
Abstract
One of the reasons hindering large-scale application of sophorolipids (SLs) is high production cost. In this study, six recombinant strains of Starmerella bombicola, sbEG1, sbEG2, sbCBH1, sbCBH1–2, sbBGL1, and sbCBH2 expressing cellulase genes eg1, eg2, cbh, cbh1–2, bgl1, and cbh2 from Penicillium oxalicum were respectively constructed. Four strains showed cellulase activities and were co-cultivated in fermentation media containing 2% glucose, 1% Regenerated Amorphous Cellulose (RAC), 2% glucose, and 1% RAC, respectively. After 7 days’ cultivation, concentration of SLs in medium with 1% RAC (g/L) reached 1.879 g/L. When 2% glucose and 1% of RAC were both contained, the titer of SLs increased by 39.5% than that of control strain and increased by 68.8% than that in the medium with only 2% glucose. Results demonstrated that cellulase genes from filamentous fungi in S. bombicola can function to degrade lignocellulosic cellulose to produce SLs.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Biosurfactants produced through microbial fermentation have attracted increasing attentions in recent years because of their excellent surface activity, good environment compatibility, biodegradability, and unique bioactivities [1]. As one of the most important biosurfactants, SLs has good biocompatibility and biodegradability and the highest fermentation production among all of biosurfactants [2]. SLs is an environmental friendly biosurfactant that can be used in food and daily chemicals, medical care, environment protection, microbial enhanced oil recovery, pesticides, and many other industrial and agricultural fields [3]. The obstacle to the large-scale applications of SLs lies in the higher production cost of SLs over chemically synthesized surfactants. High production of SLs can be achieved by adding both high concentration of hydrophilic (up to 80 g/L of glucose) and hydrophobic (such as 60 g/L rapeseed oil) carbon sources; thus, the cost of the carbon sources accounts for a large proportion of the total cost of SL production and is a major bottleneck to the development of economically viable large-scale SL production. In the last decades, researchers have made many attempts to reduce production cost of SLs, especially tried to find some cheaper alternatives of glucose and plant oils. So far, SLs has been reported to be produced by replacing the common hydrophilic carbon source (glucose) with deproteinized whey concentrate, sugar cane molasses, soybean molasses, glycerin, etc. [4,5,6]. In addition, industrial and restaurant waste oils such as biodiesel by-products, frying waste oils, industrial fatty acid residues, restaurant waste oils, and soybean black oil can be used to replace common hydrophobic carbon sources [7,8,9,10].
Lignocellulosic biomass is the most abundant renewable resource in the nature. Agricultural and forestry waste can reach 10 tons per year in China. Eighty percent of the total amount of lignocellulosic raw materials has not been well used. Lignocellulosic raw materials are mainly degraded by filamentous fungi to produce soluble sugars and then used for ethanol production; however, up to date, the reports about the production of SLs directly from lignocellulosic biomass by SL-producing yeast are still unavailable. More applications of lignocellulosic raw materials need to be explored. SL-producing yeasts are expected to be genetically engineered to utilize lignocellulosic biomass and the resulting hexose and pentose can be further used for SL synthesis.
The degradation of lignocellulosic biomass needs the synergy of the cellulase system including exoglucanases (CBH1 and CBH2), endoglucanases (EG) and β-glucosidases (BG), engineering and optimizing multiple heterologous functionalities in a single microbial strain have proven inherently challenging [11], in contrast to incorporate all required functionalities for the degradation of lignocellulosic biomass into a single strain of S. bombicola, microbial multicellular system in which individual strains with different cellulase function probably cooperate to survive and thrive together [12].
In the present study, a series of recombinant strains of S. bombicola in which several main cellulase genes from P. oxalicum were heterologously expressed were constructed. The work aimed to investigate whether different P. oxalicum cellulase genes could be respectively successfully expressed and co-culture of these recombinant S. bombicola strains with different heterologous cellulase genes can cooperate to thrive and produce SLs [13,14,15,16,17] from lignocellulosic biomass directly.
Materials and Methods
Strains, Plasmids, and Primers
Starmerlla bombicola was isolated from oily wastewater by our laboratory and was previously identified as Wickerhamiella domercqiae var. Sophorolipid by physiological and biochemical methods. After the genome of the strain was sequenced, the strain was identified as Starmerella bombicola [18]. The strain is now deposited in China General Microbiological Culture Collection Center, and the serial number is CGMCC1576.
Penicillium oxalicum 114-2 was screened and deposited by our laboratory.
E.coli DH5α is used as a plasmid cloning host strain and purchased from TransGenBiotech Beijing.
The plasmid backbone P15A was deposited in our laboratory.
The plasmid pRLMG containing hygromycin resistance marker was constructed and deposited in our laboratory.
The plasmid pPIC9K containing the signal peptide sequence α-Factor was constructed and deposited in our laboratory.
Primers used in this study are listed in Table 1.
Construction and Cultivation of Uracil Auxotroph Strain
Uracil auxotroph strain Δura of S. bombicola was constructed by homologous recombination and used as parent stain in later experiments. The back-up plasmids of uracil auxotroph strains were constructed using promoter gem-3424 and terminator Trapc. The cDNA of P. oxalicum was used as a template for PCR amplification to obtain the target gene eg1, eg2, cbh1, cbh2, cbh1–2, and bgl. The plasmid vector and the above six different cellulase genes were respectively linked together and the expression plasmids carrying different P. oxalicum cellulase genes were obtained, respectively. The above plasmids carrying different heterologous genes were respectively linearized with the restriction enzyme DraI and electrotransformed into Δura. The electrotransformation solution was spread on the FOA screening plate (5-fluoroorotic acid can combine with uracil to produce a toxic substance that kills the strain), and the uracil auxotroph positive transformant was obtained after 72 h of cultivation. The amplification was performed by using the genome of the transformant as template, the ura CDs region primers yzl-for and yzl-rev. If the ura CDs region fragment could not be amplified, it could be proved that the gene ura was successfully knocked out. Subsequently, the transcription level of gene ura was determined by RT-qPCR using actin as a reference gene and that of the starting strain was used as the control. Unless mentioned otherwise, cells were grown in YPD medium (20 g/L yeast extract, 10 g/L peptone, 20 g/L glucose) at 30 °C and 200 rpm.
Determination of Transcription Levels of the Heterologous Genes in Recombinant Strains by RT-qPCR
The transcription levels of the six genes (eg1, eg2, cbh1, cbh2, cbh1–2, and bgl) from P. oxalicum in S. bombicola were detected by RT-qPCR (SYBR Premix Ex TaqII, Takara).
Enzyme Activity Assay of the Recombinant Strains with P. oxalicum Cellulase Genes
After 3 days’ cultivation in YPD medium (50 mL in 300 flask), the fermentation broths of the six recombinant strains were applied for cellulase activity assay, respectively. The activities of endocellulase, exocellulase, and β-glucosidase of wild-type and recombinant strains were determined using carboxymethylcellulose sodium (CMC-Na), p-Nitrophenyl-cellobiose (pNPC), and p-Nitrophenyl-β-D-Glucopyranoside (pNPG) as the substrate, respectively. One unit of endocellulase, exocellulase, and β-glucosidase was respectively defined as the enzyme amount required for the release of 1 μmol of glucose by hydrolyzing 1.5 mL CMC-Na, 50 μL pNPC, and 50 μL pNPG, respectively.
Saccharification Experiments of Different Cellulose Substrates by Recombinant Strains
The recombinant strains sbEG1, sbEG2, sbCBH2, and sbBGL1 which showed higher enzyme activities were cultivated in YPD medium (50 mL in 300-mL flask) for 7 days and the enzyme activities of each day were detected. The fermentation broth of each recombinant strain was collected when reached the maximum enzyme activity and centrifuged at 10,000g for 10 min, and then the supernatant was taken for saccharification experiment. Saccharification system contained 25 mL substrate solution in 100-mL shake flasks, 0.5% Delignified Corncob Residue (DCCR), 1% DCCR, 0.5% RAC, and 1% RAC was used as substrate, respectively. The supernatant of the fermentation broth of the recombinant strains with different heterologous cellulase genes was mixed together in equal volume and applied to saccharification; the supernatant of the fermentation broth of the wild-type strain was served as the control. The saccharification was performed at 50 °C, 150 rpm for 7 days, and then the supernatant of the saccharification system was applied to determine the amount of reducing sugar to determine the optimal substrate.
Co-culture of Recombinant S. bombicola Strains with Different Cellulase Genes from P. oxalicum
Six recombinant strains with single cellulase gene from P. oxalicum were cultivated together and expected to synergistically degrade lignocellulosic biomass. The six recombinant strains with different cellulase genes were co-cultured at 30 °C, 200 rpm for 7 days in three fermentation media containing 2% glucose, 1% RAC, 2% glucose, and 1% RAC, respectively, and the wild-type strain of S. bombicola CGMCC1576 was used as the control and cultured under the same conditions as that of the recombinant strains.
Results and Discussion
Construction of Uracil Auxotrophic Strain Δura of S. bombicola
According to the principal of double-joint PCR, the hph expression box was fused with the upstream and downstream segments of ura gene to obtain the knockout box. The construction of knockout box is shown in Fig. S1.
The ura knockout box of 6000 bp was obtained by nested PCR and then was electrotransfered into S. bombicola. The colonies growing on FOA screening plate were picked up and the positive transformants without ura gene were verified by colony PCR by using the recombinant plasmid as the template and the primer pairs yzl-for and yzl-rev designed according to the CDs region sequence of ura (Fig. S2).
The transcriptional level of ura gene in uracil auxotrophic strain was further verified. The transcription of ura was significantly downregulated compared with that of the control strain, indicating the successful knockout of ura. And the selected uracil auxotrophic strain could not grow on SD plate without uracil but could grow on SD plate with uracil (Data not shown).
Construction of S. bombicola Recombinant Strains with Different Cellulase Genes from P. oxalicum
The six target genes, eg1, eg2, cbh1, cbh2, cbh1–2, and bgl1, were respectively amplified by PCR using high fidelity Pfu DNA polymerase, the cDNA of P. oxalicum 114-2 as the template. Plasmid P15A with chloramphenicol resistance gene was used as the expression vector for the expression of the heterologous cellulase genes from P. oxalicum plasmid (Fig. S3a).
The correct plasmid vector and the target genes were purified and digested by restriction enzymes SmaI and AvRII, and then the vector and the above six target cellulase genes were linked to obtain the P. oxalicum cellulase gene expression plasmid, respectively. The expression plasmids were verified by colony PCR and digested by single enzyme DraI and single enzyme by XbaI at three sites. The electrophoresis results of colony PCR are shown in Fig. S4, and size of each DNA fragment was consistent with its theoretical value.
The above recombinant plasmids were linearized by the restriction enzyme DraI and electrotransformed into Δura, respectively. The positive transformants were picked out and verified by colony PCR (Fig. S3b). Using the genome of the transformants as the template and the validation primers of each cellulase gene, six fragments were amplified and their sizes were consistent with their corresponding theoretical values as shown in Fig. S5, proving the successful insertion of each cellulase gene from P. oxalicum into S. bombicola genome. The six recombinant strains with eg1, eg2, cbh1, cbh2, cbh 1–2, and bg1, from P. oxalicum, were named as sbEG1, sbEG2, sbCBH1, sbCBH2, sbCBH1–2, and sbBGL1.
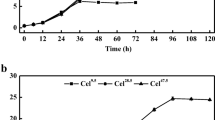
To know whether the transformed cellulase genes were expressed, the transcription level of each cellulase gene was detected. As shown in Fig. 1, the transcriptions of all P. oxalicum cellulase genes in the recombinant S. bombicola strain were all significantly upregulated compared with the control strain, which indicated the successful transcriptions of the six cellulase genes in S. bombicola recombinant strains.

Transcriptional analysis of 6 different cellulase genes respectively expressed in recombinant strains sbEG1, sbEG2, sbCBH1, sbCBH2, sbCBH1–2, and sbBGL1. Cells were grown in YPD medium at 30 °C and 200 rpm. a eg1. b eg2. c cbh1. d cbh2. e cbh1–2. f bgl1
Enzyme Activity Assay of Heterologous Cellulases in the S. bombicola Recombinant Strains
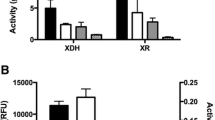
Enzyme activities of heterologously expressed cellulases in recombinant S. bombicola strains were assayed and the results are shown in Fig. 2. The recombinant strains with sbEG1 and sbEG2 showed endocellulase activities of EG1 and EG2, the recombinant strain sbCBH2 showed exocellulase activity of CBH2, the recombinant strains with bg1 genes exhibited β-glucosidase activity of BGL1, but, the activities of CBH1 and CBH1–2 in recombinant strains could not be detected. Noticeably, the wild-type strain S. bombicola exhibited weak endocellulase and β-glucosidase activities.

Cellulase activities in the recombinant strains sbEG1, sbEG2, sbCBH1, sbCBH2, sbCBH1–2, and sbBGL1. Cells were grown in YPD medium at 30 °C and 200 rpm. a Enzyme activity of EG1. b Enzyme activity of EG2. c Enzyme activity of CBH2. d Enzyme activity of BGL1
Saccharification Experiments by Cellulases in Recombinant Strains Using Different Cellulose Substrates
The recombinant strains sbEG1, sbEG2, sbCBH2, and sbBGL1 with the corresponding detectable cellulase activities were cultivated for 7 days and applied to enzyme activities assay (shown in Fig. 3). After 7 days’ cultivation, the crude enzyme solution of the above recombinant strains was used for saccharification experiment. Several cellulose materials, 0.5% DCCR, 1% DCCR, 0.5% RAC, and 1% RAC, were used as the substrates of saccharification. Since CBH1 activity in the recombinant strain sbCBH1 could not be detected, the RAC with damaged crystalized region was used as an alternative substrate. The culture broth of the wild strain was as the control, and the culture broth of the four recombinant strains sbEG1, sbEG2, sbCBH2, and sbBGL1 which exhibited their corresponding cellulase activities of EG1, EG2, CBH2, and BGL1 was added in equal volume to saccharification system. The saccharification solution after saccharification of 168 h was centrifuged at 10,000 rpm for 10 min and the supernatant was collected. The amount of reducing sugar in the supernatant was determined to find the optimal substrate. As shown in Fig. 4, DCCR could not be saccharified to produce reducing sugar by the crude enzyme solution of the four recombinant cellulases, while RAC could be used as the substrate of the four heterologously expressed cellulases and a certain amount of reducing sugar was released, and the produced reducing sugar from 1% RAC was higher than that from 0.5% RAC.

a Enzyme activity curve of recombinant strain sbEG1. b Enzyme activity curve of recombinant strain sbEG2. c Enzyme activity curve of recombinant strain sbCBH2. d Enzyme activity curve of recombinant strain with sbBGL1. The culture for the four recombinant strains lasted for 7 days

a–d Saccharification of different cellulose substrates by heterologously expressed cellulases in S. bombicola
Co-culture of Four Recombinant S. bombicola Strains with Different Cellulase Genes from P. oxalicum
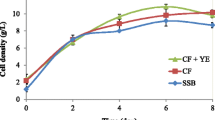
According to the results of saccharification, 1% RAC was the optimum substrate for the heterologously expressed cellulase system. Four recombinant S. bombicola strains sbEG1, sbEG2, sbCBH2, and sbBGL1 were co-cultured in three different fermentation media containing 2% glucose, 1% RAC, 2% glucose, and 1% RAC, respectively. The wild-type S. bombicola strain without heterologous genes was used as the control. The anthrone method was used to determine the content of SLs. The experimental procedure was as described by Shen [19]. The results are shown in Fig. 5. When only 2% glucose was used as the substrate, the titer of SLs produced by co-culture of the four recombinant strains was low and showed no differences with that by the control strain. The residual glucose in fermentation broth after 3 days’ cultivation was measured and glucose was found to be exhausted, which indicated that 2% glucose was mainly used for growth of these recombinant strains, and the titer of SLs by co-culture of 4 recombinant strains was almost the same as that by the control strain. When only 1% RAC was used as substrate, the concentration of SLs reached 1.878 g/L, which was 2.6 times higher than that of the control strain. When 2% glucose and 1% of RAC were both contained in fermentation medium, the titer of SLs was increased by 39.5% compared with that of the control strain and increased by 68.8% than that in the fermentation medium with only 2% glucose. The concentration of SLs by the co-culture of four recombinant strains sbEG1, sbEG2, sbCBH2, and sbBGL1 in medium with 1% RAC was higher than that in the medium with 2% glucose, this was probably attributed to reducing sugar release by the continuous degradation of RAC, while only 2% glucose was used as substrate, glucose was exhausted on the third day of fermentation.

SL production by co-culture of four different recombinant strains with different cellulase genes from P. oxalicum using RAC, glucose, and glucose+RAC as substrates. Cells were grown in medium at 30 °C and 200 rpm
Conclusions
In the study, uracil auxotroph strain was successfully obtained, and S. bombicola recombinant strains expressing heterologous cellulase single enzyme genes eg1, eg2, cbh2, and bgl1 were constructed respectively. Through saccharification test of different cellulose substrates, the optimum substrate was determined to be 1% RAC. And by co-culturing recombinant strains, the idea of directly using lignocellulosic biomass to produce SLs was successfully achieved.
Ma demonstrated for the first time the feasibility of producing SLs from the hydrolysate of DCCR [20]. Samad et al. used sweet sorghum residue hydrolysate and soybean oil as substrates to obtain 84.6 g/L of SLs [21]. Konishi et al. improved the process of synthesizing sophorolipids from hydrolysate of corncobs, reduced the amount of acid, and obtained 43.8 g/L SLs [22]. Researches above were all use enzyme-treated cellulose saccharification solution as the substrates to produce SLs. In this study, the main cellulase genes were expressed in S. bombicola and enable the recombinant strain to direct use lignocellulosic cellulose to produce SLs, which is a useful exploration to expand the substrate spectrum of SL production. The exoglucanases CBH1 could be further expressed as active enzyme in S. bombicola; it will be very likely to utilize natural lignocellulose resources to produce SLs. Moreover, by co-cultivation, more genetically modified strains can be cultivated together to increase SL production, promoting further commercialization of SLs in biosurfactant market.
Data Availability
All data generated or analyzed during this study are included in this published article (and its supplementary information files).
References
Jing, C., Xin, S., Yinbo, Q., & Aiqin, L. (2006). Conditions for biosurfactant fermentation by yeast strain Y2A. Journal of Applied and environmental Biology, 12(1), 122–124.
Jing, C., Yunrui, Z., & Xin, S. (2007). Research progress in the production and application of sophorolipids. Food Science, 28(08), 525.
Jiashan, L. (2016). Studies of the genes responsible for the synthesis and metabolism of sophorolipids in Starmerella bombicola. Doctoral Thesis of Shandong University.
Ashby, R. D., Nunez, A., Solaiman, D. K. Y., & Foglia, T. A. (2005). Sophorolipid biosynthesis from a biodiesel co-product stream. Journal of American Oil Chemistry Society, 82(9), 625–630.
Daniel, H. J., Otto, R. T., Reuss, M., & Syldatk, C. (1998). Sophorolipid production with high yields on whey concentrate and rapeseed oil without consumption of lactose. Biotechnology Letters, 20(8), 805–880.
Daverey, A., & Pakshirajan, K. (2009). Production of sophorolipids by the yeast Candida bombicola using simple and low cost fermentative media. Food Research, 42(4), 499–504.
Chen, Y., Wu, Y., Zhu, B., Zhang, G., & Weil, N. (2018). Co-fermentation of cellobiose and xylose by mixed culture of recombinant Saccharomyces cerevisiae and kinetic modeling. PLoS One, 13(6), e0199104.
Andreal, I. S., Florentino, A. P., Semerel, J., Strepis, N., Sousa, D. Z., & Stams, A. J. M. (2018). Co-culture of a novel fermentative bacterium, Lucifera butyrica gen. nov. sp. Nov., with the sulfur reducer Desulfurella amilsii for enhanced sulfidogenesis. Original research, 10, 3389.
Wang, Z., Dien, B. S., Rausch, K. D., Tumbleson, M. E., & Singh, V. (2019). Improving ethanol yields with deacetylated and two-stage pretreated corn stover and sugarcane bagasse by blending commercial xylose-fermenting and wild type Saccharomyces yeast. Bioresource Technology, 282, 103–109.
Liang, W., York, S. W., Ingram, L. O., & Shanmugam, K. T. (2019). Simultaneous fermantation of biomass-derived sugars to enthanol by a coculture of an engineered Escherichia coli and Saccharomyces cerevisiae. Bioresource Technology, 273, 269–276.
Olson, D. G., McBride, J. E., Shaw, A. J., & Lynd, L. R. (2012). Recent progress in consolidated bioprocessing. Current Opinion in Biotechnology, 23(3), 396–405.
Mee, M. T., & Wang, H. H. (2012). Engineering ecosystems and synthetic ecologies. Molecular BioSystems, 8(10), 2470–2483.
Li, H.-T., Zhou, H., Duan, R.-T., Li, H.-Y., Tang, L.-H., Yang, X. Q., Yang, Y.-B., & Ding, Z.-T. (2018). Inducing secondary metabolite production by co-culture of the endophytic fungus Phoma sp. and the symbiotic fungus Armillaria sp. Journal of Natural Products, 10, 1021.
Felse, P. A., Shah, V., Chan, J., Rao, K. J., & Gross, R. A. (2007). Sophorolipid biosynthesis by Candida bombicola from industrial fatty acid residues. Enzyme and microbialtechnology, 40(2), 316–323.
Kim, Y. B., Yun, H. S., & Kim, E. K. (2009). Enhanced sophorolipid production by feeding-rate controlled fed-batch culture. Bioresource Technology, 100(23), 6028–6032.
Shah, V., Jurjevic, M., & Badia, D. (2007). Utilization of restaurant waste oil as a precursor for sophorolipid production. Biotechnology Progress, 23(2), 512–515.
Shao, L., Song, X., Ma, X. J., Li, H., & Qu, Y. B. (2012). Bioactivities of sophorolipid with different structures against human [J]. The Journal of Surgical Research, 173(2), 286–291.
Spencer, J. F. T., Gorin, P. A., & Tulloch, A.p. (1970). Tour lops is bombicola sp.n. Antoine Van Leeuwenhoek., 36(1), 129–133.
Shen, J. (2012). Sophorolipid biosynthesis by Wickerhamiella domericqiae from medium-length alkanes. Master Thesis of Shandong University.
Ma, X. (2012). Research on the regulation of nitrogen source metabolism of sophorolipid synthesis and the production and properties of cheap substrates of sophorolipid [D]. Doctoral Thesis of Shandong University.
Samad, A., Zhang, J., & Cheng, D. (2014). Sophorolipid production from biomass hydrolysates [J]. Applied Biochemistry and Biotechnology, 175(4), 2246–2257.
Konishi, M., Yoshida, Y., & Horiuchi, J. I. (2015). Efficient production of sophorolipid by Starmerella bombicola using a corncab hydrolysate medium[J]. Biosci Bioeng, 119(3), 317–322.
Funding
This study was funded by the National Natural Science Foundation of China (No. 31971387) and Major Program of Natural Science Foundation of Shandong Province (No. ZR2019ZD19).
Author information
Authors and Affiliations
Contributions
Yue Li, Na Gao, Xinyu Zhang, and Guoqin Zhao contributed equally to this manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Ethical Statement
This article does not contain any studies with human participants or animals performed by any of the authors. The principles of ethical and professional conduct have been followed by all the authors in this study.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
ESM 1
(DOCX 384 kb)
Rights and permissions
About this article

Cite this article
Li, Y., Gao, N., Zhang, X. et al. Sophorolipid Production Using Lignocellulosic Biomass by Co-culture of Several Recombinant Strains of Starmerella bombicola with Different Heterologous Cellulase Genes from Penicillum oxalicum. Appl Biochem Biotechnol 193, 377–388 (2021). https://doi.org/10.1007/s12010-020-03433-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-020-03433-4