
Abstract
Parkinson’s disease (PD) is the second most common progressive neurodegenerative disorder characterized by the accumulation of accumulated alpha-synuclein (α-Syn) in substantia nigra. Research has shown that selenium (Se) can protect neural cells through the actions of selenoproteins, including selenoprotein P (SelP) and selenoprotein S (SelS), which participate in endoplasmic reticulum-associated protein degradation (ERAD). In this study, we investigated the potential protective role of Se in a pre-clinical PD rat model.
We aimed to evaluate the therapeutic effects of Se administration in the 6-hydroxydopamine (6-OHDA) induced unilateral rat PD model. Male Wistar rats were utilised for unilateral PD animal model which were subjected to stereotaxic surgery and injected with 20 μg 6-OHDA/5 μl 0.2% ascorbate saline. After confirming the model, the rats were intraperitoneally injected with 0.1, 0.2, and 0.3 mg/kg of sodium selenite for 7 days. We then performed behavioral tests, including apomorphine-induced rotation, hanging, and rotarod tests. Following sacrifice, we analysed the substantia nigra area of the brain and serum for protein quantification, element analysis, and gene expression analysis.
Our results indicate that the administration of 0.3 mg/kg of Se improved the motor deficiency in hanging, rotarod, and apomorphine-induced rotational tests. While there was no significant improvement in the expression of α-Syn, Se increased the expression of selenoproteins. Additionally, levels of selenoproteins, Se, and α-Syn both brain and serum were re-established by the treatment, suggesting the role of Se on the α-Syn accumulation. Furthermore, Se improved PD-induced biochemical deficits by increasing the levels of SelS and SelP (p<0.005).
In conclusion, our findings suggest that Se may have a protective role in PD. 0.3 mg/kg dosage of Se increased the expression of selenoproteins, reduced the accumulation of α-Syn in the brain, and improved PD-induced motor deficits. These results suggest that Se may be a potential therapeutic option for PD treatment.
Graphical abstract

Similar content being viewed by others
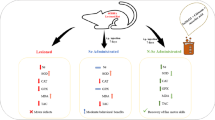
Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) is a prevalent progressive neurodegenerative disorder [1], characterized by the aggregation of alpha-synuclein (α-Syn) and a reduction in dopamine levels [2]. Pathologically, the loss of nigrostriatal dopaminergic neurons in the substantia nigra pars compacta (SNpc) is a hallmark of PD [3]. Common clinical features of PD include rigidity, tremors, bradykinesia, and postural reflexes, while non-motor symptoms such as cognitive impairments, sleep, and behavioral disorders including depression, irritability, and apathy are also common [4]. Although the precise pathogenic mechanisms of PD are not yet fully understood, congenital and environmental factors appear to play a significant role in the risk of developing PD, and nutrients have been considered to play a role in the onset and progression of PD [5]. In a balanced diet, micronutrients are essential for brain development and have a protective impact on the brain. Deficiencies or imbalances in micronutrients can be linked to neurodegenerative disorders [6].
Selenium (Se) is an essential trace element that plays a vital role in human and animal health by maintaining redox homeostasis, antioxidant defence, and anti-tumor and immune regulatory functions [7]. The average serum level of Se in adults is 13.67 mcg/dL [8], and the recommended daily intake (RDI) of Se for adults is 50–70 micrograms/day. Se is available in various forms, including multi-mineral/stand-alone supplements, selenomethionine, sodium selenite, and sodium selenate [9]. The upper tolerance level of Se is approximately 400 micrograms/day, and it is usually toxic when taken above their nutritional dosage [9, 10]. Se is essential for the brain, and brain tissue is susceptible to Se deficiency [11]. Se functions through the activation of selenoproteins in the brain [11]. There are 25 Se-containing proteins (selenoproteins (SP/SeP)) playing important roles in physiological functions such as antioxidant status and metabolism [12]. Most of the selenoproteins have only one selenocysteine (Sec) residue, while selenoprotein P (SelP) has 10 Sec residues [13]. SelP is a secreted glycoprotein composed of two domains: the larger N-terminal domain with one Sec residue in a redox motif and the smaller C-terminal domain with nine Sec [14]. SelP synthesis affects whole-body Se content through the homeostasis and distribution of Se [15]. A direct signalling function has been observed in which SelP interacts with post-synaptic apolipoprotein E receptors 2 (ApoER2) [16].
Selenoprotein S (SelS), a protein containing Sec at position 188 and a penultimate C-terminal residue, plays a significant role in the endoplasmic reticulum (ER) stress [17, 18]. SelS is an essential component of the ER-associated protein degradation (ERAD) complex located in the ER membrane. However, the pathways and roles of interacting proteins in SelS-associated disorders remain unclear [17, 18]. SelS is involved in intracellular membrane transportation and anchors protein complexes to the ER membrane [19]. The ER’s primary function is protein synthesis and quality control, where newly synthesized proteins undergo folding and post-translational modification, regulated directly by ERAD. Therefore, ERAD is crucial in relieving ER stress by degrading misfolded/unfolded proteins [19].
Recent research has revealed that SelS is essential in extracting misfolded/unfolded proteins from the ER to the cytosol during ERAD [20]. One such misfolded protein may be α-Syn, involved in Parkinson’s disease (PD) pathology. These findings highlight the association between selenoproteins and brain functions and their contribution to PD pathology, specifically α-Syn accumulation, providing further insights into the role of Se in PD.
Material and Methods
Animal and Ethics
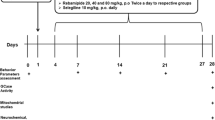
Male WISTAR rats, aged 3 months ± 17 days and weighing 420 ± 20g, were obtained from the Experimental Animal Center of Mashhad Medical University for the current study. The rats were maintained under a 12-h light/dark cycle and a constant room temperature of 22 ± 2°C. They were given normal laboratory chow (Parsfeed Co., Iran) and water ad libitum. The animals were treated following the Animal Welfare and Animal Experiment Guidelines, and the protocols were approved by the Animal Research Ethics Committee of Mashhad University of Medical Sciences (Ethic number: IR.MUMS.MEDICAL.REC.1399.345). Figure 1 provides a summary of all the procedures used in this study.

Summarized procedures of the current study
Animal Surgery
In order to anesthetizing rats before the surgery, ketamine (75 mg/kg, i.p.) and xylazine (10 mg/kg, i.p.) were administered. The rats were then placed on a stereotaxic table, with the incisor bar positioned 3.3 mm below the interaural line, to begin the surgical procedures. A midline incision (~1.5 cm length starting between the ears) was made in the skin over the head to expose the bregma and posterior by stripping the periosteum. In order to PD symptoms, 20 μg of 6-hydroxydopamine (6-OHDA) was dissolved in 5 μl of 0.2% ascorbate saline. Then, 5 μl of the solution containing 20 μg of 6-OHDA was injected into the left medial forebrain bundle (MFB) using a 0.5 mm diameter, 5-μl Hamilton syringe during 2.5 min. The injection was performed at a depth of 7.8 mm ventral to the scalp, 4.7 mm posterior to the bregma, and 1.4 mm left of the midline. The needle was left in place for further 5 min for complete absorption. In the same way, sham operated control rats were infused with 5 μl of 0.2% ascorbate saline [21].
Treatment
The purpose of the current experiment was to investigate the effects of Se administration on PD in rats. Se was administered intraperitoneally at doses of 0.1, 0.2, and 0.3 mg/kg for 7 consecutive days [22, 23]. A total of 40 rats were divided into five groups, each containing eight rats. Due to several limitations in creating the 6-OHDA model, including efflux of 6-OHDA to the lateral ventricle, backflow along the needle shaft which led to an unsuccessful lesion, and error in injecting the appropriate 6-OHDA doses, any animals were excluded from our study. Thus, six animals from each group were used for experimental analysis. The groups were designated as follows:
-
Group I: Sham-operated control group: i.p. administration of normal saline+ 0.2% ascorbate saline injection into the left MFB.
-
Group II: 6-hydroxydopamine (6-OHDA) lesioned group: i.p. administration of normal saline+ 6-OHDA injection (20 μg 6-OHDA/5 μl 0.2% ascorbate saline) into the left MFB.
-
Group III: Se 0.1 mg/kg group: i.p. administration of Se (0.1 mg/kg) + 6-OHDA injection (20 μg 6-OHDA/5 μl 0.2% ascorbate saline) into the left MFB.
-
Group IV: Se 0.2 mg/kg group: i.p. administration of Se (0.2 mg/kg) + 6-OHDA injection (20 μg 6-OHDA/5 μl 0.2% ascorbate saline) into the left MFB.
-
Group V: Se 0.3 mg/kg group: i.p. administration of Se (0.3 mg/kg) + 6-OHDA injection (20 μg 6-OHDA/5 μl 0.2% ascorbate saline) into the left MFB.
Motor and Behavior Tests
Apomorphine-Induced Rotational Test
Successfully constructing PD models in rodents requires measurable impairments in behavioral and physical activities and anatomical and biochemical modifications. The primary index for evaluating behavior is the apomorphine-induced rotation test, and apomorphine (2 mg/kg, i.p., Sigma-Aldrich, USA) was administered to the animals to perform rotation tests 14 and 21 days after surgery. The number of rotations post administration was recorded for 30 min, and the average number within 30 min was calculated. Animals with a contra-lateral rotation speed of more than 7 rotations per minute (r/min) were considered to have PD models [24, 25].
Hanging Test
The rats were placed on a wire (a 100 cm length, 40 cm height, and 2 mm diameter), and then the wire was reversed, causing the animals to swing from it. The hanging duration was recorded, and the mean duration was calculated for each group. The data were then compared between groups [26].
Rotarod Test
Prior to the surgery, rotarod training was conducted. Following the surgery, the rodents were again placed on the rotarod (10 rpm speed) for several days to train them to maintain their balance. During the experiment, the animals were placed on the rotarod, and the time they spent on it was recorded. The mean duration for each group was calculated, and the maximum time considered per rat was 600 s [27].
Sample Preparation
Twenty-one days after treatment, the animals were anesthetized with ketamine (75 mg/kg, i.p.) and xylazine (10 mg/kg, i.p.) to collect blood samples from the heart. The sera were quickly separated by centrifugation and stored at –80°C. The animals were sacrificed by decapitation, and samples were taken from the striatum area and a small amount of adjacent tissue, including the substantia nigra, and stored at –80°C.
Atomic Adsorption
Se levels were measured in both tissue liquid and serum using graphite furnace atomic absorption spectroscopy with a longitudinally heated graphite atomizer (Agilent-AA240), Zeeman-effect background correction, and Se hollow cathode lamp (Agilent Technologies). In order to prepare the liquid sample, 50 μl was diluted with 450 μl of Triton solution, which was made by dissolving 5 mg ascorbic acid (Sigma), 2.5 ml nitric acid (Sigma), and 600 μl Triton X-100 (Merck) in 500 ml of distilled water. An auto-sampler (Varian-PSD120) was used to inject 30 μl of the diluted sample and 20 μl of palladium chloride (500mg/L) as a matrix modifier into the furnace.
ELISA Test
To evaluate the levels of selenoproteins and α-Syn in both Substantia nigra specimens and serum, the ELISA technique was employed. Frozen brain samples were removed, rinsed with phosphate-buffered saline (PBS, 0.01 M), minced, mixed with PBS, and homogenized using a tissue homogenizer (all procedures were performed on ice). The homogenized samples were centrifuged (1000g for 20 min), and the supernatants were collected to detect the values following the kit's instructions (Zellbio, Germany). A microplate reader was used to detect dual wavelengths, specifically a detection wavelength of 450 nm and a calibration wavelength of 620 nm. A standard curve was generated, and the concentration of each sample was calculated. The mean level of each protein in each group was calculated and compared [28].
RNA Extraction and Real-Time Quantitative PCR (qRT-PCR)
Total RNA was extracted from the brain tissue samples using a total RNA extraction kit (Yekta Tajhiz, Iran) following the manufacturer’s instructions. A NanoDrop spectrophotometer (NanoDrop 1000™, USA) was used to quantify the RNA concentration. Next, 1 μg of RNA was reverse-transcribed to complementary DNA (cDNA) using a cDNA Synthesis Kit (Pars Toos, Iran). qRT-PCR was performed using primers for SelS, SelP, and α-Syn purchased from Bonyakhteh (Bonyakhteh Co., Tehran, Iran) (Table 1). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a housekeeping gene, and all amplifications were carried out in a Roche real-time thermo-cycler (Mannheim, Germany). The relative mRNA expression was calculated using the comparative Ct method (2–∆∆Ct) [29].
Statistical Analysis
The SPSS 21.0 program (Chicago, IL) was utilized to process the data. Firstly, normality and homogeneity of variance were checked. For normal data, one-way ANOVA with post hoc Tukey test was used to compare the mean levels and standard deviation, S.D between multiple groups. The Mann-Whitney test was used to compare medians between groups (interquartile range, IQR) for abnormal data. A p-value less than 0.05 was considered statistically significant, as disclosed in the results. For correlation analysis, the Spearman test was employed.
Results
Apomorphine-Induced Rotation Test
The apomorphine-induced rotation test was conducted 14 days after the 6-OHDA injection to confirm the PD models. Three weeks after surgery, the rotation numbers in the treatment groups were significantly lower than in group II (p<0.001). The rotation numbers in the lesion were significantly higher than in group I (p<0.001), whereas the rotation numbers significantly decreased in group V (p<0.001). Additionally, rodents in group V had a lower rate of rotations compared to groups II and III (p<0.001). At the same time, there was no significant difference between groups V and IV (p > 0.05) (see Fig. 2).

The comparison of the contralateral rotations at the end of the 3rd week. ***p < 0.001 refers to the difference between groups III, IV, and V compared to the lesion group, and *p < 0.05 shows the differences between group III and V. (Mean ± SD, n = 6); ###p< 0.001 shows the difference between groups I and II; +++p< 0.001 shows the difference between groups V and II. Group number I, sham-operated control group; II, lesion group; III, Se 0.1mg/kg; IV, Se 0.2 mg/kg; IV, Se 0.3 mg/kg
Hanging Tests
A hanging test was conducted 21 days after stereotaxic surgery. The results showed a significant decrease in hanging time for group II (p<0.001). Moreover, group IV had a higher hanging time than group III (p<0.001). The hanging time was significantly higher in treatment groups (p<0.001) compared to group II. Additionally, the hanging time was significantly higher in group V compared to groups IV and III (p<0.01) but lower than in group I (see Fig. 3).

The average wire hanging time (Sec) at the end of the 3rd week. ###p< 0.001 shows the difference between group I and II; **p < 0.05 refers to the difference between groups III and IV; ***p < 0.05 shows the differences between treatment groups and lesion (group II); +++p< 0.001 shows the difference between groups III, IV, and V; $$p < 0.05shows the difference between group III and control group (group I) (mean ± SD, n = 6). Group number I, sham-operated control group; II, lesion group; III, Se 0.1mg/kg; IV, Se 0.2 mg/kg; IV, Se 0.3 mg/kg
Rotarod Test
A rotarod test was conducted 21 days after the 6-OHDA injection. The results showed a significant impairment in performance in the rotarod test for group II (p<0.001) and group III (p<0.001) compared to group I. However, the rotarod time in group IV and group V was higher than in group II (p<0.001). Additionally, the performance time in group V was significantly higher than in group III (p<0.001) (see Fig. 4).

Rotarod performance (Sec) at the end of the 3rd week. ***p < 0.05 refers to the difference between the lesion and treatment groups compared to the control group; $$p< 0.001 shows the difference between group V compared to groups II and III; **p < 0.05 shows the differences between groups IV and V; &&p < 0.05 shows to the difference between groups IV and III; ###p< 0.001 shows the difference between groups I, II, and III (mean ± SD, n = 6). Group number I, sham-operated control group; II, lesion group; III, Se 0.1mg/kg; IV, Se 0.2 mg/kg; IV, Se 0.3 mg/kg
Selenium Level
Table 2 shows the concentrations of Se in both the brain and serum. The levels of selenium in the brain and serum of group II were significantly lower than group I (p<0.05). The most significant difference in baseline Se levels in the brain and serum was observed between groups V and I (p<0.01). Moreover, there was no significant difference in Se levels between group I and group V in both serum and brain (p<0.001). Although there was no significant difference in selenium levels in the brain between group II and group III (p> 0.05), there was a significant difference in their serum levels (p<0.05), indicating that low selenium levels could not transfer to brain cells.
Furthermore, the mean level of Se in group V was significantly higher than in other treatment groups (p≤ 0.001), but there were only slight differences between group V and the group I (p> 0.05). The mean serum Se level in both groups III and IV was approximately the same, but not in the brain (p> 0.05). Therefore, the evidence suggests that a high level of Se administration leads to a higher level of Se in the serum and brain (p <0.001). There was no significant correlation between the proportions of Se in serum and brain. However, the Se level in the serum was positively correlated with both brain (r=0.89, p≤ 0.01) and serum SelP levels (r=0.82, p≤ 0.05). Higher levels of Se resulted in higher levels of SelS (r=0.85, p≤ 0.05). Additionally, a high dosage of Se in the brain reduced the concentration of α-Syn (r=0.94, p≤ 0.05).
Selenoproteins and α-Syn Evaluations
Table 2 presents the concentrations of selenoproteins in both the brain and serum. The most notable difference in selenoprotein concentrations between groups was observed between groups V and I and the two other treatment groups (p < 0.01). While there were no significant differences in serum SelP concentrations between groups III and IV (p > 0.05), the concentration of SelP in the brain was significantly higher in group IV (p < 0.01). Additionally, the level of SelS in both the brain and serum was higher in group IV than in group III (p < 0.01). Furthermore, there was a significant positive correlation between serum SelS and SelS in the brain (r = 0.96, p < 0.01), which was followed by SelP concentration (r = 0.81, p < 0.05), and resulted in a reduction of α-Syn proportion (r = 0.76, p < 0.05).
Gene Expression
Growing evidence suggests that an increase in selenoprotein P (SEPP1/ SelP) gene expression induced by Se stimulation is associated with increased selenoprotein S (SEPS1/ SelS) gene expression (r=0.86, p≤0.01), which plays a crucial role in ERAD. To investigate the effect of Se on selenoprotein gene expression, we conducted qRT-PCR analysis in various models. As shown in Fig. 5, Se administration increased the gene expression of SEPP1 in the rat PD model brain (p≤0.001), which subsequently led to elevated SEPS1 expression (p≤0.001). However, we observed no significant change in α-Syn gene (SCNA) expression in response to Se administration when compared to the lesion group II (p≥0.05). Additionally, Se administration did not cause a substantial decrease in α-synuclein mRNA levels in brain tissue when compared to groups I and II (p≥0.05). Nevertheless, Se administration induced the expression of SeS1 (r=0.80, p <0.05) and SEPP1 (r=0.86, p <0.05). Although SEPS1 and SEPP1 did not induce the SCNA gene, SelS reduced α-Syn levels, providing evidence that SelS impacts accumulated α-Syn through ERAD.

Comparing the relative gene expression in each group at the end of the 3rd week. The levels of expression of genes were normalized by GAPDH mRNA levels.***p < 0.001 refers to the difference of SES1 expression between the lesion (group II) and group V; **p < 0.005 refers to the difference of SES1 expression between groups III and I; *p < 0.005 refers to the difference of SES1 expression between groups III and V; ##p < 0.05 refers to the difference of SEP1 expression between groups III and V; ###p < 0.001 refers to the difference of SEP1 expression between the lesion (group II) and groups IV and V; $$$p < 0.001 refers to the difference of SEP1 expression between the control (group I), lesion (group II), and group III; &&&p < 0.001 refers to the difference of SCNA expression between the control (group I), lesion (group II), and treatment group. (Mean ± SD, n = 6). Group number I, sham-operated control group; II, lesion group; III, Se 0.1mg/kg; IV, Se 0.2 mg/kg; IV, Se 0.3 mg/kg
Discussion
One significant finding of this study is that Se administration can alleviate α-Syn accumulation, which is crucial for the survival of neural cells. Accumulation of α-Syn leads to neuronal cell death and severe PD outcomes. In addition, Se can attenuate the motor dysfunctions observed in PD models. We also established a neurotoxicity model replicating several characteristics of PD disorder, including increased levels of α-Syn and 6-OHDA-induced neuronal death. It is well established that α-Syn is closely linked to PD pathogenesis since it is a crucial component of Lewy bodies, a clinical hallmark of PD [30]. However, there are conflicting reports regarding the function of α-Syn in neuronal survival or death modulation. While the expression of mutated proteins reinforces neurotoxin-induced apoptosis in primary neurons and neuronal cell lines, the overexpression of wild-type proteins has shown opposite results [31]. For instance, transgenic mice with α-Syn overexpression were found to be resistant to dopaminergic neuron neurotoxicity in an in vivo study [32]. Furthermore, α-Syn overexpression is believed to be neuroprotective and upregulates in response to neural oxidative stress [33]. Nonetheless, multiple replications of the α-Syn gene have been shown to induce PD [34,35,36]. Consistent with these findings, our results indicate that the PD models had a higher α-Syn expression, followed by increased protein accumulation in brain cells. Several preclinical therapeutic modalities targeting pathological α-Syn have revealed promising results.
The neurotoxin 6-OHDA has been widely used to induce in vivo and in vitro PD models [37,38,39]. Reactive oxygen species production has been suggested as a mechanism underlying 6-OHDA neurotoxicity [40]. Some studies have also proposed that 6-OHDA abolishes the α-Syn anti-apoptotic function by inhibiting proteasomal activities and triggering α-Syn accumulation [41]. The accumulation of α-Syn occurs through a series of actions, including ubiquitination, transition to oligomeric protofibrils, and ultimately forming fibrillar beta-sheet structures [42]. The notion that 6-OHDA induces PD-like neuropathy by promoting α-Syn accumulation is supported by the fact that 6-OHDA deposition is observed in human disorder and by in vitro experiments demonstrating that 6-OHDA, but not MPP+, initiates cell death by inhibiting proteasomal activities and causing α-Syn accumulation [35]. Moreover, 6-OHDA negatively regulates α-Syn via various pathways and synergistically leads to cell death [41].
Se is a vital dietary micronutrient that has been shown to have neuroprotective effects against nigrostriatal 6-OHDA in various forms, including sodium selenite, diphenyl di-selenide (PhSe)2, and selenium methionine [43]. In a rat model of PD induced by 6-OHDA (PhSe)2, a compound with neuroprotective properties, reversed motor impairment, and neurochemical alterations [44]. In another study, different dosages of inorganic sodium selenite and organic seleno-L-methionine (Se-Met) were fed to PD mouse models. Their neuroprotective effects and mechanisms were explored and compared. The study found that Se-Na provided better neuroprotective effects than Se-Met [45]. Our investigations also demonstrate that sodium selenate can protect substantia nigra neurons against free radical damage caused by straight administered 6-OHDA into the substantia nigra. Moreover, a long-term study has shown that prolonged exposure to dietary Se has a powerful protective effect [46]. In addition, a study by Zafar et al. has demonstrated that Se may slow down the progression of neurodegeneration in parkinsonism by upregulating the antioxidant status, reducing dopamine loss, and promoting functional recovery [23].
Other studies have reported that Se can protect against bradykinesia (locomotor damage) and DNA damage in the lymphocytes of rats in the animal model of PD induced by paraquat [47]. Khan et al. demonstrated that Se partially reversed the toxic effects (dopamine depletion) caused by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), which has the 1-methyl-4-phenyl pyridinium (MPP+) as an active metabolite, a molecule very similar to paraquat, in an animal model of PD in mice [48]. In PD mice, Sun et al. found that serum Se levels greatly increased after Se-Na supplementation, which could provide neuroprotective effects [45]. While the results of these studies are relevant, the Se exposure timeline in our animal models needs to be carefully considered. Se appeared as a toxic microelement in the early 1930s, with rodents exposed to high levels of Se suffering from selenosis disease, now known as alkali disease, and blind staggers [49]. Rodents with over 6ppm Se (as sodium selenite) supplementation for 45 days developed liver cirrhosis, enlarged spleen, and depression. Additionally, rats with a 4ppm Se diet died months earlier than their controls [49]. However, it has been proposed that selenite, the toxic form of Se, can inhibit both DNA and RNA synthesis and promote apoptosis [49]. Therefore, depending on the dosage, Se appears to have a dual role as a neuroprotective and toxic agent [50, 51].
Se possesses beneficial properties due to its incorporation into selenoproteins, some of which are expressed in the brain, including glutathione peroxidases (GPX), thioredoxin reductases (TRX), SelP, and SelS. SelP is an essential Se-binding protein [12]. We have shown a significant correlation between total Se proportions and SelP in both serum and brain samples of PD models. Additionally, SelP levels were increased in substantia nigra neurons, which are centralized in Lewy bodies, demonstrating a co-localization of expressing SelP and α-Syn genes. Since SelP brain proportions in parkinsonism brain regulation greatly depend on serum levels, and the rather small area of SelP regulation is in substantia nigra [52], the quantification of SelP can be a valuable PD biomarker that is reflected in the serum. Our novel finding in the present study is that Se usage in an appropriate dosage can increase SelP levels in both serum and brain, resulting in decreased α-Syn levels. Furthermore, there is a hypothesis that SelP provides Se for selenoprotein synthesis via selenocysteine lyase, which delivers the highly reactive Sec residues for use in other selenoprotein syntheses, such as SelS [15].
SelS, an endoplasmic reticulum (ER)-resident selenoprotein, regulates physiological procedures such as protein folding and ER stress in neurons [53]. Previous investigations have indicated that the SelS-mediated complex, composed of SelK, valosin-containing protein (VCP), Derlin, and E3 ubiquitin ligase, can transport mis/unfolded proteins to the ubiquitin-proteasome system for degradation. Thus, SelS and SelK have important functions in folding proteins and ER-associated degradation (ERAD) pathways [20, 54]. Earlier studies have shown that neuronal SelS expression boosts with ER stress, and SelS gene knockout induces ER stress-mediated apoptosis [55]. Additionally, toxic α-Syn has been introduced to cause ER stress via interacting with Bip/Grp78 in physiological situations, which SelS regulates [56,57,58]. Unfortunately, studies on the role of Se supplementation on SelS status and improved SelS expression on cognitive capacity are limited, and the pathways through which Se affects PD from the viewpoint of SelS are not yet clarified. Our experiments show a high correlation between Se intake and SelS expression in the brain, which can alleviate the accumulation of misfolded α-Syn in the brain and serum. According to our results, no significant reduction in the expression of α-Syn in the brain was found, while its level was reduced in both brain and serum, indicating that SelS is removing the translated α-Syn that is accumulated following its misfolding.
Our data also revealed that both hanging time in the hanging test and duration time on the rotarod in the treatment group animals were significantly higher than in the lesion group, proving the beneficial effects of Se on the motor functions in the rat model of PD. In line with our results, another study has shown that in the rotarod performance test, PD model mice made by chronic MPTP injection had significant motor deficits, as indicated by the decline in riding time. Treatment with a low dosage of Se-Na could significantly improve the motor functions of chronic PD mice models by increasing their riding time [45]. Furthermore, Abolarin et al. demonstrated the same results and claimed that animal PD models’ motor activity was slightly increased by Se [59].
Lastly, we recognize some limitations of our study. Once time Se treatments per se did not do realized, we cannot establish if there are beneficial or toxicological effects on the behavior and biochemical analysis in a basal situation. In this sense, further studies on the mechanisms of selenoprotein function will increase our understanding of how different forms of Se and selenoproteins could be used to treat and prevent PD.
Conclusion
In conclusion, the current study suggests that a specific dosage of Se can alleviate the accumulation of α-Syn in PD through SelS and SelP. Se induces the expression of selenoproteins but not α-Syn. Evidence indicates that the reduction of accumulated α-Syn occurs after protein translation through ERAD by SelS. Se also improves PD-induced motor dysfunctions and alleviates biochemical impairments in PD’s rat model induced by 6-OHDA.
Data Availability
All generated or analyzed data during our research are included in this published article. Future data that support the findings are available from the corresponding author, upon reasonable requests.
Abbreviations
- α-Syn:
-
Alpha-synuclein
- ApoER2:
-
Apolipoprotein E receptors 2
- cDNA:
-
Complementary DNA
- GAPDH:
-
Glyceraldehyde-3-phosphate dehydrogenase
- PD:
-
Parkinson’s disease
- PBS:
-
Phosphate-buffered saline
- Se:
-
Selenium/selenite sodium
- SelS:
-
Selenoprotein S
- SelP:
-
Selenoprotein P
- SEPS1/SES1:
-
Selenoprotein S gene
- SEPP1/SEP1:
-
Selenoprotein P gene
- SP/SeP:
-
Selenoproteins
- Sec:
-
Selenocysteine
- Se-Met:
-
Seleno-L-methionine
- SCNA:
-
α-Syn gene
- MPTP:
-
1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- i.p.:
-
Intraperitoneal
- ERAD:
-
Endoplasmic reticulum-associated protein degradation
- 6-OHDA:
-
6-Hydroxydopamine
- qRT-PCR:
-
Real-time quantitative PCR
- SNpc:
-
Substantia nigra pars compacta
- RDI:
-
Recommended daily intake
- MFB:
-
Medial forebrain bundle
- ER:
-
Endoplasmic reticulum
References
Nakajima A, Ohizumi Y (2019) Potential benefits of nobiletin, a citrus flavonoid, against Alzheimer’s disease and Parkinson’s disease. Int J Mol Sci 20(14):3380
Lo Bianco C et al (2002) α-Synucleinopathy and selective dopaminergic neuron loss in a rat lentiviral-based model of Parkinson's disease. Proc Nat Acad Sci 99(16):10813–10818
Baquet ZC et al (2009) A comparison of model-based (2D) and design-based (3D) stereological methods for estimating cell number in the substantia nigra pars compacta (SNpc) of the C57BL/6J mouse. Neuroscience 161(4):1082–1090
Balestrino R, Schapira A (2020) Parkinson disease. Eur J Neurol 27(1):27–42
Sherzai AZ et al (2016) Micronutrients and risk of Parkinson’s disease: a systematic review. Gerontol Geriatr Med 2:2333721416644286
Bianchi VE, Herrera PF, Laura R (2021) Effect of nutrition on neurodegenerative diseases. A Syst Rev Nutr Neurosci 24(10):810–834
Xu J et al (2020) Impact of selenium deficiency on inflammation, oxidative stress, and phagocytosis in mouse macrophages. Biol Trace Element Res 194:237–243
Laclaustra M et al (2010) Serum selenium and serum lipids in US adults: National Health and Nutrition Examination Survey (NHANES) 2003–2004. Atherosclerosis 210(2):643–648
Medicine, I.O (2000) Dietary reference intakes for vitamin C, vitamin E, selenium, and carotenoids: a report of the panel on dietary antioxidants and related compounds, subcommittees on upper reference levels of nutrients. National Academies Press, Washington, DC
Bai K et al (2017) Preparation and antioxidant properties of selenium nanoparticles-loaded chitosan microspheres. Int J Nanomed 12:4527
Solovyev ND (2015) Importance of selenium and selenoprotein for brain function: from antioxidant protection to neuronal signalling. J Inorg Biochem 153:1–12
Saito Y (2021) Selenium transport mechanism via selenoprotein P—Its physiological role and related diseases. Front Nutr 8:685517
Saito Y et al (2004) Domain structure of bi-functional selenoprotein P. Biochem J 381(3):841–846
Shetty S, Copeland PR (2018) Molecular mechanism of selenoprotein P synthesis. Biochim Biophys Acta (BBA)-Gen Subj 1862(11):2506–2510
Seale LA et al (2018) Relationship between selenoprotein P and selenocysteine lyase: insights into selenium metabolism. Free Rad Biol Med 127:182–189
Torres DJ et al (2021) Selenoprotein P modulates methamphetamine enhancement of vesicular dopamine release in mouse nucleus accumbens via dopamine D2 receptors. Front Neurosci 15:631825
Bubenik JL, Miniard AC, Driscoll DM (2013) Alternative transcripts and 3′ UTR elements govern the incorporation of selenocysteine into selenoprotein S. PLoS One 8(4):e62102
Turanov AA et al (2014) Selenoprotein S is involved in maintenance and transport of multiprotein complexes. Biochem J 462(3):555–565
Shchedrina VA et al (2010) Structure–function relations, physiological roles, and evolution of mammalian ER-resident selenoproteins. Antiox Redox Sig 12(7):839–849
Lee JH et al (2014) Pro178 and Pro183 of selenoprotein S are essential residues for interaction with p97 (VCP) during endoplasmic reticulum-associated degradation. J Biol Chem 289(20):13758–13768
Dolatshahi M et al (2015) Ellagic acid improves hyperalgesia and cognitive deficiency in 6-hydroxidopamine induced rat model of Parkinson’s disease. Iran J Basic Med Sci 18(1):38
Atif F, Yousuf S, Agrawal SK (2008) Restraint stress-induced oxidative damage and its amelioration with selenium. Eur J Pharmacol 600(1-3):59–63
Zafar KS et al (2003) Dose-dependent protective effect of selenium in rat model of Parkinson’s disease: neurobehavioral and neurochemical evidences. J Neurochem 84(3):438–446
Haddadi H et al (2018) Chronic treatment with carvacrol improves passive avoidance memory in a rat model of Parkinson’s disease. Arquivos de neuro-psiquiatria 76:71–77
Kumari N et al (2018) Neuroprotective effect of IDPU (1-(7-imino-3-propyl-2, 3-dihydrothiazolo [4, 5-d] pyrimidin-6 (7H)-yl) urea) in 6-OHDA induced rodent model of hemiparkinson’s disease. Neurosci Lett 675:74–82
Haeri P et al (2019) Neuroprotective effect of crocin on substantia nigra in MPTP-induced Parkinson’s disease model of mice. Anatom Sci Int 94:119–127
Li Y, Hollis ER II (2023) Nicotinic acetylcholine signaling is required for motor learning but not for rehabilitation from spinal cord injury. Neural Regen Res 18(2):364
Younesian O et al (2022) Long-term excessive selenium supplementation affects gene expression in esophageal tissue of rats. Biol Trace Element Res 201(7):3387–3394
Lorestani S et al (2018) Increased glutathione reductase expression and activity in colorectal cancer tissue samples: an investigational study in Mashhad Iran. Middle East J Cancer 9(2):99–104
Wakabayashi K et al (2013) The Lewy body in Parkinson’s disease and related neurodegenerative disorders. Mol Neurobiol 47:495–508
Blum D et al (2001) Molecular pathways involved in the neurotoxicity of 6-OHDA, dopamine and MPTP: contribution to the apoptotic theory in Parkinson's disease. Progr Neurobiol 65(2):135–172
Thomas B et al (2011) Resistance to MPTP-neurotoxicity in α-synuclein knockout mice is complemented by human α-synuclein and associated with increased β-synuclein and Akt activation. PloS One 6(1):e16706
Quilty MC et al (2006) Alpha-synuclein is upregulated in neurones in response to chronic oxidative stress and is associated with neuroprotection. Exp Neurol 199(2):249–256
Chang C-W et al (2020) Plasma and serum alpha-synuclein as a biomarker of diagnosis in patients with Parkinson’s disease. Front Neurol 10:1388
Malek N et al (2014) Alpha-synuclein in peripheral tissues and body fluids as a biomarker for P arkinson’s disease–a systematic review. Acta Neurol Scand 130(2):59–72
Wakabayashi K (2020) Where and how alpha-synuclein pathology spreads in Parkinson’s disease. Neuropathology 40(5):415–425
Chandrasekhar Y et al (2018) Gallic acid protects 6-OHDA induced neurotoxicity by attenuating oxidative stress in human dopaminergic cell line. Neurochem Res 43:1150–1160
Xi Y et al (2018) MitoQ protects dopaminergic neurons in a 6-OHDA induced PD model by enhancing Mfn2-dependent mitochondrial fusion via activation of PGC-1α. Biochim Biophys Acta (BBA)-Mol Basis Dis 1864(9):2859–2870
Yan T et al (2019) The neuroprotective effect of schisandrol A on 6-OHDA-induced PD mice may be related to PI3K/AKT and IKK/IκBα/NF-κB pathway. Exp Gerontol 128:110743
Jamal M et al (2022) COA-Cl evokes protective responses against H2O2-and 6-OHDA-induced toxic injury in PC12 cells. Neurotox Res 40(6):2061–2071
Monti B et al (2007) Alpha-synuclein protects cerebellar granule neurons against 6-hydroxydopamine-induced death. J Neurochem 103(2):518–530
Bourdenx M et al (2017) Protein aggregation and neurodegeneration in prototypical neurodegenerative diseases: examples of amyloidopathies, tauopathies and synucleinopathies. Progr Neurobiol 155:171–193
Sophiabadi M, Rastgoo N, Haghdoost-Yazdi H (2022) Dopaminergic neuronal death in substantia nigra associates with serum levels of total bilirubin, selenium, and zinc: evidences from 6-hydroxydopamine animal model of Parkinson’s disease. Biol Trace Element Res 200(9):4058–4067
Sampaio TB et al (2017) Involvement of BDNF/TrkB signaling in the effect of diphenyl diselenide on motor function in a Parkinson’s disease rat model. Eur J Pharmacol 795:28–35
Sun C et al (2023) Selenium forms and dosages determined their biological actions in mouse models of Parkinson’s disease. Nutrients 15(1):11
Rahbardar MG et al (2021) Protective effects of selenium on acrylamide-induced neurotoxicity and hepatotoxicity in rats. Iran J Basic Med Sci 24(8):1041
Ellwanger JH et al (2016) Biological functions of selenium and its potential influence on Parkinson’s disease. Anais da Acad Brasil Ciências 88:1655–1674
Khan HA (2010) Selenium partially reverses the depletion of striatal dopamine and its metabolites in MPTP-treated C57BL mice. Neurochem Int 57(5):489–491
Himeno S, Imura N (2002) Selenium in nutrition and toxicology. Heavy Metals in the Environment. Marcel Dekker Inc, New York, NY, pp 587–629
Vinceti M et al (2014) Selenium neurotoxicity in humans: bridging laboratory and epidemiologic studies. Toxicol Lett 230(2):295–303
Wei D et al (2015) Synthesis, characterization, antioxidant activity and neuroprotective effects of selenium polysaccharide from Radix hedysari. Carbohydr Polym 125:161–168
Bellinger FP et al (2012) Changes in selenoprotein P in substantia nigra and putamen in Parkinson's disease. J Parkinson's Dis 2(2):115–126
Zhang Z-H, Song G-L (2021) Roles of selenoproteins in brain function and the potential mechanism of selenium in Alzheimer’s disease. Front Neurosci 15:646518
Lee JH et al (2015) Selenoprotein S-dependent selenoprotein K binding to p97 (VCP) protein is essential for endoplasmic reticulum-associated degradation. J Biol Chem 290(50):29941–29952
Rueli RH et al (2017) Selenoprotein S reduces endoplasmic reticulum stress-induced phosphorylation of tau: potential role in selenate mitigation of tau pathology. J Alzheim Dis 55(2):749–762
Colla E (2019) Linking the endoplasmic reticulum to Parkinson’s disease and alpha-synucleinopathy. Front Neurosci 13:560
Kelly E et al (2009) Selenoprotein S/SEPS1 modifies endoplasmic reticulum stress in Z variant α1-antitrypsin deficiency. J Biol Chem 284(25):16891–16897
Remondelli P, Renna M (2017) The endoplasmic reticulum unfolded protein response in neurodegenerative disorders and its potential therapeutic significance. Front Mol Neurosci 10:187
Abolarin PO et al (2022) Selenium reduces nociceptive response in acute 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine (MPTP)-induced neurotoxicity. IBRO Neurosci Rep 12:1–11
Acknowledgements
The authors wish to extend special thanks to Kavosh laboratory for assisting with the expert advices to conducting experiments and Dr. Ali Shoeibi for his assistance.
Funding
This work was supported by Mashhad University of Medical Sciences under grant [number 980950]. Dr. Seyed Isaac Hashemy has received the grant.
Author information
Authors and Affiliations
Contributions
All of the authors contributed to the study conception. The study was designed by Dr. Seyed Isaac Hashemy. Material preparation was performed by Mahmoud Hosseini. Data collection and analysis were performed by Sanaz Salaramoli and Hamid Reza Joshaghani. The first draft of the manuscript was written by Sanaz Salaramoli and revised by Hamid Reza Joshaghani. All of the authors commented on previous versions of the manuscript. All of the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Date of Research Proposal Approval
The research proposal was approved on 2020-07-22.
Ethics Approval
The Animal Research Ethics Committee of the Mashhad university of medical sciences accepted the protocols (Ethic number: IR.MUMS.MEDICAL.REC.1399.345).
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Highlights
1. Se reduces α-Syn accumulation.
2. Se induces SelP and SelS expression.
3. 6OHDA induces α-Syn accumulation in substantia nigra.
4. SelS alleviates α-Syn accumulation.
6. Se has therapeutic effects on PD.
7. Se improves PD-induced motor dysfunctions.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Salaramoli, S., Joshaghani, H.R., Hosseini, M. et al. Therapeutic Effects of Selenium on Alpha-Synuclein Accumulation in Substantia Nigra Pars Compacta in a Rat Model of Parkinson’s Disease: Behavioral and Biochemical Outcomes. Biol Trace Elem Res 202, 1115–1125 (2024). https://doi.org/10.1007/s12011-023-03748-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12011-023-03748-3