
Abstract
A highly thermostable alkaline serine protease gene (SPSPro, MN429015) obtained from haloalkaliphilic actinobacteria, Nocardiopsis sp. Mit-7 (NCIM-5746), was successfully cloned and overexpressed in Escherichia coli BL21 under the control of the T7 promoter in the pET Blue1 vector leading to a 20-kDa gene product. The molecular weight of the recombinant alkaline protease, as determined by SDS-PAGE and the Mass Spectrometer (MALDI-TOF), was 34 kDa. The structural and functional attributes of the recombinant thermostable alkaline serine protease were analyzed by Bioinformatic tools. 3D Monomeric Model and Molecular Docking established the role of the amino acid residues, aspartate, serine, and tryptophan, in the active site of thealkaline protease.The activity of the recombinant alkaline protease was optimal at 65 °C, 5 °C higher than its native protease. The recombinant protease was also active over a wide range of pH 7.0–13.0, with a maximal activity of 6050.47 U/mg at pH 9. Furthermore, the thermodynamic parameters of the immobilized recombinant alkaline protease suggested its reduced vulnerability against adverse conditions under which the enzyme has to undergo varied applications.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The thermostable alkaline serine proteases are highly significant in biotechnological applications due to their ability to function under a wide range of pH, temperatures, and denaturing agents, including solvents. These proteases are specifically suitable for silk degumming, detergents, leather processing, food industries, dairy, and wastewater treatment [1,2,3,4].
A broader temperature range, such as 40–80 °C, facilitates the maintenance and cultivation of the microorganisms and bulk production of different metabolites and enzymes. Many studies on the variable thermostability of the proteases are reported in the literature that includes stability with a half-life of 1 h at 50 °C, 25 min at 60 °C, or, in the case of Subtilisin Carlsberg, with a half-life of 2.5 min at 60 °C [5,6,7,8]. Recombination strategies have been used to regulate and manipulate genes to modify proteases displaying specificity and enhanced stability, besides probing the structure–function relationships of the enzymes. The main objectives of gene cloning and its expression in another host are the large-scale production in bulk quantities of specific proteins and the elucidation of the structural basis of the function. The aspect is particularly fascinating and relevant concerning the cloning and expression of genes from extremophilic microorganisms. Escherichia coli is widely used as a mesophilic host to produce recombinant proteins due to its highly illustrated genetics and physiology, cost and ease of cultivation, and high transformation efficiency [9,10,11].
Specialized proteins known as chaperonins are produced by some of these organisms, which help in protein folding. Such proteins are also helpful in refolding the denatured proteins into their native form to restore their functions under stressful conditions. The cell membrane of thermophiles contains saturated fatty acids, creating a hydrophobic environment for the cell and keeping the cell rigid enough to function at elevated temperatures [12, 13].
Cloning and expression of protease genes of extremophilic microorganisms into the mesophilic host are desirable for expanding the horizons of the applications [14,15,16,17,18]. The limitation of the native protease produced in its original host is its optimal production during the late exponential growth phase. Furthermore, the complex nutritional requirement of the halophilic actinobacterium makes the process uneconomical. To overcome these problems and to achieve higher production of the proteases from the haloalkaliphilic actinobacteria, the protease gene was cloned and expressed into a mesophilic host, E. coli in this study. The gene expression was analyzed, and recombinant alkaline protease was purified and characterized. The recombinant protease was then immobilized using chitosan and glutaraldehyde-activated alginate beads, and the kinetic parameters of the immobilized enzyme were compared. The protease structure was elucidated by SWISS-MODEL, SWISS Dock, and Computed Atlas of Surface Topography of proteins (CASTp), establishing the crystal structure and surface topography of the enzyme.
The purpose of this study, therefore, was to clone the protease gene of a haloalkaliphilic actinomycete and investigate its expression in a mesophilic host. The protease included in this study was stable under alkaline pH at high temperatures and in the presence of detergents. To further investigate the properties of pH and temperature stability at the molecular level, the amino acid sequence and its composition were analyzed [19,20,21,22,23,24,25,26,27].
Materials and Methods
Bacterial Strains and Plasmids
The haloalkaliphilic actinobacteria Nocardiopsis sp. Mit-7 (NCIM-5746) was isolated from the salt-enriched soil of the Gujarat coast. A gene encoding alkaline serine protease from this organism was cloned, expressed, and characterized. Bacterial strains E. coli Novablue single and E. coli BL21 (DE3) were obtained from Novagen (USA). The plasmids pST1 and pET BlueTM1 were used in this study.
The halo-tolerant and alkaliphilic actinomycete, Mit-7, was isolated using enrichment techniques from the saline-alkaline soil of the Coastal region of Gujarat in the laboratory of Prof. Satya P. Singh, Department of Biosciences, Saurashtra University, Rajkot, India. The isolate was characterized based on the Gram reaction, colony morphology, and biochemical characteristics. The actinomycetes displayed a filamentous structure as well as the fragmentation of hyphae. The organism was further identified based on 16S rRNA gene sequencing as Nocardiopsis sp.
Extraction of the Genomic DNA
Genomic DNA was extracted from the haloalkaliphilic actinobacteria, Nocardiopsis sp. Mit-7 (KT008301.1) using a modified soft lysis method (13,18). The extracted DNA was further used as the template for amplifying the alkaline protease gene using His tagged methylated SPS 16 (forward: ATGCTTCAGTCGGTGTACGTGTACCT, reverse: CGGTGTAGAGGTTGAGAACGTTCTAA). The PCR reactions were carried out in a thermal cycler (EppendorfMaster Cycler Nexus Gradient, Germany) according to the manufacturer’s instructions (Sigma-Aldrich (USA) at varying Tm values. PCR products were purified using Gene JET™ Gel Extraction Kit (#K0691, Thermo Fisher Scientific, USA).
Amplicon Sequencing
The alkaline protease gene was sequenced using ABI automated sequencer (Applied Biosystem, Macrogen Co., South Korea). The nucleotide sequences were determined by Sanger Sequencing Method. The phylogenetic tree was constructed using the neighbor-joining (NJ) method [28] by MEGA 7.0 software. The Nocardiopsis sp. Mit-7 thermostable alkaline serine protease gene sequence (MN429015.1:1–972) and the available protease gene sequences in the GenBank database depicting the homology were aligned with the multiple alignments of the sequences using CLUSTAL_X program [29] using Kimura’s two-parameter model [30] with 1000 bootstrap replicates [31].
Cloning of the Alkaline Serine Protease Gene
The alkaline protease gene from the haloalkaliphilic actinobacteria, Nocardiopsis sp. Mit-7, was cloned using pETBlue-1 Perfectly Blunt® Cloning Kit–Novagen as per the manufacturer’s instructions. It enables cloning of DNA with any type of end because all types of ends are converted to blunt-, phosphorylated-ends during the End Conversion step. For the cloning of the protease gene, two different vectors-pSTBlue-1 and pETBlue™-1, available as AccepTor™ Vector, were used. Both provided easy visualization of the recombinants by blue/white screening using lacZ α-complementation. The pSTBlue-1 is a general-purpose vector with dual opposed T7 and SP6 promoters, both with ampicillin and kanamycin resistance and an array of flanking restriction sites. The pETBlue-1 vector is a novel plasmid specifically designed to enable high-level T7 RNA polymerase-driven expression of target genes while providing the convenience of a blue/white screening method (Fig. S5).
The pETBlue vectors are fundamentally a new category of the expression vector, combining the most desirable features of the popular cloning vectors with the full features of the T7-driven protein expression. Blue/white visual screening of the recombinants is enabled by inserting the target genes into the lacZ α-peptide coding region in an antisense orientation relative to a modified E. coli. Expression is possible by T7 transcription and translation signals correctly positioned upstream from the sense orientation of the target gene. As with the standard pET vectors, the target proteins are produced by IPTG induction.
The following components were oriented to prepare PCR products for cloning: blunt vector (50 ng/µl), positive control insert (4.5 ng/µl), end conversion mix, T4 DNA ligase, nuclease-free water, NovaBlue Singles™ Competent Cells, SOC medium, test plasmid for transformation (0.2 ng/µl), and Tuner™ DE3 PLac I competent cells. Furthermore, for the direct cloning, 2 μl of the reaction was added to the end-conversion reaction after extracting the PCR reaction with chloroform. After removal of the oil overlay, 1 volume chloroform/isoamyl alcohol (24:1) was added to the PCR reaction; the mixture was vortexed vigorously for 1 min, centrifuged at 12,000 g for 1 min, and then up to 2 μl of the aqueous phase was added to the end conversion reaction. The PCR product was precipitated and resuspended in a smaller volume of TE buffer (10 mM Tris–HCl, 1 mM EDTA, pH 8.0). To the cooled end conversion reaction, 1 μl Blunt Vector and 1 μl T4 DNA Ligase were added. The reaction mixture was incubated at 22 °C for 15 min. To transform NovaBlue Singles™ Competent Cells, 1 μl of the ligation reaction was added to ice-thawed Singles Competent Cells. To determine the transformation efficiency, 1 μl (0.2 ng) test plasmid was added to one of the tubes containing cells. After incubation on ice for 5 min, heat shock was given for 30 s in a 42 °C water bath without shaking, followed by the ice treatment for 2 min. The transformants were screened by colony PCR and blue/white screening. The proteolytic activity of the clones carrying recombinant plasmids was examined on pETBlue1 on an LB gelatin agar plate after 24 h of growth at 37 °C. The insertion of the PCR product into the competent cells was confirmed by the colony PCR using a suitable primer. DNA samples were analyzed on 1% agarose gel using a 1-kb DNA ladder (#SM1173, Thermo Fisher Scientific, USA).
For the analysis of the gene expression, 100 μl of the clone culture was inoculated in 50 ml of LB broth containing ampicillin (50 μg/ml). Cultures were grown overnight at 37 ºC at 180 rpm in an orbital shaker. The growth was monitored by the optical density at 660 nm and normalized up to 1. After normalizing the optical density, the cultures were re-inoculated into 3 different flasks containing fresh 100-ml LB + ampicillin medium. The flasks were supplemented with 0.5 mM, 1.0 mM, and 2.0 mM IPTG (isopropyl-β-D-thiogalactopyranoside), respectively, to investigate the induction effect. The culture flasks were then incubated at 37 °C at 180 rpm. The cultures after the growth were diluted to normalize O.D. up to 1 at the time intervals of 0 h, 2 h, 4 h, 6 h, 8 h, and 24 h. The culture aliquots were centrifuged at 10,000 rpm/10 min, and the pellets were washed twice with PBS buffer (7 pH). The cell suspensions were then sonicated using the Ultrasonic Homogenizer LABSONIC M, 100 W (Sartorious Stedium Biotech, France), with the intensity of 100 and 5 cycles of 5 min each. The contents were centrifuged at 10,000 rpm/15 min to separate the soluble protein fractions and inclusion bodies and the cell debris. An insoluble protein fraction was recovered by treating the pellets with 8 M urea and incubating them at 4 °C for 24 h. Proteins in inclusion bodies were denatured by urea. The urea-denatured fractions were allowed to refold after the gradual and slow removal of urea by dialysis as a renaturation process.
Media and Growth Conditions
The native alkaline protease was produced by the haloalkaliphilic actinobacterium strain, Mit-7, using gelatin broth (500 ml) that contained g/l: gelatin, 10; peptone, 5; yeast extract, 5; NaCl, 50; pH 9 adjusted by adding the appropriate volume of the separately autoclaved 20%, w/v Na2CO3. It was followed by incubation at 37 ºC on a rotary shaker at 120 rpm. The protease production was monitored for 14 days. The crude enzyme (the cell-free extract) was obtained by centrifugation at 5000 rpm for 10 min.
For recombinant protease, the actinobacterium was aerobically grown in 50 ml of the actinobacterial selective medium with 5% salt at 28 °C with 120 rpm shaking. After 2 days of cultivation, the culture was harvested, and DNA was isolated by the soft lysis method, as described by Sambrook et al., with few modifications. The detailed method is described below.
The actinomycete was grown in yeast extract-malt extract (YEME) broth supplemented with 5% salt at pH 9.0 and 28–30 °C for 2–3 days. The growth of the organism was monitored by the absorbance at 600 nm. Parallelly, for the determination of cell dry weight, 20 ml of the culture broth was centrifuged, and the pellet was washed and dried at 60 °C till constant weight. Furthermore, for the extraction of the genomic DNA, the culture aliquots were centrifugated for 15 min at 10,000 rpm to obtain cell pallets to be suspended in the STE buffer. The cell suspension was centrifuged at 10,000 rpm, and the pellets were re-suspended in GET buffer with SDS (20%) and lysozyme solution (10 mg/ml in Tris–Cl pH 8) followed by incubation for 2 h. This was followed by P/C/I (phenol/chloroform/isoamylpropanol) and C/I extraction steps. The DNA was pooled by adding 3 M potassium chloride and chilled ethanol. The pooled DNA was then suspended in an appropriate volume of distilled water and preserved at − 20 °C. During the DNA extraction process, RNAse and Proteinase K (100 μg/ml) were also used to enhance the purity and quality of the genomic DNA. The quality of the DNA was analyzed by spectrophotometry and agarose gel (0.8%) electrophoresis.
The extracted DNA was then used as a template for amplifying the alkaline protease gene using forward: ATGCTTCAGTCGGTGTACGTGTACCT and reverse: CGGTGTAGAGGTTGAGAACGTTCTAA primers. For the PCR reaction, 1 μl of the genomic DNA (~ 50 ng), 2.5 μM (1.0 μl) of each primer, 12.5 μl of the 2 × PCR Master Mix (Thermofisher), and nuclease-free water (to make up 25.0 μl) were included in a total volume of 25-μl reaction mixture. Master Mix contained an optimized concentration of Taq Polymerase, individual dNTPs, reaction buffer, and 2 mM MgCl2. The PCR reaction was carried out in a Gradient Master-Thermal Cycler (Eppendorf USA). The PCR steps included the following: 1st step 95 ºC for 1 min, 2nd step 94 ºC for 1 min, 3rd step 60 ºC for 1 min, and 4th step 72 ºC for 1.30 min. Steps 2–4 were carried out for 35 cycles, followed by the final step at 72 ºC for 10 min and at 4 ºC to hold. The PCR products were analyzed on 0.8% agarose gel electrophoresis and purified by the Gene JETTM Gel Extraction Kit (#K0691, Thermo Fisher Scientific, USA).
Escherichia coli strains were grown in LB media at 37 °C for 24 h. For protease heterologous expression, the recombinant E. coli were grown overnight in LB broth containing 50 µg/ml ampicillin and 30 µg/ml kanamycin at 37 °C and 180 rpm. The protease gene was expressed by adding 1 mM IPTG (isopropyl β-d-thiogalactopyranoside) after 2 h of growth. The effect of temperatures, incubation periods, and IPTG concentrations on the protease gene expression was studied. The bacterial growth was monitored at 600 nm. The intracellular and extracellular fractions of the clones harboring recombinant alkaline serine protease were analyzed for protease activities.
Purification of the Native Protease
The native extracellular alkaline protease from Mit-7 was purified by two steps, ammonium sulfate precipitation followed by hydrophobic interaction chromatography using phenyl sepharose 6FF. The purified protease was eluted with 0.1 M sodium phosphate buffer, pH 8.0, containing a decreasing gradient of ammonium sulfate (1000–100 mM). Fractions at a flow rate of 0.8 ml min−1 were collected and analyzed for protease activity. The final enzyme preparations from the two-step purification methods displayed a single band on the sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) [32].
Preparation and Estimation of the Extracellular and Intracellular Protein
The bacterial cultures were centrifuged at 5000 rpm for 10 min, and the supernatants were taken as an extracellular fraction. The cell pellets were then washed and resuspended in a lysis buffer (Tris–HCl buffer pH 8) and disrupted by sonication using a Lab sonic M Ultrasonicator for 15 s, 50 Hz each for 10 cycles. Finally, the cell-free proteins were separated by centrifugation at 10,000 rpm for 10 min and considered as the intracellular protein content. The protein was estimated using the Bradford method [33]. The recombinant alkaline serine protease activity was found in the intracellular fraction.
Purification of the Recombinant Protease
Native protease was purified by conventional methods, while a provision for the polyhistidine tag was used for the purification of the recombinant protease. Thus, the recombinant protease from the intracellular fraction of E. coli BL21(DE3)-pET Blue™ 1 was purified with the help of a His-Tag purification column (#06,781,535,001, Sigma-Aldrich, USA) and was used at a faster flow rate of 2.5 ml min−1, eluted with phosphate buffer (0.1 M, pH 7.5) at a flow rate of 2.5 ml/min. The active fractions were monitored for protease activity. Complete His-Tag Purification Columns included a Sepharose-based, pre-charged, ready-to-use Ni2+ chelate resin to purify His-tagged proteins. It yielded highly pure protease from the crude lysates in a single step.
The purity of the native and recombinant alkaline proteases was judged on SDS-PAGE according to Laemmli’s (1970) method [32]. The protein bands were visualized by Coomassie brilliant blue R-250 staining, and the molecular weight of the protease was determined using protein markers (#11,802,124, Fermentas).
MALDI Analysis
Matrix assisted laser desorption ionization imaging mass spectrometry (IMS) is a powerful tool to rapidly and accurately analyze and identify proteins. In the MALDI experiment, peptides were generated by digesting the protease with sequence-specific trypsin. The peptides were then analyzed by MALDI-TOF Mass Spectrometer to get insight into the peptide masses. The data of the experimental analysis was compared with the database containing peptide masses of a given organism generated by the same sequence-specific protease. Due to soft-ionization, the ionized molecules show fragmentation of the analytes, leading to the identification of the molecular ions in the mixtures. The Mascot Score for the proteins generated by the MALDI TOF MS/MS analysis predicted the unique peptides match with the significant query coverage.
The analysis was carried out using a Thermo Scientific Orbitrap Elite Mass Spectrometer and Bruker Autoflex MALDI-TOF Mass Spectrometer at the Proteomics and Mass Spectrometry Facility, University of Georgia, Athens, Georgia, USA.
Structural Attributes of the Recombinant Alkaline Serine Protease
The three-dimensional structure of a protein determines its specific function. Experimental determination of the protein structure is an expensive and time-consuming process. Therefore, the computational structure modeling of the genomic data was used to generate a 3D model structure of the alkaline serine protease by the SWISS Model. The SWISS Model was developed by the Computational Structural Biology Group at the SIB Swiss Institute of Bioinformatics at the Biozentrum, University of Basel (https://swissmodel.expasy.org) [34]. Since a protein model building is error-prone and thus can affect the conclusions regarding the structural stability of the protein, the Ramachandran plot was generated to analyze the stability of the protein structure [35]. Furthermore, the most favored structure was analyzed using CASTp (Computed Atlas of Surface Topography of proteins). It analyzes four aspects of the query protein: (i) imprints of the negative volumes of pockets, cavities, and channels; (ii) topographic features of biological assemblies in the Protein Data Bank; (iii) improved visualization of the protein structures and pockets; and (iv) more intuitive structural and annotated information, including information of secondary structure, functional sites, variant sites, and other annotations of protein residues [36].
Protease Assay
To estimate protease activity, the cloned E. coli was cultured on a nutrient agar medium containing 3% gelatin. The addition of Frazier reagent gives a clear zone of gelatin hydrolysis against a white background. Protease was quantitatively assayed by the modified Hanson-Hagihara method [37]. One unit of protease was defined as the amount required to liberate 1 µg/ml of tyrosine per min.
Biochemical and Kinetic Studies of the Native and Recombinant Proteases
Protease activity was monitored at various temperatures in the range of 30–80 °C. The effect of pH was determined by using different buffer systems (20 mM phosphate buffer, pH 6–8; 20 mM glycine–NaOH buffer, pH 9; 20 mM borax-NaOH buffer, pH 10–11, and 20 mM KCl-NaOH buffer, pH 12–13). Enzyme activities were expressed in relative terms. For the kinetic analysis, the reactions were carried out at different concentrations of casein in the range of 0–10 mg. Km and Vmax were calculated from the Lineweaver–Burk plots. The activation energy was calculated from the Arrhenius plot.
Catalysis and Stability of Native, Recombinant, and Immobilized Proteases
pH
The catalysis of the native, recombinant, and immobilized proteases was investigated using sodium phosphate buffer (pH-7 and 8), glycine–NaOH buffer (pH-9), borax-NaOH buffer (pH-10), and KCl-NaOH buffer (pH-12 and 13). Casein (0.6%, w/v) prepared in buffer systems of different pH was used as substrate, and the protease assay was carried out at 60 °C for 10 min.
The stability of the native, recombinant, and immobilized proteases was assessed by incubating the enzymes in different buffer systems for 6 h. The residual protease activities were monitored at 1-h intervals for 6 h. The deactivation rate constant (k) was calculated from the semi-logarithmic plot of the residual activities as a function of time.
Temperatures
For temperature profiling, native, recombinant, and immobilized proteases were assayed at temperatures in the range of 30–80 °C. The reaction mixtures containing 0.6%, w/v, casein solution prepared in 20 mM Borax–NaOH buffer, pH 10, and 0.1 ml of the protease samples were incubated at various temperatures for 10 min.
The activation energy (Ea) for the thermal inactivation was calculated from the slope of the Arrhenius plot according to the equation:
where k is the rate of inactivation at T, R, gas constant (8.314 J mol−1 K−1), and T is the absolute temperature in Kelvin. The operational stability of the native, recombinant, and immobilized proteases at various temperatures was examined as described above. The deactivation rate constant (k) was calculated from the semi-logarithmic plot of the residual activities as a function of time.
Effect of Solvents, Inhibitors, and Surfactants on the Protease Activity
The influence of various effectors on the activities of the native and recombinant proteases was studied under standard assay conditions, where the reaction mixture was supplemented with 50% (v/v) benzene (log Pow 2.1) and methanol (log Pow -0.75), 10 mM of inhibitors: phenyl methyl sulfonyl fluoride (PMSF), ethylene diamine tetra acetate (EDTA), dithiothrionine (DTT), and β-mercaptoethanol. The surfactants, triton X-100 and tween-80 at 0.5% (v/v) and SDS at 0.5% (w/v), were used. The effect was assessed by considering the control (without inhibitors and surfactants) as 100%.
Immobilization of the Recombinant Protease
One gram of chitosan was added to 0.1 M HCl (2.5 ml) containing glutaraldehyde (2.5%, v/v) and shaken for 2 h at 30 °C. The solubilized chitosan was mixed with a sodium alginate solution loaded with 500 U of protease (A). After incubation for 30 min, with the help of a syringe, tiny beads were prepared by dropping the alginate solution in 0.1 M CaCl2. The resultant beads (I) were washed and stored in 20 mM Borax-NaOH buffer (pH-10) at 4 °C for 24 h to remove the unbound protease (B). Recovery activity compares the activity yield of the immobilized enzyme and the activity yield of the free enzyme. The immobilization yield and respective recovery activity were calculated by the equations: \(Immobilization yield=A-B/A*100\) and \(Recovery activity= (I/A-B*100)\), where A, B, and I were designated as loaded, unbounded, and immobilized proteases [38].
Results
Amplification and sequencing of the alkaline serine protease gene
The alkaline serine protease gene was PCR amplified and sequenced. The sequence was submitted to GenBank with the accession number MN429015.
Structural Attributes of the Alkaline Serine Protease
The protease gene sequence shows high similarity with trypsin-like serine proteases from Nocardiopsis sp. TSRI0078, Nocardiopsis sinuspersici, Nocardiopsis halotolerans, Nocardiopsis arvandica, and MULTISPECIES: trypsin-like serine protease.
A 3D monomeric model structure of the reported thermostable alkaline protease (MN429015) was generated using SWISS Model (Fig. S3 (a)).The structure was created using 3WY8.1.A serine protease monomer and 3cp7.1. An alkaline serine protease AL20 homodimer from an extremophilic microorganism was used as a reference. Based on the analysis of the Ramachandran Plot, the stability of the proposed structure was established. The proposed model suggests that most residues belong to the most favored and additionally allowed region, while six residues were in the least allowed regions, and only two residues were located in the disallowed region (Fig. S3 (b), suggesting a stable protein structure. Furthermore, molecular docking of the most favored model of the alkaline serine protease revealed that the active site contained aspartate, serine, and tryptophan residues (Fig. 3).
Figure S3(c) illustrates the FTIR of the immobilized recombinant protease. FTIR has elucidated the structural changes during the immobilization of protease and trypsin. Furthermore, the amide I and amide II bands were least affected during the immobilization retaining their functionality [38].
Expression of Alkaline Serine Protease Gene from Nocardiopsis alba Mit-7.
The pET blue™ 1 plasmid-harboring 1000 bp DNA fragment of the alkaline serine protease gene (Gene Bank accession number MN429015) with promoter and terminator region was expressed in E. coli BL21 (DE3) on LB agar plates containing 3% gelatin (Fig. S1 (a), S1 (b)). Furthermore, the colony PCR confirmed approximately 1000 bp of the insert in the recombinant plasmids (Fig. S1 (c)). A maximum protease activity of 223.80 U/ml was detected in the intracellular fraction with 0.1 mM IPTG induction and 4 h of growth at 37 °C, further depicted in the form of percent residual activities (Fig. S1(d).
Purification of the Native and Recombinant Protease
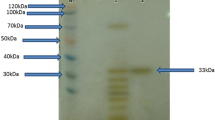
The optimum growth and protease production were achieved after 7 days of the incubation. The protease in the culture filtrate was precipitated by ammonium sulfate followed by the hydrophobic interaction chromatography using Phenyl Sepharose 6FF. The two-step purification method yielded 51.17-fold purification, 21.62% yield, and a specific activity of 6050.48 U/mg for the native alkaline protease (Table 1). The recombinant alkaline protease was extracted by sonication of E. coli BL21 (DE3)-pET Blue™ 1. A purification fold of 56.41 was achieved by His tag purification, with 82.64% yield (Table 1). The protein profile of the purification was analyzed by SDS-PAGE stained by Coomassie blue. The purified protease exhibited a single band on SDS-PAGE with approximate molecular weight of 34 kDa. The zymogram confirmed the location of the band of the protease activity (Fig. 1). The comparative properties of both proteases are displayed in Table 2.

Visualization of the alkaline serine protease on SDS-PAGE stained with Coomassie brilliant blue R-250. Lane 1 protein ladder, Lane 2 purified native protease, Lane 3 crude native protease, Lane 4 purified cloned protease from Escherichia coli BL21, Lane 5 total protein profiles of cloned E. coli BL21 harboring pET plasmid
MALDI Analysis
MALDI analysis revealed that the functional groups of the present alkaline serine protease corresponded with the proteases/peptidases of Nocardiopsis sp. and Bacillus sp. with the mascot score of 268.99 and 194.52, respectively. A total of 14 peptide spectra matched with the peptidase/protease, and the theoretical peptide mass was estimated as 34.2 kDa (Fig. S2).
Characteristics of the Native and Recombinant Proteases
The native and recombinant proteases had reasonably good activities over a wide range of temperatures, 30–90 °C (Table 3). Both, the native and recombinant proteases, exhibited significant maximum activities of 234 U/ml and 682.5 U/ml, respectively, at pH 10 in borax-NaOH buffer. The relative activities of the native alkaline protease were 19.23, 26.92, 80.45, 51.60, 32.05, and 28.84% at pH 7, 8, 9, 11, 12, and 13, respectively. Similarly, for the recombinant alkaline serine protease, the relative activities were 28.57, 42.86, 87.36, 62.53, 44.72, and 30.22% at pH 7, 8, 9, 11, 12, and 13, respectively (Fig. 2).

Relative activities of native (closed dot with sleeping line), recombinant (closed square with sleeping line), and immobilized recombinant (closed triangle with sleeping line) proteases at different pH
The effects of various denaturing agents on the native and recombinant proteases are illustrated in Fig. S6. PMSF almost completely inhibited the native and recombinant proteases, suggesting its serine nature. On the other hand, in the presence of EDTA, DTT, and β-mercaptoethanol both, native and recombinant proteases, were significantly stable with an average of 76%, 68%, and 69% residual activities, respectively. While in the presence of triton X-100, tween 80, SDS, benzene, and methanol, the native and recombinant proteases had average of 94%, 79%, 66%, 86%, and 55% residual activities, respectively (Fig. S6).
Immobilization of the Recombinant Protease
The immobilized recombinant protease showed a lower deactivation rate constant at higher temperatures, suggesting its enhanced thermostability. At 70 °C, half-life of the free and immobilized recombinant proteases was 84 and 480 min, respectively (Table 3). At pH 13, free recombinant protease had 30% residual activity, while the immobilized protease displayed above 50% residual activity (Fig. S6).Immobilized protease had higher activities in various denaturing agents, such as solvents, surfactants, and inhibitors. More than 80% relative activity was recorded in 50% methanol (Log Pow -0.75), which is higher than free recombinant protease. The immobilized protease retained 80% relative activity in 0.5 g/ml SDS, as compared to 66% relative activity of the free recombinant protease under the same condition (Fig. S6).
The immobilization efficiency was calculated as per the equation mentioned in the methods. The immobilized protease had 95% immobilization yield with 75% recovery activity. Loaded protease A = 100 (U/15 ml gel), unbound protease B = 5 (U/15 ml gel), immobilized protease (U/15 ml gel) I = 75 (U/15 ml gel), lost protease activity = A-B-I = 20 (U/15 ml gel). \(Immobilization yield=A-B/A*100=95(\%)\), \(Recovery activity (I/A-B*100)=75 (\%)\).
Kinetic Parameters of the Native, Recombinant, and Immobilized Protease
The Km of the native protease was estimated as 0.0115 mg/ml and maximum velocity as 134 µM/s based on the Lineweaver–Burk plots. For the recombinant protease, these values enhanced to 0.02 mg/ml and 195 µM/s hydrolyzed casein, respectively. While immobilized recombinant protease had Km 0.09 and Vmax 265 µM/s. The standard deviations were 0.09, 0.75, and 0.07 for the native, recombinant and immobilized proteases respectively. The values of standard deviation indicated a limited spread of data.
Discussion
An alkaline protease gene from haloalkaliphilic actinobacteria was cloned, sequenced, and expressed in the mesophilic host, E. coli. The recombinant protease was characterized by its expression, catalysis, stability, and structural traits. The structure of the protease was elucidated using bioinformatics tools. This study represents the first of its kind for an alkaline serine protease from haloalkaliphilic actinobacteria Nocardiopsis. Earlier, SapRH protease gene encoding a serine alkaline protease, SAPRH (GenBank: KR706502.1), from Bacillus safensis RH12, was sequenced and analyzed [39]. The deduced amino-acid sequence had strong homology with other Bacillus proteases [40].
The knowledge of the structure of a protein is instrumental to gain information about the molecular basis of its function [39]. The three-dimensional structures of the recombinant protease in this study were created using SWISS MODEL, which relies on the open computational structural biology framework [41]. The Ramachandran Plot provided fundamental information on the stability of the predicted protein structures and their validation. The conformational versatility of the protein backbone was generated by the possibility of modulating the dihedral angles φ, ψ, and ω with a diversified level of variability. In the line of a typical good model, the selected models possess the most residues in the most favored region, with only a few residues within the disallowed regions [42]. Protein homology modeling and molecular docking were useful in predicting the protein structure and function [43]. The docking simulations data allowed insight into the involvement of some key amino acids in the substrate binding accounting for the selectivity [44]. Since the purified Mit-7 protease was identified as a serine alkaline protease, serine residue played a central role in the breakdown of the peptide bonds. In the cleavage of the peptide bonds, serine functions as the nucleophilic region at the protease active site [45], while some other residues, such as a pair of histidine residues bond with the serine residues at the active site [46]. The presence of a serine residue in the active site proved the serine nature of the protease, while the direct involvement of aspartate was established earlier with serine protease activity [47]. For the first time, the involvement of arginine and tryptophan in the active site of the alkaline serine protease is established based on the structural model generated by molecular docking.
The 3D structural model of the protease was validated by the structure analysis tool of the SWISS-MODEL used to search templates for homology modeling. Swiss Model and Docking simulate the data and allow the involvement of certain pivotal amino acid/s in substrate specificities and binding abilities along with the quality estimation based on the single model [48] (Fig. 3).

Active site prediction of the alkaline serine protease SPSPro by Molecular Docking
Mass spectrometry imaging (MSI) provided label-free, non-targeted molecular and spatial information on the biomolecules. Matrix-assisted laser desorption/ionization (MALDI) imaging mass spectrometry (IMS) proved as a powerful tool for the analysis of the protease structure [49]. The in vitro analysis can provide information about substrate recognition signals and conditional effects. The protein imaging provides significant clues on protein identification and substrate trapping capabilities [50].
Molecular cloning is an essential technique in molecular biology to construct recombinant DNA molecules [51]. The pET Vector system having gene encoding T7 RNA polymerase was used for the protease expression in E. coli BL21 (DE3). Various factors affect and control the level of the expressed protein and its cellular solubility [52]. The factors affecting the expression of the protease gene were investigated concerning the level of induction, growth temperatures, and growth kinetics. The highest expression of the protease gene was found after 4 h of growth at 37 °C with 0.1 mM IPTG induction. The 34 kDa protease of haloalkaliphilic actinobacteria, Nocardiopsis sp., was cloned and overexpressed in E. coli BL21 (DE3) in pET vector. In a similar context, a 38-kDa protease from Bacillus sp. was expressed in E. coli BL21 (DE3) using pET vector [53]. Furthermore, AmyLa α-amylase gene from Laceyella sp. DS3 was expressed in E. coli BL21, maximally after 4 h of adding 0.02 mM IPTG at 37 °C [54].
The Km and Vmax of the recombinant thermostable alkaline serine protease were 0.02 mg/ml and 195 µM/s, respectively. A deactivation rate constant of 3.0689E − 3 at high temperatures suggests the enzyme’s thermostability with t1/2 of 225.21 min at 70 °C (Table 3). The values were significantly higher than a recently reported recombinant serine alkaline protease, which has a t1/2 of 120 min at 70 °C [55]. Both, the native and recombinant proteases, were most active in the alkaline range of pH 9–10, with significant residual activities at pH 8 and 11. In nutshell, the native and recombinant proteases were active over a wide range of pH 7–13, the highest activity being at pH 9–10 (Fig. 2). In a similar context, the expression of an alkaline serine protease in E. coli BL21 (DE3) with pET28a( +) vector revealed the optimum activity at 55 °C and pH 11.0 [56]. In general, the recombinant enzymes are expected to retain similar properties to their native counterparts. Overall, it can be stated that both native and recombinant proteases displayed high activity and stability under various conditions.
With various surfactants, triton X-100, tween 80, and SDS, the native and recombinant proteases were quite stable. Both proteases were also stable in solvents of different log Pow. The solvent stability of the proteases makes them a promising candidate for biodegradation and bioremediation [57]. With triton X-100, the residual activities of the native and recombinant proteases were above 90%. Similarly, Tween-80 did not significantly affect the stability of both forms of the proteases. However, SDS at higher concentrations projecting strongly basic character negatively affected the native as well as recombinant proteases. The solvents with higher log Pow values, such as benzene and surfactants, only marginally affected the activity and stability of the proteases. Nevertheless, the enzymes were marginally inhibited by methanol of log Pow − 0.75. With the increasing log Pow of the solvents, the toxicity adversely affects the alkaline serine proteases [58].
Immobilization of enzymes is expected to enhance the shelf-life, stability, and catalytic features of the enzyme. [59]. Immobilization enhances enzyme activity and stability by making it less vulnerable to adverse environmental changes. The recombinant free protease and immobilized protease have Km values of 0.02 mg/ml and 0.09 mg/ml, respectively. In a similar context, Km of an alkaline serine protease from halophilic actinobacteria did not significantly change probably due to the substrate recognition signal with the specified substrate [60]. The half-life of the immobilized recombinant Mit-7 protease was 480, 329, and 116 min at 60, 70, and 90 °C, respectively. However, the half-life of the enzyme in this study was quite higher than a recently reported thermophilic lipase from Serratia rubidaea strain Nehal-mou [61].
The recombinant and immobilized alkaline serine proteases in this study had high activities and stability in various solvents, detergents, and denaturing agents, which make them suitable candidates for detergent formulations and other applications. Alkaline proteases are considered promising candidates for industrial sectors due to their activity and stability under alkaline and harsh environment [7, 62, 63]. Halotolerant proteases from marine bacterium Vibrio sp. LA-05 are useful in liquid detergent formulations [62]. Furthermore, the use of immobilized protease for dairy and wastewater treatment can be widely applicable for industrial applications [7]. A subtilisin-like serine protease displayed effective properties suitable for detergent industries [64]. In recent times, there appears to be a shift from the purification of enzymes to cloning and protein engineering to meet the expanding needs.
This study, in nutshell, describes the heterologous expression of a highly thermostable alkaline serine protease from Nocardiopsis sp. in E. coli. The recombinant enzyme was purified, immobilized, and characterized. The recombinant protease had improved thermostability compared to the native enzyme. The kinetic and thermodynamic parameters were elucidated for the immobilized recombinant protease suggesting its enhanced stability. The Swiss Model and Docking simulations data provided insight and suggested the involvement of certain key amino acids in substrate binding and selectivity. The study assumes significance as only limited information is available on the diversity and phylogeny, in general, and kinetics and structural aspects of the proteases, particularly of the haloalkaliphilic microorganisms from the saline habitats [65,66,67,68].
Data Availability
All data generated or analyzed during this study are included in this published article.
References
Raiyani, N., & Singh, S. P. (2020). Taxonomic and functional profiling of the microbial communities of Arabian Sea: a metagenomics approach. Genomics, 112, 4361–4369.
Rathore, D. S., Sharma, A. K., Dobariya, A., Ramavat, H., & Singh, S. P. (2022). Cultivation and diversity of marine actinomycetes: Molecular approaches and bioinformatics tools. In L. Karthik (Ed.), Actinobacteria-microbiology to synthetic biology (vol. 10, pp. 215–240). Springer.
Dwivedi, P., Sharma, A. K., & Singh, S. P. (2021). Biochemical properties and repression studies of an alkaline serine protease from a haloalkaliphilic actinomycete, Nocarpdiopsis dassonvillei subsp albirubida OK-14. Biocatalysis and Agricultural Biotechnology, 35, 1–7.
Rathore, D. R., & Singh, S. P. (2021). Kinetics of growth and co-production of amylase and protease in novel marine actinomycete, Streptomyces lopnurensis KaM5. Folia Microbiologica, 66, 303–316.
Bhosale, S. H. L. (1995). Thermostability of high-activity alkaline protease from Conidiobolus coronatus(NCL 86.8.20). Enzyme and Microbial Technology, 17, 136–139.
Durham, D. R., Stewart, D. B., & Stellwag, E. J. (1987). Novel alkaline- and heat-stable serine proteases from alkalophilic Bacillus sp strain GX6638. Journal of Bacteriology, 169, 2762–2768.
Sharma, A. K., Kikani, B. A., & Singh, S. P. (2020). Biochemical, thermodynamic and structural characteristics of a biotechnologically compatible alkaline protease from a haloalkaliphilic, Nocardiopsis dassonvillei OK-18. International Journal of Biological Macromolecules, 153, 680–696.
Arya, P. S., Yagnik, S. M., Rajput, K. N., Panchal, R. R., & Raval, V. H. (2021). Understanding the basis of occurrence, biosynthesis, and implications of thermostable alkaline proteases. Applied Biochemistry and Biotechnology, 1–38.
Suberu, Y., Akande, I., Samuel, T., Lawal, A., & Olaniran, A. (2019). Cloning, expression, purification, and characterization of serine alkaline protease from Bacillus subtilis RD7. Biocatalysis, and Agricultural Biotechnology, 10, 101264.
Mechri, S., Jaouadi, N. Z., Bouacem, K., Allala, F., Bouraoui, A., Ferard, C., & Jaouadi, B. (2021). Cloning and heterologous expression of subtilisin SAPN, a serine alkaline protease from Melghiribacillus thermohalophilus Nari2AT in Escherichia coli and Pichia pastoris. Process Biochemistry, 105, 27–41.
Mechri, S., Bouacem, K., Allala, F., Khaled, M., Bouanane-Darenfed, A., Hacene, H., & Jaouadi, B. (2021). Cloning, expression, and structural modeling of two alkaline serine protease genes from extremophilic Bacillaceae-related species: Application in valorization of invasive crustaceans. Revue Nature et Technologie, 13(02), 19–19.
Everly, C., & Alberto, J. (2000). Stressors, stress, and survival: An overview. Frontiers in Bioscience, 5, 780–786.
Herbert, R., & Sharp, R. (1992). Molecular biology and biotechnology of extremophiles. Chapman and Hall.
Purohit, M., & Singh, S. P. (2014). Cloning, over expression and functional attributes of serine proteases from Oceanobacillus iheyensis O.M.A18 and Haloalkaliphilic bacterium O.M.E12. Process Biochemistry, 49, 61–68.
Chauhan, J. V., Mathukiya, R., Singh, S. P., & Gohel, S. D. (2021). Two steps purification, biochemical characterization, thermodynamics and structure elucidation of thermostable alkaline serine protease from Nocardiopsis alba strain OM-5. International Journal of Biological Macromolecules, 169, 39–50.
Gohel, S. D., & Singh, S. P. (2012). Cloning and expression of alkaline protease genes from two salt-tolerant alkaliphilic actinomycetes in E. coli. International Journal of Biological Macromolecules, 50, 664–671.
Abbasi-Hosseini, S., Eftekhar, M., Yakhchali, F. B., & Minai-Tehrani, D. (2011). Cloning and enhanced expression of an extracellular alkaline protease from a soil isolate of Bacillus clausii inBacillus subtilis. Iranian Journal of Biotechnology, 9, 275.
Sambrook, J., & Russell, D. W. (2001). Molecular cloning-Sambrook & Russel-vol. 1, 2, 3. Cold Springs Harbor Lab Press: Long Island, NY, USA.
Purohit, M. K., & Singh, S. P. (2013). A metagenomic alkaline protease from saline habitat: Cloning, over-expression and functional attributes. International Journal of Biological Macromolecules, 53, 138–143.
Manachini, P. L., Fortina, M. G., & Parini, C. (1988). Thermostable a1- kaline protease produced by Bacillus thermoruber - a new species of Bacillus. Applied Microbiology and Biotechnology, 28, 409–413.
Takami, H., Akiba, T., & Horikoshi, K. (1989). Production of extremely thermostable alkaline protease from Bacillus sp. no. AH-101. Applied Microbiology and Biotechnology, 30, 120–124.
Kumar, C. G. (2002). Purification and characterization of a thermostable alkaline protease from alkalophilic Bacillus pumilus. Letters in Applied Microbiology, 34(1), 13–17.
Peña-Montes, C., González, A., Castro-Ochoa, D., & Farrés, A. (2008). Purification and biochemical characterization of a broad substrate specificity thermostable alkaline protease from Aspergillus nidulans. Applied Microbiology and Biotechnology, 78, 603–612.
Madhavi, J., Srilakshmi, J., Rao, M. R., & Rao, K. R. S. S. (2011). Efficient leather detailing by bacterial thermostable protease. Int J Bio-Sci Bio-Technol, 3(1), 11–26.
Ibrahim, N., Harun, H. C., & Ibrahim, N. A. (2022). Cloning and expression of thermostable alkaline protease 50a in E. coli BL21 (DE3) and TOP10. In AIP Conference Proceedings, 2454(1), 030005.
Lee, J. K., Kim, Y. O., Kim, H. K., Park, Y. S., & Oh, T. K. (1996). Purification and characterization of a thermostable alkaline protease from Thermoactinomyces sp E79 and the DNA sequence of the encoding gene. Bioscience Biotechnology and Biochemistry, 60(5), 840–846.
Saggu, S. K., & Mishra, P. C. (2017). Characterization of thermostable alkaline proteases from Bacillus infantis SKS1 isolated from garden soil. Plos One, 12(11), e0188724.
Ghorbel, S., Kammoun, M., Soltana, H., Nasri, M., & Hmidet, N. (2014). Streptomyces flavogriseus HS1: Isolation and characterization of extracellular proteases and their compatibility with laundry detergents. BioMed Research International, 2014, 345980.
Saha, S., Dhanasekaran, D., Shanmugapriya, S., & Latha, S. (2013). Nocardiopsis sp. SD5: A potent feather degrading rare actinobacterium isolated from feather waste in Tamil Nadu India. Journal of Basic Microbiolology, 53(7), 608–616.
Moreira, K. A., Porto, T. S., Teixeira, M. F. S., Porto, A. L. F., & Lima Filho, J. L. (2003). New alkaline protease from Nocardiopsis sp. partial purification and characterization. Process Biochemistry, 39, 67–72.
Yamamura, H., Ohkubo, S., Ishida, Y., Otoguro, M., Tamura, T., & Hayakawa, M. (2010). Nocardiopsis nikkonensis sp. nov., isolated from a compost sample. International Journal of Systematics and Evolutionary Microbiology, 60, 2967–2971.
Laemmli, U. K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227, 680–685.
Bradford, M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochemistry, 72(1–2), 248–254.
Waterhouse, A., Bertoni, M., Bienert, S., Studer, G., Tauriello, G., Gumienny, R., Heer, F. T., de Beer, T. A. P., Rempfer, C., Bordoli, L., & Lepore, R. (2018). SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Research, 46, 1–7.
Balasco, N., Smaldone, G., Vigorita, M., Del Vecchio, P., Graziano, G., Ruggiero, A., & Vitagliano, L. (2019). The characterization of Thermotogamaritima arginine binding protein variants demonstrates that minimal local strains have an important impact on protein stability. Science and Reports, 9(1), 6617.
Garikapati, V., Karnati, S., Bhandari, D. R., Baumgart-Vogt, E., & Spengler, B. (2019). High-resolution atmospheric-pressure MALDI mass spectrometry imaging workflow for lipidomic analysis of late fetal mouse lungs. Science and Reports, 9(1), 3192.
Hagihara, B. (1960). Bacterial and mold proteases. The enzymes, 4, 193–213.
Thakrar, F. J., & Singh, S. P. (2019). Catalytic, thermodynamic and structural properties of an immobilized and highly thermostable alkaline protease from a haloalkaliphilic actinobacteria, Nocardiopsisalba Tata-5. Bioresource Technology, 278, 150–158.
Rekik, H., Frikha, F., Jaouadi, N. Z., Gargouri, F., Jmal, N., Bejar, S., & Jaouadi, B. (2019). Gene cloning, expression, molecular modeling and docking study of the protease SAPRH from Bacillus safensis strain RH12. International Journal of Biological Macromolecules, 125, 876–891.
Furhan, J., Awasthi, P., & Sharma, S. (2019). Biochemical characterization and homology modelling of cold-active alkophilic protease from Northwestern Himalayas and its application in detergent industry. Biocatalysis and Agricultural Biotechnology, 17, 726–735.
Studer, G., Tauriello, G., Bienert, S., Waterhouse, A. M., Bertoni, M., Bordoli, L., Schwede, T., & Lepore, R. (2019). Modeling of protein tertiary and quaternary structures based on evolutionary information in computational methods in protein evolution. Humana Press, New York, 1851, 301–316.
Biasini, M., Schmidt, T., Bienert, S., Mariani, V., Studer, G., Haas, J., Johner, N., Schenk, A. D., Philippsen, A., & Schwede, T. (2013). Open Structure: An integrated software framework for computational structural biology. Acta Crystallographica D, 69(5), 701–709.
Daud, N. H., Leow, T. C., Oslan, S. N., & Salleh, A. B. (2019). A novel mini protein design of Haloalkane Dehalogenase. Molecular Biotechnology, 61(7), 477–488.
Kikani, B. A., & Singh, S. P. (2022). Amylases from thermophilic bacteria: Structure and function relationship. Critical Reviews in Biotechnology, 42(3), 325–341.
Thumar, J. T., & Singh, S. P. (2022). Antimicrobial potential and metabolite profiling of marine actinobacteria, ‘Actinobacteria-microbiology to synthetic biology.’ Springer, Singapore, 45, 241–264.
Scheidig, A. J., Horvath, D., & Szedlacsek, S. E. (2019). Crystal structure of a xylulose 5-phosphate phosphoketolase insights into the substrate specificity for xylulose 5-phosphate. Journal of Structural Biology, 10, 85–102.
Gurumallesh, P., Alagu, K., Ramakrishnan, B., & Muthusamy, S. (2019). A systematic reconsideration on proteases. International Journal of Biological Macromolecules, 128, 254–267.
Masui, A., Fujiwara, N., & Imanaka, T. (1994). Stabilization and rational design of serine protease AprM under highly alkaline and high-temperature conditions. Applied and Environment Microbiology, 60(10), 3579–3584.
Tian, W., Chen, C., Lei, X., Zhao, J., & Liang, J. (2018). CASTp 3.0: computed atlas of surface topography of proteins. Nucleic Acids Research, 46(W1), 363–367.
Ryan, D. J., Spraggins, J. M., & Caprioli, R. M. (2019). Protein identification strategies in MALDI imaging mass spectrometry: A brief review. Current Opinion in Chemical Biology, 48, 64–72.
Liao, J. Y. R., & van Wijk, K. J. (2019). Discovery of AAA+ protease substrates through trapping approaches. Trends in Biochemical Sciences, 44(6), 528–545.
Bhatt, H. B., & Singh, S. P. (2020). Cloning, expression and structural elucidation of a biotechnologically potential alkaline serine protease from a newly isolated Haloalkaliphilic Bacillus lehensis JO-26. Frontiers in Microbiology, 41, 1–16.
Gurunathan, R., Huang, B., & Ponnusamy, V. K. (2021). Jiang-Shiou Hwang & Hans-Uwe Dahms: Novel recombinant keratin degrading subtilisin like serine alkaline protease from Bacillus cereus isolated from marine hydrothermal vent crabs. Scientific Reports, 1, 1–12.
Ariyaei, A., Farhadi, A., Moradian, F., & Mianji, G. R. (2019). Cloning, expression and characterization of a novel alkaline serine protease gene from native Iranian Bacillus sp.; a producer of protease for use in livestock. Gene, 693, 10–15.
El-Sayed, A. K., Abou-Dobara, M. I., El-Fallal, A. A., & Omar, N. F. (2019). Heterologous expression, purification, immobilization and characterization of recombinant α-amylase AmyLa from Laceyella sp. DS3. International Journal of Biological Macromolecules, 132, 1274–1281.
Joshi, S., & Satyanarayana, T. (2013). Characteristics and applications of a recombinant alkaline serine protease from a novel bacterium Bacillus lehensis. Bioresource Resource Technology, 131, 76–85.
Devi, S. G., Fathima, A. A., Sanitha, M., Iyappan, S., Curtis, W. R., & Ramya, M. (2016). Expression and characterization of alkaline protease from the metagenomic library of tannery activated sludge. Journal of Bioscience and Bioengineering, 122(6), 694–700.
Purohit, M. K., Rathore, D. S., Koladiya, G., Pandey, S., & Singh, S. P. (2022). Comparative analysis of the catalysis and stability of the native, recombinant and metagenomic alkaline proteases in organic solvents. Environmental Science and Pollution Research, 7, 1–15.
Moser, M., Menz, G., Blaser, K., & Crameri, R. (1994). Recombinant expression and antigenic properties of a 32-kilodalton extracellular alkaline protease, representing a possible virulence factor from Aspergillus fumigatus. American Society for Microbiology, 62, 936–942.
Gohel, S. D., & Singh, S. P. (2018). Thermodynamics of a Ca2+ dependent, highly thermostable and detergent compatible purified alkaline serine protease from Nocardiopsis xinjiangensis strain OM-6. International Journal of Biological Macromolecules, 113, 565–574.
Nehal, F., Sahnoun, M., Dab, A., Sebaihia, M., Bejar, S., & Jaouadi, B. (2019). Production optimization, characterization, and covalent immobilization of a thermophilic Serratia rubidaea lipase isolated from an Algerian oil waste. Molecular Biology Reports, 46(3), 3167–3181.
Zhang, H., Liu, H., Lang, D. A., Xu, H., & Zhu, H. (2019). The application of a halotolerant metalloprotease from marine bacterium Vibrio sp LA-05 in liquid detergent formulations. International Biodeterioration & Biodegradation, 142, 18–25.
Shukla, R., & Singh, S. P. (2016). Structural and catalytic properties of immobilized α- amylase from Laceyella sacchari TSI-2. International Journal of Biological Macromolecules (IJBIOMAC), 85, 208–216.
Salwan, R., & Sharma, V. (2019). Trends in extracellular serine proteases of bacteria as detergent bioadditive: Alternate and environmental friendly tool for detergent industry. Archives of Microbiology, 201(7), 1–15.
Sharma, A. K., & Singh, S. P. (2016). Effect of amino acids on the repression of alkaline protease synthesis in haloalkaliphilic Nocardiopsis dassonvillei. Biotechnology Reports, 12, 40–51.
Jeong, Y. K., Yu, J., & Bae, S. (2019). Construction of non-canonical PAM-targeting adenosine base editors by restriction enzyme-free DNA cloning using CRISPR-Cas9. Science and Reports, 9(1), 4939.
Raiyani, N., & Singh S. P. (2023). Microbial community and predictive functionalities associated with the marine sediment of Coastal Gujarat. Environmental Science & Pollution Research. Published On-Line 18 January. https://doi.org/10.1007/s11356-023-25196-1
Dobariya A., Mankad G., Ramavat H., & Singh, S. P. (2023). Efficacy of the fruit and vegetable peels as substrates for the growth and production of α-amylases in marine actinobacteria. Applied Biochemistry and Biotechnology.
Acknowledgements
FJT and GK are grateful for the UGC-BSR Meritorious Fellowship and the financial assistance under the UGC-BSR Faculty Fellow Project, respectively. SPS acknowledges International Travel Fellowships from DST-SERB, CSIR, DBT, and UGC to present his work in Hamburg (Germany), Brisbane (Australia), Cape Town (South Africa), and Kyoto (Japan). Besides, SPS is grateful for the award of the UGC BSR Faculty Fellowship. Furthermore, we are grateful to Dr. Peter J. Rogers (Professor and now Adjunct Professor, School of Science, Griffith University, Brisbane, Australia, and Ex-Manager, New Technologies, Carlton United Breweries, Melbourne, Australia) for the language editing and suggestions to improve the manuscript.
Funding
The work was partly supported under the UGC-BSR Faculty Program (No.F.4–5(11) 2019 (BSR)). Facilities created under the DBT-Multi-Institutional Project (BT/PR8269/AAQ/03/307/2006), DST-FIST Program (No.SR/FST/LSI-033/2006), MoES Net Working Project (MoES/16/06/2013-RDEAS), and UGS-CAS Program (F.5–4.2012(SAP-II) of the Government of India were utilized.
Author information
Authors and Affiliations
Contributions
FJT has conducted the experiments, analyzed data, and prepared the first draft of the manuscript. GK was involved in the analysis and presentation of the data, integration of the new citations, and organization of the manuscript. SPS conceived the idea, designed the experiments, and supervised the entire work, besides contributing to the analysis and interpretation of the data and editing and finalizing the manuscript. All the authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics Approval
Not applicable.
Consent to Participate
All the authors agreed to participate in the preparation and submission of the manuscript.
Consent for Publication
All the authors agree to publish the data.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Highlights
• Heterologous expression of Protease gene from haloalkaliphilic Nocardiopsis.
• Characteristics of native, recombinant, and immobilized proteases.
• MALDI-TOF-MS peptide spectra matched with the peptidase/protease.
• 3D structure revealed insight into the active site by docking.
• Immobilization enhanced the stability and activity of recombinant protease.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Thakrar, F.J., Koladiya, G.A. & Singh, S.P. Heterologous Expression and Structural Elucidation of a Highly Thermostable Alkaline Serine Protease from Haloalkaliphilic Actinobacterium, Nocardiopsis sp. Mit-7. Appl Biochem Biotechnol 195, 7583–7602 (2023). https://doi.org/10.1007/s12010-023-04472-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-023-04472-3