
Abstract
5-Aminolevulinic acid (ALA) is an important metabolic intermediate compound with high value and has recently been used in agriculture and medicine. In this study, we have constructed six recombinant Escherichia coli (E. coli) strains that are involved in pET system under the regulation of the T7 promoter and LacI to express codon-optimized hemA gene from Rhodobacter capsulatus (RchemA) for ALA production via the C4 pathway. Due to codon optimization, hemA has a high transcriptional level; however, most RcHemA proteins were formed as inclusion body. To improve expression in soluble form, the vector with TrxA fusion tag was successfully used and co-expressed with partner GroELS as chaperone in another vector. As a result, ALA production increased significantly from 1.21 to 3.67 g/L. In addition, optimal ALA production was developed through adjustment of induction time and isopropyl β-D-1-thiogalactopyranoside (IPTG) concentration, as well as substrate addition conditions. By adopting a two-stage induction strategy, the highest ALA reached 5.66 g/L when 0.1 mM of IPTG was added at early exponential phase (i.e., OD600 was equal to 0.7 to 0.8), while 6 g/L of glycine, 2 g/L of succinate, and 0.03 mM of pyridoxal 5′-phosphate (PLP) were provided in the mid-exponential phase in fermentation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
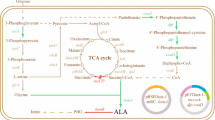
5-Aminolevulinic acid (ALA) is an endogenous non-protein amino acid that is an important intermediate in the biosynthesis pathway of tetrapyrrole compounds, such as purpurin, blood matrix, chlorophyll, and vitamin B12. Tetrapyrrole compounds provide the essential cofactors for redox reaction, growth of photoautotrophic organisms, and several enzymatic reactions, which makes ALA a key metabolic intermediate in microorganisms [1, 2]. ALA is a hydrophilic molecule that does not easily penetrate into the cell membrane and periplasm, thus needs to be derivatized to increase the lipophilicity of the application [3]. In recent years, a lot of endeavors have paid on ALA-based photodynamic treatment, which are widely used in diagnosis and therapy of cancer diseases, and also work as biodegradable and non-toxic herbicides in agricultures [4]. The current production of ALA relies on chemical methods; however, this method involves several complicated steps, resulting in low yields and high costs. Therefore, biosynthesis by microbial production of ALA has been considered to be a simple, cheaper, and more sustainable approach [1, 5, 6].
Two different natural pathways for ALA biosynthesis have been reported, namely, C4 and C5 pathways. The C4 pathway is a one-step synthesis that relies on 5-aminolevulinic acid synthase (ALAS) to condense succinyl-CoA and glycine in animals, fungi, and some α-proteobacteria (i.e., Rhodobacter species) [7, 8]. The active site of ALAS utilizes three key amino acid side chains: Arg-85, Thr-430, and Lys-313, and also involved in pyridoxal 5′-phosphate (PLP) as a cofactor. The PLP cofactor binds to the lysine side chain prior reaction to form a Schiff base and then reacts with glycine. ALAS acts as a homodimer in the reaction and converts glycine and succinyl-CoA into coenzyme A, carbon dioxide, and ALA [9, 10]. The C5 pathway is an upstream metabolism of the heme synthesis pathway found in plants, algae and bacteria. ALA in C5 pathway is catalyzed by three enzymes: glutamyl-tRNA synthetase, glutamyl-tRNA reductase, and glutamate-1-semialdehyde aminotransferase. Glutamyl-tRNA synthetase is encoded by gltX and converts glutamate to glutamyl adenylate; then, glutamine-tRNA is formed. The glutamine-tRNA is reduced by hemA encoding glutamyl-tRNA reductase to form the active intermediate glutamate-1-semialdehyde (GSA), which is unstable and quickly converted. The final reaction is a rapid conversion of GSA to ALA by the key enzyme glutamate-1-semialdehyde aminotransferase which is encoded by hemL [11, 12]. However, both pathways contain a 5-aminolevulinic acid dehydratase (ALAD) encoded by hemB in the downstream of ALA. Therefore, ALA will be converted to porphobilinogen (PBG) by ALAD, which would be inhibited by glucose. Although inhibition of ALAD increases ALA production, the resulting decrease in cell growth will result in a significant decrease in total yield. Besides, previous studies have found that hemB is an essential gene for microorganisms that cannot be knocked out, so it is more effective to reduce ALAD activity by adding glucose or competitive inhibitors [13,14,15].
Since the C4 pathway is much simpler than the C5 pathway, it is more promising to obtain higher ALA production through the C4 pathway. The expression of a key enzyme, i.e., ALAS, by recombinant E. coli has attracted many appeals due to its high accessibility for regulation and operation. However, most of the recombinant proteins produced in E. coli are insoluble aggregates (i.e., inclusion bodies), and are considered to be non-functional. Therefore, the participation of chaperon or fusion proteins is essential for the production of soluble protein [16]. Functional analysis of the cylindrical chaperone protein complex provides for the development of cellular protein folding as a chaperone-assisted process, due to the mechanism of cellular evolution and the prevention of undue interactions between non-natural polypeptides, thereby primarily aiding protein folding [17]. The function of GroEL and its cofactor GroES provides a nanocage for the folding of individual protein that can occur independently and prevent aggregation without damage. In general, co-expression of molecular chaperone could minimize aggregation and increase proteins solubility.
In a recent study, the highest ALA production was obtained by expressing ALAS from Rhodobacter capsulatus under optimizing fermentation in the recombinant E. coli Rosetta (DE3), which is including a pRARE vector for rare codon transcription [18]. It implies the contribution of codon-optimized ALAS. However, higher transcriptional rate result in insoluble ALAS in E. coli. Thus, strategies to increase the expression of ALAS from R. capsulatus by chaperones will be explored and enhanced in this study. Since the T7 system is commonly for protein overexpression in E. coli, pET21a(+) and pET32a(+) are used to express ALAS, while the partner GroELS is present in a compatible vector (i.e., pT7). Finally, the operating condition of inducer and substrate are further optimized to achieve the highest yield of ALA.
Materials and Methods
Materials
The codon-optimized RchemA DNA fragment was designed by online software and synthesized by Integrated DNA Technologies (IDT, USA). The Ex-taq DNA polymerase was purchased from the Clontech Laboratories (Clontech USA) while plasmid DNA extraction and DNA clean up kits were purchased from FAVORGEN Biotech Corp. The Taq DNA polymerase was obtained from the GeneDireX. All the restriction enzyme was purchased from New England Biolabs (NEB, UK). Rapid T4 DNA ligase was ordered from the Leadgene Biomedical, Inc. Chemicals of ALA and acetylacetone were purchased from the Sigma, while 4-dimethylaminobenaldehyde (DMAB) and perchloric acid were ordered from ACROS Organics™ and SHOWA, respectively. Other chemicals are used at the analytical grade.
Cloning and Transformation of RchemA to Different Vectors and Host
All of the strains, plasmids, and primers used in this study were shown in Table 1. RchemA fragment was amplified with primers of EcoRI-RchemA-F and HindIII-RchemA-R. pET21a(+), pET32a(+), and PCR product of RchemA fragment were digested with EcoRI and HindIII. Ligation was performed under 22 °C for an hour with fast T4 DNA ligase. After heat-shock transformation of constructed plasmid to DH5α, the inserted fragment was confirmed by colony PCR and enzyme digestion. The successful-constructed plasmids were extracted from DH5α and transformed to BL21(DE3) to obtain strains 21-RC or 32-RC, while co-transform plasmid of pT7-groELS for strains as 21-GRC and 32-GRC, respectively.
Culture Conditions
Luria Bertani (LB) medium and modified minimal medium (MM9) were used in preculture and production of 5-ALA, respectively. The components of LB included 10 g/L tryptone, 5 g/L yeast extract, and 10 g/L sodium chloride, while MM9 contained 16 g/L (NH4)2SO4, 16 g/L Na2HPO4•12H2O, 3 g/L KH2PO4, 2 g/L yeast extract, 1 g/L MgSO4•7H2O, 0.01 g/L MnSO4•7H2O, and 20 g/L glucose. All genetic strains were preculture in LB medium with antibiotic of 100 μg/mL ampicillin (Amp) or 50 μg/mL ampicillin and 12.5 μg/mL chloramphenicol (Cm) for single- and duTo test substrate-fed effect, extraal-plasmid strain respectively in 15 mL tubes at 37 °C and 200 rpm for 12 h. The culture was inoculated with 3% (v/v) seed culture in 30 mL MM9 medium at 37 °C. After cultivating for 4, 6, 8, or 10 h, IPTG was added with final concentration of 0.1 mM for induction. After induction, further cultivation was progressed at 30 °C. Final concentration of 3 g/L glycine, 1 g/L succinate, and 30 μM pyridoxal phosphate (PLP) were added as substrate and cofactor for 5-ALA production when OD600 reached to 0.8, 1.0, or 1.7. Lastly, cells were harvested at 24 h. For substrate feeding effect, an extra 3 g/L glycine and 1 g/L succinate were supplemented when OD600 reached to 1.7, and the cells were harvested at 24 h and 36 h, respectively.
SDS-PAGE Analysis of Protein Expression
The cells were harvested by centrifuging at 10,000×g for 10 min and then washed with deionized water. The cell density was adjusted to an OD600 of 3 as whole cell (WC) and further disrupted with a pressure of 30 kpsi for one cycle (Constant system, One-Shot, UK). The soluble cytoplasmic protein (S) was then collected by centrifuged at 12,000×g for 10 min. Finally, WC and S were analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) with 10% separating gel and 4% stacking gel. Proteins were visualized by staining with Coomassie blue R-250 and scanned with an image scanner.
Bio-Rad Bradford Protein Assay
Dye Reagent Concentrate (Bio-Rad) was diluted to one-fifth with deionized water and allowed to stand at room temperature for no more than 2 weeks. Pipette 10 μL of each sample into the wells of microplate, then add 200 μL diluted dye reagent to each well and mix well. Incubate the microplate for 5 min at room temperature. Afterwards, absorbance at 595 nm was measured with spectrophotometer (SpectraMax 340, Molecular Devices, USA).
Analysis of ALA by Ehrlich’s Reagent Assay
A 0.5 mL sample or standard solution was well mixed with 0.5 mL of 1 M acetate buffer (pH 4.6) and 0.1 mL of acetylacetone. Subsequently, the mixture was heated at 100 °C for 10 min. After cooling to room temperature, 0.1 mL of the reaction solution was mixed with 0.1 mL of freshly prepared modified Ehrlich’s reagent [7] in 96-well plates under minimal light. Finally, absorbance at 553 nm was measured after 10-min reaction with a spectrophotometer (SpectraMax 340, Molecular Devices, USA).
Quantitative PCR Analysis for Evaluation of Plasmid Copy Number
Different strains were cultured in 15 mL tube as described above and harvested after 12 h. The cells were collected by centrifugation at 10,000×g for 10 min and washed twice with deionized water. Afterwards, the cell density was adjusted to an OD600 of 3, followed by incubation at 100 °C for 10 mins. After centrifugation, the supernatant was used as template DNA. Quantitative PCR analysis (qPCR) was performed using the EvaGreen qPCR System-ROX I (GeneDireX) and StepOnePlus™ Real-Time PCR System (Applied Biosystems, Foster City, Calif). The Amp and Cm resistance genes were selected as the target genes for pET and pT7-groELS to evaluate the plasmid copy number (PCN), and the T7 RNA polymerase gene was selected as the reference gene for one copy number in BL21(DE3). Primers used to amplify partial fragment were also listed in Table 1. The PCN was calculated by formula as 2-ΔΔCT.
Results and Discussion
Gene Screening and Codon Optimization
As shown in Fig. 1a, the phylogenetic tree indicates that R. capsulatus is located in a different branch from R. sphaeroides. ALAS from R. capsulatus is more closely related to Rhodobacter viridis (95% of identity), Thioclava sp., and Paracoccus sp. than to R. sphaeroides (74.6% of identity), but far from the Ochrobacterium sp., Serratia plymuthica, Saccharomyces cerevisiae, and Rattus norvegicus which the similarity and identity are below 50% (Fig. 1a). The ranking of ALAS activity in the BRENDA database shows that purified R. sphaeroides accounts 2.2 U/min/mg > Rattus norvegicus (i.e., 1.22 U/min/mg) > Saccharomyces cerevisiae (0.65 U/min/mg) > crude R. sphaeroides (i.e., 0.375~0.558 U/min/mg) > Serratia plymuthica (i.e., 0.35 U/min/mg) [19,20,21,22,23]. From this point of view, ALAS activity is not related to phylogenetic tree. Most of the previous studies focused on ALAS encoded by hemA genes from R. sphaeroides (RS-ALAS) [19,20,21]. However, higher specific activity of ALAS was found in R. capsulatus, and the current ALA production in recombinant E. coli has reached 8.8 g/L [18]. Over two decades, it has been reported that R. capsulatus ALAS is the first step in the biosynthesis of heme [24], and has recently completed its crystal structure [25]. Therefore, we selected R. capsulatus ALAS encoded by RchemA and synthesized with codon optimization for E. coli for the first time. As shown in Fig. 1b, the identity between the native sequence and codon-optimized sequence was 76.6%. In fact, an effective DNA synthesis method provided by IDT company is the gBlocks® gene fragments coupled with codon optimization tools. Following the design principle, the GC content of the entire sequence should be less than 60%, and the content of repeat sequence less than 40% of the entire sequence. GC content of the 40 base pair domain region should be less than 90%. Afterwards, RchemA was cloned into different pET vectors and tested for expression of ALA.

a Phylogenetic tree analysis of 5-aminolevellunic acid synthase (ALAS) from different species with the neighbor-joining methods constructed with the MEGA 6.0 software. GenBank accession numbers for all species are given in parenthesis. The numbers at the branch nodes are bootstrap values based on 1000 resamplings. Scale bar indicates amino acid divergence as 20%. b DNA sequence of ALAS between Rhodobacter capsulatus SB1003 and codon optimized for Escherichia coli. The identity of both sequences is 76.6%
Effect of TrxA and Chaperone GroELS for ALA Production
As shown in Table 1, four strains of E. coli BL21(DE3) harboring pET21a(+)-RchemA or pET32a(+)-RchemA and with or without pT7-GroELS plasmid were labeled as 21-RC, 21-GRC, 32-RC, and 32-GRC, and were used to compare the plasmid copy number (PCN), protein expression by SDS-PAGE analysis, Bio-rad assay, biomass, and ALA. The results were illustrated in Fig. 2. Actually, the pBR322 replication origin of pET21a(+) and pET32a(+) should be within 20 to 40 of PCN (Fig. 2a). On the other hand, the replication origin of pT7-GroELS was p15A which PCN were reported to be 5 to 10. Interestingly, PCNs of both vectors were up-regulating in 32-GRC strain. Thus, the PCN of pET32a(+)-RchemA and pT7-GroELS increased to 49 and 38, respectively. Due to the gene was codon optimized, the protein expressions observed by SDS-PAGE were greatly improved. However, most RcHemA (indicated by WC) was inclusion body in 21-RC and 32-RC (Fig. 2b). Therefore, the key point was translation, not transcription. Thereby, how to express the soluble protein would be very important. By co-expressing GroELS in 21-GRC and 32-GRC, the soluble form of RcHemA or TrxA-RcHemA (represented by S) was increased (Fig. 2b). Additionally, when TrxA was fused on the upstream of RcHemA, the partner GroELS was very helpful in protein folding, suggesting that 32-GRC is the best strain for obtaining more functional proteins. The total protein amount is shown in Fig. 2c and biomass and ALA production are presented in Fig. 2d. With co-expression of GroELS, the total amount of soluble proteins was up to 800 mg/L for 21-GRC and 600 mg/L for 32-GRC, respectively. The biomass was affected by ALA accumulation which triggered an acidic environment caused by the reactive oxygen in cell [26]; therefore, the higher ALA production in 32-GRC and lower biomass was observed (i.e., OD600 was 2.2). The highest ALA production in 32-GRC reached to 3.67 g/L, owing to GroEL and GroES assisting folding and obtaining more soluble proteins. The group I chaperone, GroEL of E. coli is a cylindrical complex of two heptameric loops. GroES is a dome-shaped heptameric loop that binds to the ends of the GroEL cylinder, and then forms cages for protein folding [27]. The GroEL–GroES cage has enough space to accommodate up to 60 kDa in size. The hydrophobic binding site of the protein is buried in the GroEL–GroES complex, and the inner wall of the cage is hydrophilic in nature, providing a free environment for folding [28]. pET32a contains trxA gene, which may form disulfide bond and can be regarded as a redox switch [29], thus avoiding the formation of inclusion body and increase of soluble protein expression. Similarly, the groEL and groES genes help to isolate the substrate and protein into a native state in the cage compartment [30]. We also attempted to co-express DnaKJ in different strains and determine the effect on RcHemA expression and ALA production. As shown in Fig. S1, the soluble form of RcHemA or TrxA-RcHemA is similar as to the strain without DnaKJ. The recombinant proteins remained at the same level (Fig. S1b) and final ALA production was 1.85 g/L in 21-DRC and 0.18 g/L in 32-DRC (Fig. S1c). When DnaKJ is involved in the pET32a(+) vector, the critical low yield of ALA illustrates the different mechanism of GroELS and DnaKJ. The supposed mechanism of DnaKJ that accelerates ATP hydrolysis, and will not be beneficial for the enhancement of the amount of soluble ALAS, while GroELS coupled with TrxA is directly affecting the protein folding and increased the soluble ALAS.

a Examine plasmid copy number of different strains. Amp stands for the ampicillin in pET21a and pET32a and Cm stands for the chloramphenicol in pT-groELS. b SDS-PAGE of BL21(DE3) harboring of different plasmids which including pET21a-RchemA, pET21a-RchemA+groELS, pET32a-RchemA or pET32a-RchemA and pT-groELS. Comparison of different strains which with or without chaperone TrxA and GroELS to produce. c Soluble ALAS and d cell growth (●), extracellular 5-ALA (■) and intracellular 5-ALA ( )
)
Optimal Concentration of IPTG and Induction Time
Since recombinant protein levels are affected by IPTG induction, expression levels are determined by SDS-PAGE as showed in Fig. 3a. Higher concentrations of IPTG induced more TrxA-RcHemA protein, but the optimal conditions for producing soluble forms of GroELS and TrxA-RcHemA occurred at 0.1 mM IPTG (Fig. 3a). As seen in Fig. 3a, both GroEL and TrxA-RcHemA are at lower expression levels due to the fact that the LacI repressor in the pET system dominates the transcriptional level of T7RNA polymerase. On the other hand, 1 mM IPTG was toxic to cell growth consistent with previous results [31], so lower biomass was observed (Fig. 3b). Intracellular ALA is much less than extracellular ALA. Among different concentrations of IPTG, the highest ALA production and biomass at 0.1 mM IPTG were 3.32 g/L and OD600 of 3.25, respectively. In addition to the IPTG concentration, the addition time is also critical in the pET system. The effect of induction time on RcHemA expression was determined by SDS-PAGE in 32-GRC is shown in Fig. 4a and b. Whether 24 h (Fig. 4a) or 36 h (Fig. 4b), subsequent induction at 8 h and 10 h resulted in a decrease in recombinant protein expression, and further caused lower ALA production. As a result, the most appropriate induction time was after 4 h of culture (i.e., OD600 at 0.7 to 0.9), which was at the early logarithmic growth phase. For biomass and ALA production in extracellular and intracellular, the sampling times at 24 h and 36 h are shown in Fig. 4c, where they are labeled 24-4 or 36-4, 24-6 or 36-6, 24-8 or 36-8, and 24-10 or 36-10 indicate the results of 24 h or 36 h with induction at 4 h, 6 h, 8 h, and 10 h, respectively. The highest ALA achieved 2.8 g/L at 24 h of cultivation and induction at 4 h, while obtained 3.0 g/L at 36 h of cultivation and induction at 4 h, respectively. Interestingly, biomass and ALA production are produced in the opposite direction, and the ALA production index is representing the toxicity to cell growth. Actually, the excess reactive oxygen species (ROS) will cause oxidative damage to proteins and DNA in the cell, and further impede high-level of ALA production. Therefore, the strategy by engineering the antioxidant defense system as a higher tolerance of ROS and enhancing the ALA production in E. coli has been reported recently [26].

Effect of IPTG concentration on (a) RchemA expression determined by SDS-PAGE, and (b) cell growth (●), extracellular 5-ALA (■), and intracellular 5-ALA ( ) of pET32a-RchemA and pT-groELS in BL21(DE3) after 24 h of cultivation
) of pET32a-RchemA and pT-groELS in BL21(DE3) after 24 h of cultivation

Effect of induction time on RchemA expression determined by SDS-PAGE in pET32a-RchemA and pT-groELS in BL21(DE3) after a 24 h and b 36 h of cultivation. c Effect of induction time on cell growth ( ), extracellular 5-ALA (■), and intracellular 5-ALA (□) of pET32a-RchemA and pT-groELS in BL21(DE3) at cultivation at 24 h and 36 h, where 24-4 or 36-4, 24-6 or 36-6, 24-8 or 36-8 and 24-10 or 36-10 representing results of 24 h or 36 h by induction at 4 h, 6 h, 8 h, and 10 h
), extracellular 5-ALA (■), and intracellular 5-ALA (□) of pET32a-RchemA and pT-groELS in BL21(DE3) at cultivation at 24 h and 36 h, where 24-4 or 36-4, 24-6 or 36-6, 24-8 or 36-8 and 24-10 or 36-10 representing results of 24 h or 36 h by induction at 4 h, 6 h, 8 h, and 10 h
Optimal Concentration and Additional Time of Substrates
Because ALA may cause toxicity to the cell and glycine and succinate are the two substrates in the C4 pathway, effects of the substrates additional time on cell growth and ALA production were critical. As shown in Fig. 5, cell growth (a) and ALA production (b) were enhanced when substrates were added between OD600 of 1.0 and 1.7 rather than at OD600 of 0.7 to 0.9. According to this result, it was proposed that the rate of the ALA synthesis was fast when transformed succinyl-CoA to ALA with glycine and PLP. The theoretical yield of ALA from 3 g/L glycine and 1 g/L succinate should be 5.24 g/L, so it was over 80% conversion under the strategy of postponed substrates additional time. Therefore, in order to increase the driving force, more glycine and succinate substrates were designed and tested via using batch or fed-mode supply. As shown in Fig. 6, the substrates provided with 3 g/L succinate and 1 g/L glycine were insufficient, and increasing the substrates concentration to 6 g/L of glycine and 2 g/L of succinate enhanced the ALA production up to 5.66 g/L. However, the fed-mode of glycine and succinate had no effect on cell growth and ALA accumulation. Previous studies used a strategy of continuously replenishing substrates during fermentation to maintain a substrate concentration within a certain range, and the ALA production eventually reached 11.5 g/L [26]. However, feeding in flask is still a problem because cell growth is restricted without controlling dissolved oxygen and pH. In summary, increasing the biomass by reducing the pressure on cells from the ALA production does not increase the ALA production. Under flask condition, ALA production increased to 5.66 g/L with delayed addition of more substrates, which is an effective strategy.

Fig. 5 Effect of substrates additional time on 5-aminolevulinic acid and cell growth of pET32a-RchemA and pT-groELS in BL21(DE3) after 12 h of cultivation with 3 g/L glycine, 1 g/L succinate, 30 µM PLP. a Time course, and b Biomass ( ), 5-aminolevulinic acid in extracellular (■) and intracellular (□), respectively
), 5-aminolevulinic acid in extracellular (■) and intracellular (□), respectively

Effect of substrates concentration and additive mode on biomass ( ), 5-aminolevulinic acid in extracellular (■) and intracellular (□) of pET32a-RchemA and pT-groELS/BL21(DE3) after 24 and 36 h at optimized condition of cultivation. G means glycine, S means succinate. 3G1S represents 3 g/L and 1 g/L for G and S, 6G2S represents 6 g/L and 2 g/L for G and S. Both additions are after 8 h of cultivation. 6G2S(fed) means 3 g/L and 1 g/L for G and S, which addition at 8 h and feed the same substrate after 24 h of cultivation separately
), 5-aminolevulinic acid in extracellular (■) and intracellular (□) of pET32a-RchemA and pT-groELS/BL21(DE3) after 24 and 36 h at optimized condition of cultivation. G means glycine, S means succinate. 3G1S represents 3 g/L and 1 g/L for G and S, 6G2S represents 6 g/L and 2 g/L for G and S. Both additions are after 8 h of cultivation. 6G2S(fed) means 3 g/L and 1 g/L for G and S, which addition at 8 h and feed the same substrate after 24 h of cultivation separately
Conclusion
In this study, harboring a codon-optimized hemA from R. capsulatus has explored higher yields of ALA. The key factors are the plasmids of pET32a-RchemA and pT7-groELS whose trxA and groELS genes will promote the expression of soluble proteins. The critical mechanism of soluble RcHemA expression is translation, not transcription. We also found that the results were different from the combination of chaperones by TrxA with GroELS or TrxA with DnaKJ. Finally, the optimal strategy for producing ALA was established and reached a maximum of 5.66 g/L in flask culture. Further enhancement of ALA will be obtained by scaling up in a bioreactor.
References
Sasaki, K., Watanabe, M., & Tanaka, T. (2002). Biosynthesis, biotechnological production and applications of 5-aminolevulinic acid. Applied Microbiology and Biotechnology, 58(1), 23–29.
Liu, S., Zhang, G., Li, X., & Zhang, J. (2014). Applied Microbiology and Biotechnology, 98(17), 7349–7357.
Uehlinger, P., Zellweger, M., Wagnières, G., Juillerat-Jeanneret, L., et al. (2000). Journal of Photochemistry and Photobiology, B: Biology, 54(1), 72–80.
Cornelius, J. F., Slotty, P. J., El Khatib, M., Giannakis, A., Senger, B., & Steiger, H. J. (2014). Photodiagnosis and Photodynamic Therapy, 11(1), 1–6.
Noh, M. H., Lim, H. G., Park, S., Seo, S. W., & Jung, G. Y. (2017). Metabolic Engineering, 43, 1–8.
Li, T., Guo, Y. Y., Qiao, G. Q., & Chen, G. Q. (2016). Microbial synthesis of 5-aminolevulinic acid and its coproduction with polyhydroxybutyrate. ACS Synthetic Biology, 5(11), 1264–1274.
Shemin, D., & Russell, C. S. (1953). Journal of the American Chemical Society, 75(19), 4873–4874.
Hunter, G. A., & Ferreira, G. C. (1999). Biochemistry, 38(12), 3711–3718.
Ferreira, G. C., Neame, P. J., & Dailey, H. A. (1993). Heme biosynthesis in mammalian systems: evidence of a Schiff base linkage between the pyridoxal 5'-phosphate cofactor and a lysine residue in 5-aminolevulinate synthase. Protein Science, 2(11), 1959–1965.
Shoolingin-Jordan, P. M., Al-Daihan, S., Alexeev, D., et al. (2003). Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics, 1647(1–2), 361–366.
Kang, Z., Wang, Y., Gu, P., Wang, Q., et al. (2011). Metabolic Engineering, 13(5), 492–498 21.
Schauer, S., Chaturvedi, S., Randau, L., Moser, J., Kitabatake, M., Lorenz, S., Verkamp, E., Schubert, W. D., Nakayashiki, T., Murai, M., Wall, K., Thomann, H. U., Heinz, D. W., Inokuchi, H., Söll, D., & Jahn, D. (2002). Escherichia coli glutamyl-tRNA reductase. Trapping the thioester intermediate. Journal of Biological Chemistry, 277(50), 48657–48663.
Chung, S. Y., Seo, K. H., & Rhee, J. I. (2005). Process Biochemistry, 40(1), 385–394 26.
Liu, X. X., Wang, L., Wang, Y. J., & Cai, L. L. (2010). D-glucose enhanced 5-aminolevulinic acid production in recombinant Escherichia coli culture. Applied Biochemistry and Biotechnology, 160(3), 822–830.
Zhang, J., Kang, Z., Ding, W., Chen, J., & du, G. (2016). Integrated optimization of the in vivo heme biosynthesis pathway and the in vitro iron concentration for 5-aminolevulinate production. Applied Biochemistry and Biotechnology, 178(6), 1252–1262.
Yasukawa, T., Kanei-Ishii, C., Maekawa, T., Fujimoto, J., Yamamoto, T., & Ishii, S. (1995). Increase of solubility of foreign proteins in Escherichia coli by coproduction of the bacterial thioredoxin. Journal of Biological Chemistry, 270(43), 25328–25331.
Quan, S., & Bardwell, J. C. (2012). Chaperone discovery. Bioessays, 34(11), 973–981.
Lou, J. W., Zhu, L., Wu, M. B., Yang, L. R. Lin, J. P., &, Cen, P. L. (2014) Journal of Zhejiang University. Science. B 15(5), 491–499.
Fanica-Gaignier, M., & Clement-Metral, J. (1973). 5-AMINOLEVULINIC-acid synthetase of Rhodopseudomonas spheroides Y. Purification and some properties. European Journal of Biochemistry, 40(1), 13–18.
Yubisui, T., & Yoneyama, Y. (1972). Aminolevulinic acid synthetase of Rhodopseudomonas spheroides: purification and properties of the enzyme. Archives of Biochemistry and Biophysics, 150(1), 77–85.
Jordan, P. M., & Laghai-Newton, A. (1986). Methods in enzymology, vol. 123 (pp. 435–443). Academic Press.
Nakakuki, M., Yamauchi, K., Hayashi, N., & Kikuchi, G. (1980). Purification and some properties of delta-aminolevulinate synthase from the rat liver cytosol fraction and immunochemical identity of the cytosolic enzyme and the mitochondrial enzyme. Journal of Biological Chemistry, 255(4), 1738–1745.
Volland, C., & Felix, F. (1984). Isolation and properties of 5-aminolevulinate synthase from the yeast Saccharomyces cerevisiae. European Journal of Biochemistry, 142(3), 551–557.
Ferreira, G. C., & Gong, J. (1995). 5-Aminolevulinate synthase and the first step of heme biosynthesis. Journal of Bioenergetics and Biomembranes, 27(2), 151–159.
Stojanovski, B. M., & Ferreira, G. C. (2015). Murine erythroid 5-aminolevulinate synthase: adenosyl-binding site Lys221 modulates substrate binding and catalysis. FEBS Open Bio, 5, 824–831.
Zhu, C., Chen, J., Wang, Y., Wang, L., Guo, X., Chen, N., Zheng, P., Sun, J., & Ma, Y. (2019). Enhancing 5-aminolevulinic acid tolerance and production by engineering the antioxidant defense system of Escherichia coli. Biotechnology and Bioengineering, 116(8), 2018–2028.
Hayer-Hartl, M., Bracher, A., & Hartl, F. U. (2016). Trends in Biochemical Sciences, 41(1), 62–76.
Xu, Z., Horwich, A. L., & Sigler, P. B. (1997). Nature, 388(6644), 741.
Collet, J. F., & Messens, J. (2010). Antioxidants & Redox Signaling, 13(8), 1205–1216.
Hayer-Hartl, M., Bracher, A., & Hartl, F. U. (2016). The GroEL-GroES chaperonin machine: a nano-cage for protein folding. Trends in Biochemical Sciences, 41(1), 62–76.
Fu, W., Lin, J., & Cen, P. (2010). Applied Biochemistry and Biotechnology, 160(2), 456–466.
Su, T., Guo, Q., Zheng, Y., Liang, Q., et al. (2019). Frontiers in Microbiology. https://doi.org/10.3389/fmicb.2019.01731.
Funding
This work received financial support from the Ministry of Science and Technology (MOST 108-2221-E-006-004-MY3, MOST 108-2218-E-006-006, and MOST-108-2621-M-006-015) in Taiwan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics Approval and Consent to Participate
All the authors have read and agreed the ethics for publishing the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
ESM1
(DOCX 508 kb)
Rights and permissions
About this article

Cite this article
Yu, TH., Yi, YC., Shih, IT. et al. Enhanced 5-Aminolevulinic Acid Production by Co-expression of Codon-Optimized hemA Gene with Chaperone in Genetic Engineered Escherichia coli. Appl Biochem Biotechnol 191, 299–312 (2020). https://doi.org/10.1007/s12010-019-03178-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-019-03178-9