 2174–2185.
Acknowledgments
This work was supported by National Nature Science Foundation of China (Nos. 81570318, 81300149, 81270263) and Shaanxi Social Development and Scientific Problem Tackling Program (2015SF097).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Zhang, M., Pan, X., Zou, Q. et al. Notch3 Ameliorates Cardiac Fibrosis After Myocardial Infarction by Inhibiting the TGF-β1/Smad3 Pathway. Cardiovasc Toxicol 16, 316–324 (2016). https://doi.org/10.1007/s12012-015-9341-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12012-015-9341-z