
Abstract
Tannic acid (TA) is a metal chelating polyphenol that plays a crucial role in metal detoxification, but its modulatory role in co-exposure of these heavy metals’ exposure needs to be explored. Cadmium (Cd) and nickel (Ni) are inorganic hazardous chemicals in the environment. Humans are prone to be exposed to the co-exposure of Cd and Ni, but the toxicological interactions of these metals are poorly defined. Present study was undertaken to study the preventive role of TA in Cd–Ni co-exposure-evoked hepato-renal toxicity in BALB/c mice. In the current investigation, increased oxidative stress in metal intoxicated groups was confirmed by elevated peroxidation of the lipids and significant lowering of endogenous antioxidant enzymes. Altered hepato-renal serum markers, DNA fragmentation, and histological alterations were also detected in the metal-treated groups. Present study revealed that Cd is a stronger toxicant than Ni and when co-exposure was administered, additive, sub-additive, and detrimental effects were observed. Prophylactic treatment with TA significantly reinstated the levels of lipid peroxidation (LPO), non-enzymatic, and enzymatic antioxidants. Moreover, it also restored the serum biomarker levels, DNA damage, and histoarchitecture of the given tissues. TA due to its metal chelating and anti-oxidative properties exhibited cyto- and genoprotective potential against Cd–Ni co-exposure-induced hepatic and renal injury.
Graphical Abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Heavy metals are defined as naturally occurring elements that have a high atomic weight and a density at least five times higher than that of water [1]. While few heavy metals are essential for biological processes in trace amounts, their excessive exposure can lead to adverse health effects in humans. Humans are exposed to these metals through anthropogenic activities and occupational exposure and can bioaccumulate via air, water, and soil [2,3,4,5]. The exposure of heavy metals extends their critical concerns to the environment owing to their complex nature that often involves co-exposure of these metals [6]. Their concurrent presence can lead to unpredictable and potential supra-additive, additive, or sub-additive effects on human health, which may exacerbate the adverse outcomes associated with individual metal exposures [5, 7].
Understanding the toxicity of co-exposure to Cd and Ni is crucial due to their widespread environmental presence. Cadmium (Cd) is an industrial and environmental pollutant, which is poorly excreted due to its long biological half-life of 10–35 years [8]. The main routes of its exposure are tobacco smoking, drinking contaminated water, metal plating, batteries, and air pollution silently wreaking havoc on human health [9]. Likewise, nickel (Ni) is a well-known essential trace element, but at the same time is hazardous due to its wide use in electroplating, alloy production, jewelry, stainless steel, and electrical batteries [10]. Electrical and electronic waste (e-waste), which is the largest source of Ni–Cd contamination, poses a tremendous threat and concern to human health [11]. According to the Global E-waste Monitor, ~ 53.6 million metrics tons (mMt) of e-waste were produced in 2019 globally, and Asia was the largest generator of e-waste in 2019 (24.9 mMt) [12].
Both Ni and Cd can bioaccumulate in different organs and tissues of the body causing hepatorenal dysfunction, carcinogenicity, neurotoxicity, reproductive toxicity, and developmental and gastrointestinal abnormalities [9, 10, 13]. These observed toxicities are most plausibly caused by heavy metal-induced oxidative stress and imbalance of pro- and anti-oxidant system [8, 10]. Therefore, examination of toxicant interaction is important to understand co-exposure-mediated complex mechanisms underlying their toxicity. Moreover, the hepato-renal system, necessary for the metabolism and excretion of xenobiotics, necessitates the examination of co-exposure in this system [14].
To date, no effective approach exists to counteract the adverse consequences of individual or co-exposure of heavy metals, making it essential to uncover the potential of nutraceuticals in this field [13, 15]. Plant-based polyphenols, flavonoids, and alkaloids exhibit antioxidant potential due to their hydrogen-donating and metal-chelating properties [16,17,18]. Tannic acid (TA) is a polyphenolic plant compound, typically found in tea, wood barks, walnut, berries, and Chinese galls with excellent free radical scavenging, antioxidant, metal-chelating, and quenching properties [19]. The antioxidant property of TA is attributed to its hydroxyl groups, which allow them to act as reducing agent, hydrogen donor, and quencher of singlet oxygen making it a suitable candidate to be explored against metal toxicity [20]. Thus, we hypothesized that TA could be a potent therapeutic agent against Cd–Ni co-exposure-induced biochemical and histological alterations and genotoxicity in hepato-renal tissues. Therefore, this research was aimed to investigate the antioxidant and cyto/genoprotective effects of TA against hepato-renal toxicity induced by individual and co-exposure of cadmium and nickel in BALB/c mice.
Materials and Methods
Chemicals
Cadmium sulfate (CdSO4, CAT #7790–84-3) with 98% purity was obtained from Loba Chemie, Mumbai and nickel chloride (NiCl2, CAT #7791–20-0) with 98% purity was purchased from Central drug house (P) Ltd., New Delhi. The tannic acid (TA, CAT #151013) of analytical research grade was used for the present study was supplied by Thomas baker (Chemicals) Pvt. Ltd., Mumbai. All other chemicals of analytical grade specifications were obtained from Himedia Ltd., SRL, CDH, and Merck.
Experimental Animals
Forty-eight adult female BALB/c mice of 2–3 months age weighing ~ 25–40 g were procured from the central animal house of Panjab University, Chandigarh (approval number PU/45/99/CPCSEA/IAEC/2015/678). Animals were housed under a standard controlled temperature of 25 ± 3 °C, 12 h light–dark cycle, and fed a standard rodent pellet diet (Ashirwad Industries, Punjab, Hindustan Lever, India; Cat #23,099,010) and water ad libitum. All the animals were acclimatized for 7 days prior to experimentation and used as per the “Guide for the Care and Use of Experimental Animals” approved by the Institutional Animal Ethics Committee, Panjab University.
Effective Dose Selection and Experimental Design
Cadmium was administered intraperitoneally (i.p.) with 0.7 mg/kg b.wt. (low dose) which is 1/10th of LD50 in mice models for 30 days [21]. To obtain the toxicologically effective nickel dose, LD50 of the nickel chloride was calculated by employing probit analysis, which came out to be 22.98 mg/kg b.wt. Based on that observation, the sub-chronic dose of 7.06 mg/kg b.wt. of Ni (1/3rd of LD50) was given intraperitoneally for 30 days to induce observable toxicity in mice. Pre-treatment of 100 mg/kg b.wt. of tannic acid was given through gavage for 15 days, and this dose was selected from the data available in the literature [20]. The oral route was chosen to mimic the commonly used mode of administration of TA to humans.
The adult mice were randomly divided into the following eight groups following acclimatization for 1 week. One control group and seven experimental groups comprised six mice in each group (Table 1).
All mice were euthanized 24 h after the last exposure, and liver and kidney tissues from experimental animals were collected for biochemical analysis and histopathological studies. Bone marrow from the femur bone of mice was flushed and collected for the micronuclei assay, and the comet assay was performed on the blood lymphocytes.
Oxidative Stress Biomarkers
Homogenates (10%) of liver and kidney tissues were prepared in 50 mM Tris–HCl buffer (pH 7.4, 4 °C) using the homogenizer. The homogenates and further the supernatants obtained were then used for spectrophotometric determination of lipid peroxidation (LPO), reduced glutathione (GSH), total protein concentration by method of Lowry et al. (1951) with slight modifications, catalase (CAT) activity, superoxide dismutase (SOD), glutathione-S-transferase (GST), and glutathione reductase (GR) [22,23,24,25,26,27,28,29].
Liver and Kidney Function Markers
For liver function, serum glutamate pyruvate transaminase (SGPT, CAT #CC2-ALT.17N), serum glutamate oxaloacetate transaminase (SGOT, CAT #CC3-AST.16N), alkaline phosphatase (ALP; CAT #CC2-ALK.02U), and total bilirubin (CAT #CC3-BIG.004) were estimated. Urea (CAT #CC2-UAB.019) and creatinine (CAT #CC3-CEN.024) concentrations were analyzed for kidney function. These serum assays were done by using commercially available kits from Reckon Diagnostics Pvt. Ltd., Gujarat (India).
Histological Studies
For histological assessment, double staining was done with hematoxylin and eosin (H&E) [30]. Briefly, the mice liver and kidney were isolated, fixed in 10% formaldehyde, dehydrated using different grades of alcohol, and embedded in paraffin. After that 5-µM-thick sections were cut, dewaxed. These sections were then hydrated with alcohol gradient and stained with hematoxylin for 1 min. The samples were then washed with phosphate buffered saline (PBS) and stained with eosin for 15 s before dehydration with an alcohol gradient. The sections were treated with xylene, mounted in DPX, and viewed under a light microscope. Then, tissue samples were evaluated with a light microscope to observe cellular damage. To determine an appropriate scoring system for liver and kidney tissue alterations, the scores were derived semi-quantitatively using light microscopy [31].
Genotoxic Parameters
Micronuclei Assay
Post-sacrifice the femur was cleared, and bone marrow was flushed out of the femur using a syringe filled with 1 mL of fetal calf serum into centrifuge tubes. Cells were dispersed by repeated aspiration and pipetting and collected by centrifugation at 1200 rpm for 10 min at 4 °C. A 5 µL of bone marrow cell suspension was placed on the centre of the acridine orange (10 µL of 1 mg/mL) coated slide. This protocol was done according to the method of Hayashi et al. (2000) [32]. Stained cells were examined by fluorescent microscope, and micronuclei frequency in polychromatic erythrocytes (PCE) was evaluated by scoring 1000 PCE and normo-chromatic erythrocytes (NCE) per animal (n = 6) per group.
Comet Assay
The extent of DNA damage was quantified in lymphocytes by studying DNA migration patterns through single gel electrophoresis [33]. Briefly, ~ 1.5 mL blood from the jugular vein was collected and centrifuged (1400 rpm for 30 min) in histopaque to separate lymphocytes. These lymphocytes were lysed, electrophoresed, neutralized, and fixed on slides in different buffers. Slides were stained with EtBr and viewed under the fluorescence microscope. A total number of 150 cells per animal (n = 6) were analyzed using CometScoreTM (version 1.5) software. Three replicates were prepared per slides and three slides were produced per animal in each group. Fifty randomly selected cells were analyzed per replicate per sample. The parameters investigated to determine the DNA damage level were % DNA in the comet tail and tail moment.
DNA Fragmentation
For genomic DNA fragmentation, agarose gel electrophoresis was performed using standard phenol:chloroform:isoamyl alcohol method. Briefly, liver and kidney tissues were homogenized in Tris-EDTA (TE) buffer and after centrifugation, the pellet was dissolved in lysis buffer. After addition of 5% sodium dodecyl sulfate (SDS) and proteinase K, the phenol:chloroform:isoamyl alcohol (25:24:1) mixture was added and centrifuged. Further, isopropanol was added to the obtained aqueous layer and incubated for 2–3 h. After centrifugation, the pellet was washed with 70% ethanol and the final pellet was dissolved in 20 µL of TE buffer and stored at 20 °C until further use. DNA integrity was checked on 0.8% agarose gel.
Interaction Index for Cadmium-Nickel Co-exposure
The combined effect of Cd and Ni was measured quantitatively using Bliss independence method or also called as relative-effect multiplicative model. Using this method, the relative levels (θX) of a parameter X were calculated and further, the interaction index (γ) value was determined using the equation
where θA = normalized level of parameter X in Cd exposed group, θB = normalized level of parameter X in Ni exposed group, and θAB = normalized level of parameter X in Cd + Ni co-exposed group.
The calculated value of γ < 0, γ = 0, or γ > 0 indicates a sub-additive, additive, or supra-additive interaction of Cd and Ni co-exposure, respectively [34, 35]. These calculated interaction index values (γ) for combinational exposure of Cd and Ni are given in Table 2.
Statistical Analysis
The data was expressed as mean ± standard deviation. The comparison of control, experimental, and TA-treated groups was statistically analyzed by one-way ANOVA (analysis of variance) followed by Tukey’s post hoc test. Values with p < 0.05 were considered statistically significant.
Results
Oxidative Stress Biomarkers
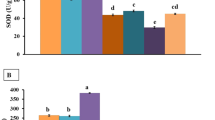
Thirty days of cadmium (0.7 mg/kg b.wt.) and nickel (7.07 mg/kg b.wt.) exposures (individual and co-exposure) instigated statistically significant (p < 0.05) intensification in the levels of LPO in hepatic and renal tissues of mice in comparison with the control group. Co-exposure of these inorganic metals caused ~ 1.4-fold and threefold increase in LPO in hepato-renal tissues, respectively. The oral pre-treatment of TA for 15 days lowered the LPO levels by ~ 1.1-fold in hepatic and twofold in renal tissue, respectively, compared to Cd- and Ni-exposed groups, respectively (p < 0.05). While in the co-exposure groups, TA caused significant reduction of LPO levels by 1.6-fold and twofold in liver and kidney tissues, respectively (p < 0.05; Fig. 1a). Cd and Ni intoxication significantly decreased the reduced glutathione levels (GSH) in the liver and kidney (p < 0.05). While TA pre-treatment efficiently restored the GSH levels in both the organs to near control values in the case of Cd, Ni, and co-exposed groups (p < 0.05) (Fig. 1b).

a Lipid peroxidation (LPO) (n moles/mg protein). b Reduced glutathione (GSH)-μ moles/mg protein. c–f Levels of endogenous antioxidant enzymes SOD (units/min/mg protein), CAT (μ moles of H2O2 decomposed/min/mg protein), GST (μ moles GST adduct formed/min/mg protein), and GR (moles NADPH oxidised/min/mg protein) in the liver and kidney tissues of rats in CON-, Cd-, Ni-, Cd + Ni-, TA-, TA + Cd-, TA + Ni-, and TA + Cd + Ni-treated groups, respectively. Values are shown as mean ± S.D. (n = 6); levels of significance, * p < 0.05 (statistically significant); a, comparison with control group; b, comparison with cadmium treated group; c, comparison with nickel treated group; d, comparison with Cd + Ni co-exposure treated group. ANOVA followed by Tukey’s honestly significant difference test
Administration of Cd and Ni significantly declined the levels of CAT, SOD, GST, and GR in liver and kidneys, respectively (p < 0.05). Cd alone was found to be more toxic; it caused more reduction of antioxidants than Ni alone. The co-exposure of both the metals exhibited sub-additive effect in enhancing LPO (γ = − 0.06, − 0.38) and supra-additive interaction in reducing GSH levels (γ = 0.11, 0.26) in liver and kidney tissues, respectively. Furthermore, supra-additive effect was observed in all other oxidative stress markers of liver and kidney tissues. TA treatment significantly elevated the levels of CAT, SOD, GST, and GR antioxidants in all the Cd-, Ni-, and co-exposure-treated groups (γ values are given in Table 2) (p < 0.05) (Fig. 1c–f).
Liver and Kidney Function Markers
Thirty days of cadmium and nickel (individual and co-exposure) intoxication caused a statistically significant (p < 0.05) increase in the levels of SGOT, SGPT, and ALP, whereas there was a significant decline in the levels of serum bilirubin (p < 0.05). Pre-treatment of tannic acid effectively restored the levels of these hepatic markers (p < 0.05) (Fig. 2a). Principle kidney function markers, i.e., urea and creatinine, were elevated in blood serum after sub-chronic exposure to Cd, Ni, and their co-administration (p < 0.05). However, TA decreased the levels of urea by 1.3-fold and of creatinine by 1.1-fold in blood serum in co-exposure group (p < 0.05) (Fig. 2b).

a Levels of liver function markers SGPT (U/L), SGOT (U/L), ALP (U/L), and total bilirubin (mg/dL) in control, Cd, Ni, Cd + Ni, TA, TA + Cd, TA + Ni, and TA + co-exposure treated groups. b Levels of kidney function markers urea (mg/dL) and creatinine (mg/dL) in CON-, Cd-, Ni-, Cd + Ni-, TA-, TA + Cd-, TA + Ni-, and TA + Cd + Ni-treated groups. Values are shown as mean ± S.D. (n = 6); a, = comparison with control group; b, comparison with cadmium treated group; c, comparison with nickel treated group; d, comparison with cadmium + nickel treated group. ANOVA followed by Tukey’s honestly significant difference test
Histopathology
Individual exposures of Cd and Ni for 30 days caused histopathological alterations in liver like vacuolization (25%, 18%), few foci of Kupffer cell infiltration (26%, 19%), widening of sinusoids (18%, 16%), damaged hepatocytes (13%, 10%), and vascular congestion around the central vein (Fig. 3a). Thirty-day co-exposure of Cd and Ni exhibited pronounced histoarchitectural alterations in liver revealing evident vacuolization (30%), Kupffer cell infiltration (32%), sinusoidal widening (25%), and highly damaged hepatocytes (14%) indicative of sub-additive effect of co-exposure. Pre-treatment of mice with TA effectively attenuated the individual cadmium and nickel exposure-induced histopathological alterations in the liver as indicated by normal hepatocytes, reduction in sinusoidal widening, decreased vacuolization, and reduced Kupffer cell infiltration (p < 0.05), whereas in the co-exposure group, moderate ameliorative efficacy of TA was observed (Fig. 3a, b).

a Comparative light micrographs of normal and metal treated liver of CON, Cd, Ni, Cd + Ni, TA, TA + Cd, TA + Ni, and TA + Cd + Ni groups of mice (× 400). b Graphical representation of the percent of histological alterations in liver tissue in each group. Abbreviations: CV, central vein; PT, portal triad; H, hepatocytes; S, sinusoids; Nc, necrosis; Pk, pyknosis; KC, Kupffer cell; VL, vacuolization; CI, cellular infiltration; Hm, hemorrhage; SW, sinusoidal widening. c Comparative light micrographs of normal and metal treated kidney of CON, Cd, Ni, Cd + Ni, TA, TA + Cd, TA + Ni, and TA + Cd + Ni groups of mice (× 400). d Graphical representation of percent histological alterations in kidney of each group. Abbreviations: BC, Bowman’s capsule; G, glomerulus; M, mesangiolysis; PC, protein cast; C, congestion; SG, shrinked glomerulus; VL, vacuolization; HM, hemorrhage; TC, tubular congestion; GN, glomerular necrosis; TN, tubular necrosis. Values are shown as mean ± S.D. (n = 3); a, comparison with control group; b, comparison with cadmium treated group; c, comparison with nickel treated group; d, comparison with cadmium + nickel treated group. ANOVA followed by Tukey’s honestly significant difference test
After 30 days of Cd and Ni individual exposures, kidneys demonstrated significant histoarchitectural changes like enlargement of mesangial space (24%, 21%), congestion in proximal and distal convoluted tubules (11%, 9%) due to protein casts (27%, 26%), glomerular shrinkage (10%, 8%), and tubular degeneration (14%, 11%), revealing the nephrotoxic nature of Cd and Ni, respectively. Cd exposure was found more toxic than Ni individual exposure as mesangiolysis and infiltration of mononuclear cells were visible at multiple foci in Cd treated mice kidneys. Cd–Ni co-exposure has more pronounced toxic effects in the renal tissue exhibiting sub-additive effect. A large number of shrunken glomeruli (14%), mesangiolysis, enlargement in mesangial space (27%), interstitial inflammation, and necrotic lesions in tubular cells (17%) along with dense protein cast in PCT and DCT (33%) were observed due to co-exposure of metals. Pre-treatment with TA significantly restored the histoarchitecture of individual Cd and Ni exposure groups indicated by normal glomeruli, PCT, and DCT (p < 0.05). Pre-treatment with TA in the co-exposure group decreased the impact of heavy metals, but mononuclear cell infiltration and few foci of tubular congestion were visible, indicating moderate amelioration of TA pre-treatment in this case (Fig. 3c, d).
Micronuclei Induction
The Cd and Ni exposures enhanced the levels of micronucleation in bone marrow cells by approximately 26- and 22-fold, respectively, as compared to the control (p < 0.05). The combinational exposure of Cd and Ni showed sub-additive effects (γ = − 1.14), i.e., a 44-fold increase in the micronucleation in bone marrow cells was observed (Fig. 4a, b). Pre-administration of TA significantly (p < 0.05) reduced micronucleation as compared to toxicant groups. TA more efficiently reduced the micronucleation in bone marrow cells in Cd and Ni alone treated groups as compared to the co-exposure due to heavy load of genetic damage.

a Photomicrographs illustrating micronuclei formation in bone marrow cells of a CON-, Cd-, Ni-, Cd + Ni-, TA-, TA + Cd-, TA + Ni-, and TA + Cd + Ni-treated mice. b Percentage frequency of micronuclei/1000 cells in bone marrow cells of CON-, Cd-, Ni-, Cd + Ni-, TA-, TA + Cd-, TA + Ni-, and TA + Cd + Ni-treated mice. Values are shown as mean ± S.D. (n = 6); a, comparison with control group; b, comparison with cadmium treated group; c, comparison with nickel treated group; d, comparison with cadmium + nickel-treated group. ANOVA followed by Tukey’s honestly significant difference test
Comet Assay
A significant (p < 0.05) increase in comet assay parameters, such as % DNA in tail (8.58 ± 0.92, 7.27 ± 0.89) and tail moment (0.85 ± 0.12, 0.80 ± 0.11), was observed in blood lymphocytes of mice treated with Cd and Ni, respectively, when compared with normal control mice (Fig. 5a, b). After co-exposure of these heavy metals, sub-additive effect in DNA damage, i.e., % DNA in tail (10.41 ± 0.99; γ = − 0.43) and tail moment (0.93 ± 0.13; γ = − 0.34), was observed. Pre-treatment of TA for 15 days to Cd and Ni individual as well as co-exposed mice demonstrated a substantial DNA protective efficacy by significantly reducing % DNA damage and tail moment (4.93 ± 0.47, 0.60 ± 0.11) (Fig. 5a, b).

a Photomicrographs of comet assay in peripheral blood lymphocytes of CON-, Cd-, Ni-, Cd + Ni-, TA-, TA + Cd-, TA + Ni-, and TA + Cd + Ni-treated groups. b Graphical representation of DNA migration patterns, i.e., percent DNA in tail and tail moment in lymphocytes of mice. Values are shown as mean ± S.D. (n = 6); a, comparison with control group; b, comparison with cadmium treated group; c, comparison with nickel-treated group; d, comparison with cadmium + nickel-treated group. ANOVA followed by Tukey’s honestly significant difference test
DNA Fragmentation
Agarose gel electrophoretograms revealed intact DNA bands in control and TA groups (lanes 1 and 5) of hepato-renal tissues. While, 30 days Cd and Ni exposure revealed shearing of DNA in liver and kidney tissues (lanes 2 and 3). DNA shearing was more evident in co-exposure group revealing maximum DNA fragmentation in both the tissues as indicated in lane 4. Pretreatment of TA to Cd, Ni, and co-exposed groups markedly protected DNA damage as demonstrated in lanes 6, 7, and 8, respectively, of electrophoretograms revealing genoprotective efficacy of TA. Moderate DNA preventive efficacy of TA was seen in Cd–Ni co-exposure group (Fig. 6a, b).

Representative agarose-gel electrophoretograms of extracted DNA from a liver and b kidney of BALB/c mice. Control (lane 1), cadmium (lane 2), nickel (lane 3), Cd + Ni (lane 4), TA (lane 5), TA + Cd (lane 6), TA + Ni (lane 7), and TA + Cd + Ni (lane 8) treated mice
Discussion
Co-exposure to Cd and Ni may produce additive or supra- or sub-additive interactions that could enhance their toxic effects. Evaluating these interactions is essential for risk assessment and management of toxicity related to their co-exposure [36]. In the present study, 30-day cadmium and nickel exposures exhibited a marked elevation in lipid peroxidation in hepatic and renal tissues. These observations comply with the observations of other studies, who have documented the cytotoxic effects of cadmium and nickel in rats [37,38,39]. Similarly, the co-exposure of Cd and Ni caused an additive effect in LPO in hepato-renal organs. Moreover, a significant decline of GSH levels was also recorded in the liver and kidney of Cd, Ni individual and co-treated groups. These results are validated by the studies of Micali et al. (2018) and Yu et al. (2018), who observed decreased levels of GSH after co-exposure intoxication [40, 41]. The most probable reason for augmented LPO and decreased GSH could be the enhanced generation of free radical species, like superoxide anions, hydroxyl radicals, and hydrogen peroxide by nickel and high affinity of cadmium toward the thiol group of GSH [38, 42]. Enhanced levels of free radicals cause oxidation of cellular macromolecules, which formed the basis of redox imbalance, genotoxicity, and histological alterations [43].
Oxidative stress-mediated suppression of cellular antioxidant defense was confirmed by declined levels of CAT, SOD, GST, and GR, in individual and co-exposed groups. Most plausibly, augmented consumption of these enzymatic antioxidants in scavenging free radicals instigated unalterable inhibition in their activities [8, 10, 14]. Many authors have documented similar observations of heavy metal-induced suppression in antioxidant enzymes and support the present study [44,45,46]. Moreover, present study has evidently exhibited that combinational exposure of Cd and Ni has supra-additive effect on cellular antioxidant suppression. In current study, TA-mediated reduction in LPO could be due to free radical scavenging or electron transferring ability to electrophilic radicals. Phenolic groups of TA might chelate the metal ions and stop the progression of free radical formation via complexing ferrous ions and inhibiting steps of the Fenton reaction [19, 47]. These properties of TA allow it to act as a reducing agent by converting Fe (II) to Fe (III), hydrogen donor, and quencher of singlet oxygen [19, 21]. TA scavenges reactive metabolites due to the presence of the galloyl moiety, allowing efficient H•-atom transfer to the free radicals and converting them to less reactive metabolites [48]. In the present study, tannic acid reduced LPO and restored the levels of antioxidants in hepato-renal tissues and re-established the balance of pro- and antioxidants.
Oxidative stress and subsequent lipid peroxidation might have formed the basis for observed histoarchitectural alterations in hepatic and renal tissues following Cd–Ni individual and co-exposures. These observations are in accordance with the study of Yu et al. (2018) and Zou et al. (2020) who have documented cadmium- and nickel-induced histological changes in hepatic tissue [41, 49]. Furthermore, observed increase in levels of AST and ALT frequently utilized indicators of hepatocellular necrosis, correlates and validates Cd–Ni co-exposure-induced hepatocellular damage. Multiple reports support these observations and highlight the adverse effects of heavy metal co-exposure in liver [14, 31, 39, 50]. Kidneys being the principle excretory organ exhibits evident metal co-exposure-induced tissue damage. Plausibly, these metals get attached to the lipid membranes of the glomerulus and result in the accumulation of lipid droplets in the voids of the glomerular membrane, which results in decreased glomerular filtration rate and increase in nitrogenous waste products (urea and creatinine) in the serum in intoxicated metal groups [9, 44]. Observed elevation in renal LPO, kidney function markers, and histological alterations are in consensus with above mentioned reports. However, TA treatment significantly lowered the levels of serum hepato-renal function biomarkers in Cd–Ni individual and co-exposure groups, indicating its redox balancing and ameliorative potential. This study is in close agreement with the observations of Akomolafe et al. (2014), who have studied the modulatory potential of TA against cisplatin-induced nephrotoxicity in rats [51].
Moreover, the present study revealed Cd–Ni co-exposure-induced sub-additive effect in causing genotoxicity. Genotoxic potential of these metals is confirmed by DNA fragmentation, and enhanced micronucleation and comet formation in bone marrow cells. Cd and Ni exposures lead to increased free radicals, which can induce DNA cross-links, DNA strand breaks, and modification of DNA bases. The present observations of enhanced comet tailing and micronucleation in Cd–Ni co-exposure are in accordance with studies of Kaushal et al. (2019) and El-Habit and Moneim (2014), who have also documented heavy metal-induced genotoxicity [52, 53]. Cd–Ni individual and co-exposure demonstrate genotoxic potential of these moieties plausibly by inhibiting DNA repair enzymes [44, 49, 52]. Co-exposure of these metals can cause DNA modifications such as fragmentation, micronuclei formation, chromosomal aberrations, and aneuploidogenicity by direct binding to DNA and/or enhancing the effects of other mutagens to generate genetic lesions [14, 54,55,56,57]. In the present study, TA has exerted its genoprotective potential possibly by two mechanisms: first, by preventing the oxidative damage of DNA by directly quenching metal ions and harmful free radical species, and second, by binding to DNA and reducing its susceptibility to damaging effects of heavy metals [58, 59]. However, its moderate efficacy against Cd–Ni co-exposure is plausibly due to competition among heavy metals for binding sites on TA, potentially hindering metal ion sequestration. Overall, this study suggests that free radical scavenging and metal chelating properties of TA have helped in restoring cellular redox imbalance, integrity of DNA, and normal histoarchitecture of hepato-renal tissues in Cd–Ni individual and co-exposure groups. Thus, it is noteworthy to add that use of TA containing drinks is an effective, economical, and convenient source of antioxidants, if added in daily routine.
Taken together, present observations suggest that Cd is a more potent toxicant than Ni metal in individual exposures, while their combinational exposure exhibits either supra-additive, additive, or sub-additive effects. Owing to antioxidant and genoprotective properties, TA exhibits remarkable ameliorative efficacy against individual metal exposure and moderate efficacy in co-exposure group. For optimizing TA efficacy in co-exposure scenario, exploring various TA dosages, potential structural modifications to enhance its selectivity or capacity, and understanding its interactions with metal ions at molecular level are pivotal. Although TA-based nano-formulations are worthy of further investigation.
Data Availability
The data that support the findings of this study are available from the corresponding author, Chopra M, upon reasonable request.
References
Tchounwou PB, Yedjou CG, Patlolla AK, Sutton DJ (2012) Heavy metal toxicity and the environment. In: Luch, A. (eds) Molecular, Clinical and Environmental Toxicology. Experientia Supplementum, vol 101:133–64. Springer, Basel. https://doi.org/10.1007/978-3-7643-8340-4_6
Das K, Reddy R, Bagoji I, Das S, Bagali S, Mullur L, Khodnapur J, Biradar M (2019) Primary concept of nickel toxicity – an overview. J Basic Clin Physiol Pharmacol 30(2):141–152. https://doi.org/10.1515/jbcpp-2017-0171
Balali-Mood M, Naseri K, Tahergorabi Z, Khazdair MR, Sadeghi M (2021) Toxic mechanisms of five heavy metals: mercury, lead, chromium, cadmium, and arsenic. Front Pharmacol 12:227. https://doi.org/10.3389/fphar.2021.643972
Ahmed SF, Kumar PS, Rozbu MR, Chowdhury AT, Nuzhat S, Rafa N, Mahlia TM, Ong HC, Mofijur M (2022) Heavy metal toxicity, sources, and remediation techniques for contaminated water and soil. Environ Technol Innov 25:102114. https://doi.org/10.1016/j.eti.2021.102114
Mitra S, Chakraborty AJ, Tareq AM, Emran TB, Nainu F, Khusro A, Idris AM, Khandaker MU, Osman H, Alhumaydhi FA, Simal-Gandara J (2022) Impact of heavy metals on the environment and human health: Novel therapeutic insights to counter the toxicity. J King Saud Univ Sci 34(3):101865. https://doi.org/10.1016/j.jksus.2022.101865
Fagbenro OS, Alimba CG, Bakare AA (2019) Experimental modeling of the acute toxicity and cytogenotoxic fate of composite mixtures of chromate, copper and arsenate oxides associated with CCA preservative using Clarias gariepinus (Burchell 1822). Environ Anal Health Toxicol 34(3):e2019010. https://doi.org/10.5620/eaht.e2019010
Wu X, Cobbina SJ, Mao G, Xu H, Zhang Z, Yang L (2016) A review of toxicity and mechanisms of individual and mixtures of heavy metals in the environment. Environ Sci Pollut Res Int 23(9):8244–8259. https://doi.org/10.1007/s11356-016-6333-x
Wang Z, Sun Y, Yao W, Ba Q, Wang H (2021) Effects of cadmium exposure on the immune system and immunoregulation. Frontiers Immunol 12:695484. https://doi.org/10.3389/fimmu.2021.695484
Genchi G, Sinicropi MS, Lauria G, Carocci A, Catalano A (2020) The effects of cadmium toxicity. Int J Environ Res Public Health 17(11):3782. https://doi.org/10.3390/ijerph17113782
Genchi G, Carocci A, Lauria G, Sinicropi MS, Catalano A (2020) Nickel: human health and environmental toxicology. Int J Environ Res Public Health 17(3):679. https://doi.org/10.3390/ijerph17030679
Parvez SM, Jahan F, Brune MN, Gorman JF, Rahman MJ, Carpenter D, Islam Z, Rahman M, Aich N, Knibbs LD, Sly PD (2021) Health consequences of exposure to e-waste: an updated systematic review. Lancet Planet Health 5(12):e905–e920. https://doi.org/10.1016/S2542-5196(21)00263-1
Forti V, Balde C, Kuehr R, Bel G (2020) The global e-waste monitor 2020: quantities, flows and the circular economy potential. United Nations University (UNU)/United Nations Institute for Training and Research (UNITAR) – co-hosted SCYCLE Programme, International Telecommunication Union (ITU) & International Solid Waste Association (ISWA). Bonn/Geneva/Rotterdam
Briffa J, Sinagra E, Blundell R (2020) Heavy metal pollution in the environment and their toxicological effects on humans. Heliyon 6(9):e04691. https://doi.org/10.1016/j.heliyon.2020.e04691
Owumi SE, Olayiwola YO, Alao GE, Gbadegesin MA, Odunola OA (2020) Cadmium and nickel co-exposure exacerbates genotoxicity and not oxido-inflammatory stress in liver and kidney of rats: Protective role of omega-3 fatty acid. Environ Toxicol 35(2):231–241. https://doi.org/10.1002/tox.22860
Sharifi-Rad M, Anil Kumar NV, Zucca P, Varoni EM, Dini L, Panzarini E, Rajkovic J, Tsouh Fokou PV, Azzini E, Peluso I, Prakash Mishra A, Nigam M, El Rayess Y, Beyrouthy ME, Polito L, Iriti M, Martins N, Martorell M, Docea AO, Setzer WN, Calina D, Cho WC, Sharifi-Rad J (2020) Lifestyle, oxidative stress, and antioxidants: back and forth in the pathophysiology of chronic diseases. Front Physiol 11:694. https://doi.org/10.3389/fphys.2020.00694
Vona R, Pallotta L, Cappelletti M, Severi C, Matarrese P (2021) The impact of oxidative stress in human pathology: focus on gastrointestinal disorders. Antioxidants 10(2):201. https://doi.org/10.3390/antiox10020201
Varesi A, Chirumbolo S, Campagnoli LIM, Pierella E, Piccini GB, Carrara A, Ricevuti G, Scassellati C, Bonvicini C, Pascale A (2022) The role of antioxidants in the interplay between oxidative stress and senescence. Antioxidants (Basel) 11(7):1224. https://doi.org/10.3390/antiox11071224
Ahsan AU, Sharma VL, Wani A, Chopra M (2020) Naringenin upregulates AMPK-mediated autophagy to rescue neuronal cells from β-amyloid (1–42) evoked neurotoxicity. Mol Neurobiol 57(8):3589–3602. https://doi.org/10.1007/s12035-020-01969-4
Yeo J, Lee J, Yoon S, Kim WJ (2020) Tannic acid-based nanogel as an efficient anti-inflammatory agent. Biomater Sci 8(4):1148–1159. https://doi.org/10.1039/C9BM01384A
Zhang R, Dang M, Qiu S, Gu H, He P, Gang G, Zhang T (2019) Ameliorative effects of tannic acid on lipopolysaccharide-induced sepsis and acute lung injury in mice. Pharmacog Mag 15(61):238–243. https://doi.org/10.4103/pm.pm_364_18
Lukawski K, Nieradko B, Sieklucka-Dziuba M (2005) Effects of cadmium on memory processes in mice exposed to transient cerebral oligemia. Neurotoxicol Teratol 27(4):575–584. https://doi.org/10.1016/j.ntt.2005.05.009
Majeed M, Majeed S, Nagabhushanam K, Lawrence L, Novel ML (2020) Combinatorial regimen of garcinol and curcuminoids for non-alcoholic steatohepatitis (NASH) in mice. Sci Rep 10:7440. https://doi.org/10.1038/s41598-020-64293-w
Olugbodi JO, Lawal B, Bako G, Onikanni AS, Abolenin SM, Mohammud SS, Ataya FS, Batiha GE (2023) Effect of sub-dermal exposure of silver nanoparticles on hepatic, renal and cardiac functions accompanying oxidative damage in male Wistar rats. Sci Rep 13(1):10539. https://doi.org/10.1038/s41598-023-37178-x
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193(1):265–275
Nagree MS, Rybova J, Kleynerman A, Ahrenhoerster CJ, Saville JT, Xu TM et al (2023) Spinal muscular atrophy-like phenotype in a mouse model of acid ceramidase deficiency. Commun Biol 6:560. https://doi.org/10.1038/s42003-023-04932-w
Kosar F, Akram NA, Ashraf M, Ahmad A, Alyemeni MN, Ahmad P (2021) Impact of exogenously applied trehalose on leaf biochemistry, achene yield and oil composition of sunflower under drought stress. Physiol Plant 172(2):317–333. https://doi.org/10.1111/ppl.13155
Kono Y (2022) Reprint of: Generation of superoxide radical during autoxidation of hydroxylamine and an assay for superoxide dismutase. Arch Biochem Biophys 726:109247. https://doi.org/10.1016/j.abb.2022.109247
Almasri H, Liberti J, Brunet JL, Engel P, Belzunces LP (2022) Mild chronic exposure to pesticides alters physiological markers of honey bee health without perturbing the core gut microbiota. Sci Rep 12(1):4281. https://doi.org/10.1038/s41598-022-08009-2
Sequeira S, Rao AV, Rao A (2012) Increased oxidative stress and altered antioxidants status in patients with chronic allergic rhinitis. Adv Biosci Biotechnol 3(07):951–956. https://doi.org/10.4236/abb.2012.327117
Noor AR, Shakil A, Hoque NF, Rahman MM, Akter S, Talukder A, Ahmad-Al-Nahid S, Wahab MA, Nahiduzzaman M, Rahman MJ, Asaduzzaman M (2021) Effect of eco-physiological factors on biometric traits of green mussel Perna viridis cultured in the south-east coast of the Bay of Bengal. Bangladesh Aquac Rep 19:100562. https://doi.org/10.1016/j.aqrep.2020.100562
Seth E, Ahsan AU, Bamrara P, Kaushal S, Sharma VL, Chopra M (2021) Cytoprotective and antioxidant potential of Aegle marmelos on cadmium-induced hepato-renal toxicity: an in vivo study. Biologia 76(6):1859–1872. https://doi.org/10.1007/s11756-021-00733-w
Hayashi M, MacGregor JT, Gatehouse DG, Adler ID, Blakey DH, Dertinger SD, Krishna G, Morita T, Russo A, Sutou S (2000) In vivo rodent erythrocyte micronucleus assay. II. Some aspects of protocol design including repeated treatments, integration with toxicity testing, and automated scoring. Environ Mol Mutagen 35(3):234–252
Ogunsuyi OM, Fasakin PT, Ajibiye OP, Ogunsuyi OI, Adekoya KO (2023) Perfluoroundecanoic acid induces DNA damage, reproductive and pathophysiological dysfunctions via oxidative stress in male Swiss mice. Chemosphere 338:139491. https://doi.org/10.1016/j.chemosphere.2023.139491
Bliss C (1939) The toxicity of poisons applied jointly. Ann Appl Biol 26(3):585–615. https://doi.org/10.1111/j.1744-7348.1939.tb06990.x
Roell KR, Reif DM, Motsinger-Reif AA (2017) An introduction to terminology and methodology of chemical synergy-perspectives from across disciplines. Front Pharmacol 8:158. https://doi.org/10.3389/fphar.2017.00158
Heys KA, Shore RF, Pereira MG, Jones KC, Martin FL (2016) Risk assessment of environmental mixture effects. RSC Adv 6(53):47844–47857. https://doi.org/10.1039/C6RA05406D
Kumar A, Siddiqi NJ, Alrashood ST, Khan HA, Dubey A, Sharma B (2021) Protective effect of eugenol on hepatic inflammation and oxidative stress induced by cadmium in male rats. Biomed Pharmacother 139:111588. https://doi.org/10.1016/j.biopha.2021.111588
Lamtai M, Ouakki S, Zghari O, El Hamzaoui A, Benmhammed H, Azirar S, El Hessni A, Mesfioui A, Ouichou A (2020) Neuroprotective effect of melatonin on nickel-induced affective and cognitive disorders and oxidative damage in rats. Environ Anal Health Toxicol 35(4):e2020025. https://doi.org/10.5620/eaht.2020025
N, Wallace D, Bulat Z (2019) Toxic effect of acute cadmium and lead exposure in rat blood, liver, and kidney. J Environ Res Public Health 16(2):274https://doi.org/10.3390/ijerph16020274
Micali A, Pallio G, Irrera N, Marini H, Trichilo V, Puzzolo D, Pisani A, Malta C, Santoro G, Laurà R, Santoro D, Squadrito F, Altavilla D, Germanà A, Minutoli L (2018) Flavocoxid, a natural antioxidant, protects mouse kidney from cadmium-induced toxicity. Oxid Med Cell Longev 2018:9162946. https://doi.org/10.1155/2018/9162946
Yu S, Liu F, Wang C, Zhang J, Zhu A, Zou L, Han A, Li J, Chang X, Sun Y (2018) Role of oxidative stress in liver toxicity induced by nickel oxide nanoparticles in rats. Mol Med Rep 17(2):3133–3139. https://doi.org/10.3892/mmr.2017.8226
Seth E, Chopra M (2022) Neuroprotective efficacy of berberine following developmental exposure to chlorpyrifos in F1 generation of Wistar rats: apoptosis-autophagy interplay. Sci Total Environ 834:155292. https://doi.org/10.1016/j.scitotenv.2022.155292
Phaniendra A, Jestadi DB, Periyasamy L (2015) Free radicals: properties, sources, targets, and their implication in various diseases. Indian J Clin Biochem 30(1):11–26. https://doi.org/10.1007/s12291-014-0446-0
Bouhalit S, Kechrid Z, Elfeki A (2017) Effect of silymarin extracted from Silybum marianum on nickel hematotoxicity and nephrotoxicity in male albino Wistar rats. Int J Pharm Pharm Sci 9(8):84. https://doi.org/10.22159/ijpps.2017v9i8.18293
Sani A, Darma AI, Abdullahi IL, Musa BU, Imam FA (2023) Heavy metals mixture affects the blood and antioxidant defense system of mice. J Hazard Mater Adv 11:100340. https://doi.org/10.1016/j.hazadv.2023.100340
Orji OU, Awoke JN, Harbor C, Igwenyi IO, Obasi OD, Ezeani NN, Aloke C (2020) Ethanol leaf extract of Psychotria microphylla rich in quercetin restores heavy metal induced redox imbalance in rats. Heliyon 6(9):e04999. https://doi.org/10.1016/j.heliyon.2020.e04999
Ashafaq M, Tabassum H, Vishnoi S, Salman M, Raisuddin S, Parvez S (2016) Tannic acid alleviates lead acetate-induced neurochemical perturbations in rat brain. Neurosci Lett 617:94–100. https://doi.org/10.1016/j.neulet.2016.02.001
Alechinsky L, Favreau F, Cechova P, Inal S, Faye PA, Ory C, Thuillier R, Barrou B, Trouillas P, Guillard J, Hauet T (2020) Tannic acid improves renal function recovery after renal warm ischemia–reperfusion in a rat model. Biomolecules 10(3):439. https://doi.org/10.3390/biom10030439
Zou H, Sun J, Wu B, Yuan Y, Gu J, Bian J, Liu X, Liu Z (2020) Effects of cadmium and/or lead on autophagy and liver injury in rats. Biol Trace Elem Res 198(1):206–215. https://doi.org/10.1007/s12011-020-02045-7
Kim DW, Ock J, Moon KW, Park CH (2021) Association between Pb, Cd, and Hg exposure and liver injury among Korean adults. Int J Environ Res Public Health 18(13):6783. https://doi.org/10.3390/ijerph18136783
Akomolafe SF, Akinyemi AJ, Anadozie SO (2014) Phenolic acids (gallic and tannic acids) modulate antioxidant status and cisplatin induced nephrotoxicity in rats. Int Sch Res Notices 2014:1–8. https://doi.org/10.1155/2014/984709
El-Habit OH, Abdel Moneim AE (2014) Testing the genotoxicity, cytotoxicity, and oxidative stress of cadmium and nickel and their additive effect in male mice. Biol Trace Elem Res 159(1–3):364–372. https://doi.org/10.1007/s12011-014-0016-6
Kaushal S, Ahsan AU, Sharma VL, Chopra M (2019) Epigallocatechin gallate attenuates arsenic induced genotoxicity via regulation of oxidative stress in balb/C mice. Mol Biol Rep 46(5):5355–5369. https://doi.org/10.1007/s11033-019-04991-5
Tucker PG (2011) Agency for toxic substances and disease registry. Environmental Health and Medicine Education. Cadmium Toxicity 1–63. https://www.atsdr.cdc.gov/csem/cadmium/docs/cadmium.pdf
Inglot P, Lewinska A, Potocki L, Oklejewicz B, Tabecka-Lonczynska A, Koziorowski M, Bugno-Poniewierska M, Bartosz G, Wnuk M (2012) Cadmium-induced changes in genomic DNA-methylation status increase aneuploidy events in a pig Robertsonian translocation model. Mutat Res Genet Toxicol Environ Mutagen 747(2):182–189. https://doi.org/10.1016/j.mrgentox.2012.05.007
Guo H, Liu H, Wu H, Cui H, Fang J, Zuo Z, Deng J, Li Y, Wang X, Zhao L (2019) Nickel carcinogenesis mechanism: DNA damage. Int J Mol Sci 20(19):4690. https://doi.org/10.3390/ijms20194690
Dumala N, Mangalampalli B, Chinde S, Kumari SI, Mahoob M, Rahman MF, Grover P (2017) Genotoxicity study of nickel oxide nanoparticles in female Wistar rats after acute oral exposure. Mutagenesis 32(4):417–427. https://doi.org/10.1093/mutage/gex007
Guo Z, Xie W, Lu J, Guo X, Xu J, Xu W, Chi Y, Takuya N, Wu H, Zhao L (2021) Tannic acid-based metal phenolic networks for bio-applications: a review. J Mater Chem B 9(20):4098–4110. https://doi.org/10.1039/D1TB00383F
Baer-Dubowska W, Szaefer H, Majchrzak-Celińska A, Krajka-Kuźniak V (2020) Tannic acid: specific form of tannins in cancer chemoprevention and therapy-old and new applications. Curr Pharmacol Rep 6(2):28–37. https://doi.org/10.1007/s40495-020-00211-y
Funding
The authors are thankful to the Department of Science and Technology-Fund for Improvement of Science and Technology Infrastructure (DST-FIST), Government of India, for providing financial assistance to the Department of Zoology, Panjab University, Chandigarh, India, for carrying out the present work.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study's conception and design. Material preparation, data collection, and analysis were performed by M.S., P.D., S.K., A.u.A., S.M., M.B., and M.C. The first draft of the manuscript (along with figures) was prepared by M.S., and all the authors commented on all versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics Approval
This study was performed as per the “Guide for the Care and Use of Experimental Animals” approved by the Institutional Animal Ethics Committee (IAEC), Panjab University (PU/45/99/CPCSEA/IAEC/2015/678).
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sharma, M., Devi, P., Kaushal, S. et al. Cyto and Genoprotective Potential of Tannic Acid Against Cadmium and Nickel Co-exposure Induced Hepato-Renal Toxicity in BALB/c Mice. Biol Trace Elem Res (2024). https://doi.org/10.1007/s12011-024-04117-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12011-024-04117-4