
Abstract
An extracellular polysaccharide TP1A was purified from the fermented broth of Trichoderma sp. KK19L1 by combination of Q Sepharose fast flow and Sephacryl S-300 chromatography. TP1A was composed of Man, Gal, and Glc in a molar ratio of about 3.0:5.1:8.1. The molar mass of TP1A was about 40.0 kDa. Methylation and NMR analysis indicated that the probable structure of TP1A was [→4,6)-α-D-Glcp(1→6)-β-D-Galf(1→6)-β-D-Galf(1→2,6)-β-D-Galf(1→2,6)-β-D-Galf(1→2,6)-β-D-Galf(1→2,6)-α-D-Manp(1→2,6)-α-D-Manp(1→] with [α-D-Glcp(1→] and [α-D-Manp(1→6)-α-D-Glcp(1→6)-α-D-Glcp(1→] as branches. The antitumor study showed that TP1A was able to inhibit the cell viability of HeLa and MCF-7 cells. TP1A could arrest HeLa cells in G2/M phase and induce HeLa cell apoptosis. These findings suggest that fungal polysaccharides could be a potential source for antitumor agents.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Microorganism is abundantly distributed in nature and could produce a variety of homo- and heteropolysaccharides [1–4]. Polysaccharides from microorganism exhibited the biological activities of antitumor [3, 5], antioxidant [6], and immunomodulant [7]. Therefore, microorganism is a good resource of polysaccharides with novel structures and excellent biological activities.
Trichoderma species belong to the genus Hypocrea (Ascomycota, Pyrenomycetes, Hypocreals, Hypocreaceae) and are generally found on decaying wood and in soil [8]. Trichoderma species are widely used for industrial enzyme production to hydrolyze the plant cell wall polysaccharides such as chitinase [9], xylanase [10], cellulose, and hemicellulose [11]. But besides the industrial importance of producing enzymes, there are limited reports on the production of polysaccharides by Trichoderma species. The polysaccharide from cell walls of Trichoderma reesei was a heteroglycan composed of mannose (45 ± 5%), galactose (30 ± 3%), glucose (11 ± 3%), galacturonic acid (9 ± 4%), and glucuronic acid (5 ± 3%) [12]. The heteroglycan had a mannan core, which was a linear α-(1,6)-manno-pyranan [13]. In order to better understand the polysaccharides in the fermented broth of Trichoderma and enhance the economic interest, the extracellular polysaccharides produced by Trichoderma were studied in this text. A fungus named Trichoderma sp. KK19L1 was isolated from the soil of Hebei University (Baoding, China). A water-soluble polysaccharide was extracted from the fermented broth of Trichoderma sp. KK19L1 for the first time, and its chemical structure was characterized. The in vitro antitumor activity of the exopolysaccharides was investigated as a preliminary evaluation of its possible application in the pharmaceutical aspects.
Materials and Methods
Materials
D-mannose (Man), D-galactose (Gal), D-galacturonic acid (GalUA), D-glucose (Glc), D-glucuronic acid (GlcUA), L-rhamnose (Rha), D-xylose (Xyl), L-arabinose (Ara), and 3-(4,5-dimethylthiazol-2yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Molar mass standards (pullulan standards 133.8, 84.4, 41.1, 21.4, and 10.0 kDa) were from Shodex (Tokyo, Japan). Dialysis membranes (molecular weight cutoff 3500) were from Viskase (Darien, IL, USA). Acetonitrile was HPLC grade and purchased from Merck (Darmstadt, Germany). Dulbecco’s modified Eagle’s medium (DMEM) was from Gibco (Grandlsland, NY, USA). Other chemicals and reagents used in this study were the highest quality available from commercial vendors.
Microbial Strain and Culture Conditions
The strain named SF-2 was isolated from about 20-cm deep soil collected from Hebei University (Baoding, China). SF-2 was identified according to its morphology and its DNA sequence; the GeneBank accession number was JF736513. The strain SF-2 was activated on potato dextrose agar (PDA) slants and then cultured at 28 °C for 21 days. The ingredients of the culture medium were mannitol 20 g/L, yeast extract 3 g/L, glucose 10 g/L, maltose 20 g/L, monosodium glutamate 5 g/L, peptone 5 g/L, and juice of potato 2000 g/L; they were formulated with artificial sea water by adjusting the pH to 6.0.
Preparation of the Exopolysaccharides
After the removal of mycelium, the fermented broth was concentrated in a rotary evaporator. Polysaccharides were precipitated with 75% ethanol at 4 °C for 12 h in order to acquire complete precipitation. The crude polysaccharides were dialyzed against deionized water with dialysis membrane for three successive days and lyophilized. Then, the polysaccharides were dissolved in distilled water and applied to an anion-exchange column packed with Q Sepharose Fast Flow (QFF, GE Healthcare, Piscataway, NJ, USA) coupled to a Biotage One rapid preparation liquid chromatography system (Uppsala, Sweden). The eluent was 0, 0.2, 0.4, and 0.6 mol/L NaCl. The phenol-sulfuric acid detection method [14] was used to detect the polysaccharide content after eluting by QFF column. The main polysaccharide fraction eluted with 0.2 mol/L NaCl (TP1) was pooled, concentrated, and dialyzed against deionized water. TP1 was further purified on a Sephacryl S-300 column (S-300, GE Healthcare, Piscataway, NJ, USA) eluted with 0.2 mol/L NaCl, and the polysaccharide fraction was detected by the phenol-sulfuric acid method and then pooled according to the phenol-sulfuric acid detection curve. NaCl was removed by dialysis with distilled water, and the polysaccharide TP1A was obtained.
Physiochemical Properties
Total carbohydrate content was assayed by phenol-sulfuric acid method [14]. Protein content was determined by the method of Bradford [15]. The homogeneity and average molar mass of TP1A were detected by an Agilent 1260 high performance liquid chromatography (HPLC, USA) system equipped with an OHpak SB-804 HQ (7.8 × 300 mm) column. The mobile phase was 0.1 mol/L Na2SO4, and the sample was detected with an Agilent refractive index detector G1362A at 35 °C. The molar mass of TP1A was estimated according to the retention time by reference to the calibration curve made by the molecular standards.
Monosaccharide Composition Analysis
HPLC (Aglilent 1260, USA) was used for identification and quantification of the monosaccharide composition of TP1A after precolumn derivatization. TP1A was hydrolyzed with 2 mol/L TFA at 105 °C for 6 h in a sealed glass tube, then TFA was removed by repeatedly stripping off by adding methanol. The completely hydrolyzed sample and monosaccharide standards were derivatized with 1-phenyl-3-methyl-5-pyrazolone (PMP) as reported by the literature [16]. The derivative was detected with an UV detector at 245 nm, and Agilent Zorbax SB-C18 column (5 μm, 4.6 × 250 mm, USA) was used by eluting with 0.1 mol/L KH2PO4 (pH 6.7)-acetonitrile (83:17) at 1.0 mL/min. Monosaccharide composition and molar ratio of the sample were identified and calculated by comparing the retention time and peak area with the monosaccharide standards detected by HPLC chromatography spectrum.
Structure Analysis
Specific Optical Rotation Analysis
Specific optical rotation of the polysaccharide TP1A was performed on AA-55 digital polarimeter (Optical activity, UK) at 20 ± 0.1 °C and calculated as follows: \( {\left[\alpha \right]}_{\lambda}^t=\frac{\alpha }{L\times C} \). α is the optical rotation, t is the temperature, λ is the wavelength of light source, L is the length of polariscope tube, and C is the polysaccharide concentration (g/100 mL).
FT-IR Analysis
For Fourier transform infrared (FT-IR) spectrum analysis, TP1A was pressed with KBr powder and then measured on a Bruker Vertex 70 spectrometer (Bruker Optik Gmbh, Ettlingen, Germany) in the wavenumber of 4000–400 cm−1.
Methylation and GC/MS Analysis
Methylation analysis of polysaccharides was conducted according to Hakomori method with minor modifications [17]. One milligram of TP1A was dissolved in 2 mL dimethyl sulfoxide. After adding 150 mg NaH, the mixture was reacted at room temperature for 1 h. Then, 1 mL CH3I was added, and the mixture was stirred for a further 1.5 h. The methylated polysaccharide was hydrolyzed by 2 mol/L TFA at 105 °C for 6 h, reduced by NaBH4, acetylated by acetic anhydride, and then analyzed by an Agilent GC-MS system (HP6890II, Agilent, USA) coupled with a DB 225 fused silica capillary column (0.25 mm × 30 m). The temperature program was as following: the column temperature increased at 5 °C/min from 100 to 220 °C and kept at 220 °C for 15 min. Identification of the linkage patterns was carried out on the mass spectra by comparing to Complex Carbohydrate Research Center Database (http://www.ccrc.uga.edu/).
NMR Analysis
For NMR analysis, TP1A was lyophilized with D2O and then dissolved in 0.5 mL D2O (99.98%) containing a trace of acetone as internal standard (1H 2.225 ppm; 13C 31.07 ppm). 1H (40 °C), DEPT (135°), TOCSY (100 ms), HSQC, NOESY, and HMBC NMR spectra were recorded on a Bruker Avance III (600.19 MHz for 1H and 150.91 MHz for 13C) spectrometer. 2D NMR spectra were obtained using the standard Bruker software and processed with the MestReNova-5.3.1 program.
Antitumor Activity
Human cervical carcinoma cells (HeLa) and human breast carcinoma cells (MCF-7) were maintained in DMEM medium supplemented with 10% newborn bovine serum (NBS), 100 U/mL penicillin, and 100 μg/mL streptomycin. Cells were cultured in a humidified atmosphere with 5% CO2 at 37 °C. The polysaccharide TP1A was dissolved by 0.1 mol/L PBS with pH 7.4.
Cell Viability Assay
MTT assay was used to determine the inhibition effect of polysaccharides on cancer cells [18]. Ninety microliters of cells at a density of 2 × 104 cells/mL was added into 96-well plate and cultured overnight. A final concentration of 25, 50, 100, 200, and 400 μg/mL TP1A was applied to the cancer cells and incubated for 24 and 48 h. Ten microliters of MTT was added to each well and incubated for another 4 h, then dissolved with 150 μL DMSO. Absorbance was determined on a microplate reader at 570 nm (SpectraMax M4, USA). The inhibition rate of the polysaccharide on tumor cells was calculated according to the formula below: Inhibition rate (%) = [1 − (A sample − A blank)/(A control − A blank)] × 100, where A blank was the absorbance of the blank and A sample and A control were the absorbance of the sample treated and untreated cells, respectively.
Cell Cycle Analysis
HeLa cells (1 × 105 cells/mL) were seeded in six-well plates. After cultured for 12 h, a gradient concentration of polysaccharide (100, 200, 400 μg/mL) was added and incubated for 24 h. Then, cells were collected, washed with cold PBS, and fixed with cold 70% ethanol (PBS) at 4 °C for 12 h. After removing ethanol with PBS, cells were stained with propidum iodide (PI) at 25 °C in the dark for 15 min. A flow cytometer (BD FACS, USA) was used to analyze the cell cycle.
Apoptosis Analysis
Apoptosis induction of polysaccharides was assessed with Annexin V-FITC/PI double fluorescence apoptosis detection kit (Invitrogen, Life technologies) on a flow cytometer (BD FACS, USA). HeLa cells (1 × 105 cells/mL) were incubated with different concentrations of polysaccharide (100, 200, 400 μg/mL) for 48 h. Then, cells were collected and stained with Annexin V-FITC and PI in annexin-binding buffer according to the supplier’s instructions. The fluorescence of cells was assayed by a flow cytometry within 1 hour.
Statistical Analysis
The results of the antitumor activity were expressed as mean ± SD. p < 0.05 was considered statistically significant.
Results and Discussion
Preparation and Physicochemical Properties of the Polysaccharide TP1A
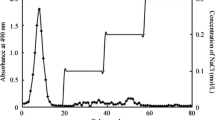
The crude polysaccharides produced by Trichoderma sp. KK19L1 were isolated by an anion-exchange chromatography column, and the main polysaccharide fraction TP1 eluted with 0.2 mol/L NaCl was obtained (Fig. 1a). TP1 was further purified by a Sephacryl S-300 chromatography column; the polysaccharide fraction TP1A was obtained (Fig. 1b). As detected by HPGPC chromatography, there was one single symmetrical peak for TP1A (Fig. S1a), which indicated TP1A was a homogeneous polysaccharide. According to the standard curve made by the logarithm of molar mass and retention time of the molecular standards (Fig. S1b, y = − 0.509× + 9.8382, R 2 = 0.9952, y was the logarithm of molar mass, x was the retention time), the retention time of TP1A was 10.287 min and the average molar mass of TP1A was about 40.0 kDa. The total sugar content of TP1A was 97.6%, and no protein was detected in TP1A. HPLC chromatography analysis indicated that TP1A consisted of glucose, galactose, and mannose in a molar ratio of 8.1:5.1:3.0. The specific optical rotation of the polysaccharide TP1A was +36.9°, which indicated that TP1A had a right-handed form.

Preparation of the polysaccharide TP1A from Trichoderma sp. KK19L1. a Elution profile of the crude polysaccharides by Q Sepharose fast flow chromatography column. b Elution profile of TP1 by Sephacryl S-300 chromatography column
Structure Analysis
The FT-IR spectrum of TP1A was depicted in Fig. 2. The signals at 3431 and 2939 cm−1 were the vibration of O-H and C-H bonds, respectively. The bands at 1645 and 1429 cm−1 were attributed to the bending vibrations of HOH and O-H, respectively. The peak at 1049 cm−1 was assigned to the stretch vibration of C-O-C linkage.

FT-IR spectrum of TP1A
TP1A was methylated, hydrolyzed, reduced, acetylated, and then analyzed by GC/MS. The methylated products 1,5-di-O-acetyl-2,3,4,6-tetra-O-methyl-glucitol, 1,5,6-tri-O-acetyl-2,3,4-tri-O-methyl-glucitol, and 1,4,5.6-tetra-O-acetyl-2,3-di-O-methyl-glucitol were detected in TP1A, which suggested that TP1A possessed the linkages of [Glcp(1→], [→6)-Glcp(1→] and [→4,6)-Glcp(1→]. The presence of 1,5-di-O-acetyl-2,3,4,6-tetra-O-methyl-mannitol and 1,2,5,6-tetra-O-acetyl-3,4-di-O-methyl-mannitol suggested that TP1A had the linkages of [Manp(1→] and [→2,6)-Manp(1→]. 1,4,6-tri-O-acetyl-2,3,5-tri-O-methyl-galactitol and 1,2,4,6-tetra-O-acetyl-3,5-di-O-methyl-galactitol indicated that TP1A was composed of [→6)-Galf(1→] and [→2,6)-Galf(1→]. The molar ratio of [Glcp(1→], [→6)-Glcp(1→], [→4,6)-Glcp(1→], [Manp(1→], [→2,6)-Manp(1→], [→6)-Galf(1→], and [→2,6)-Galf(1→] was about 5.1:1.9:1.2:1.0:2.1:2.2:3.0 (Table S1).
NMR is a powerful technique that is used to characterize the structure of polysaccharides. 1H and DEPT (135°) signals of TP1A were assigned according to the 2D NMR. TP1A showed the presence of seven anomeric proton signals at 5.52, 5.20, 5.18, 5.16, 5.11, 5.09, and 4.93 ppm (Fig. 3a). For the convenience of assignment, the anomeric protons were labeled as A, B, C, D, E, F, and G (Fig. 3a), respectively. The proton signals at 3.40∼4.40 ppm were designated to the protons of C2∼C6. In the DEPT of TP1A (Fig. 3b), there were seven anomeric carbon signals labeled as A, B, C, D, E, F, and G with the chemical shifts at 99.9, 107.9, 103.8, 107.3, 99.7, 98.5, and 99.9 ppm, respectively. The carbon signals at 107.9 and 107.3 ppm were assigned to be the C-1 of β-D-galactofuranose residues as their extremely low field shifts [19, 20], and the signal at 64.4 ppm indicates the presence of unsubstituted C-6 of Galf.

The NMR spectra and structure of TP1A. a 1H NMR. b DEPT spectrum. c Structure of TP1A. A [→4,6)-α-D-Glcp(1→], B [→6)-β-D-Galf(1→], C [α-D-Manp(1→], D [→2,6)-β-D-Galf(1→], E [→2,6)-α-D-Manp(1→], F [→6)-α-D-Glcp(1→], G [α-D-Glcp(1→]
According to the 1H-1H TOCSY (Fig. S2a) and 1H-13C HMQC (Fig. S2b) spectra, the proton and carbon signals of different linkage patterns were assigned. The anomeric proton signals at 5.52, 5.09, and 4.93 ppm with the anomeric carbons at 99.9, 98.5, and 99.9 ppm corresponded to the H-1 and C-1 of [→4,6)-α-D-Glcp(1→], [→6)-α-D-Glcp(1→], and [α-D-Glcp(1→], respectively, which were in good agreement with the literatures [21, 22]. The anomeric protons at 5.18 and 5.11 ppm related to the carbons at 103.8 and 99.7 ppm, which were assigned to the H-1 and C-1 of [α-D-Manp(1→] and [→2,6)-α-D-Manp(1→], respectively [23]. The anomeric protons at 5.20 and 5.16 ppm with the anomeric carbons at 107.9 and 107.3 ppm could be attributed to the H-1 and C-1 of [→6)-β-D-Galf(1→] and [→2,6)-β-D-Galf(1→] [24, 25]. The proton and carbon chemical shifts of different linkage patterns were assigned in Table 1.
According to the 1H-1H NOESY spectrum (Fig. S2c) of TP1A, the sequence of different sugar residues was assigned. The signals of H1 of C (5.18 ppm) with H6 of F (3.99 ppm), H1 of F (5.09 ppm) with H6 of F (3.76 ppm) and A (3.76 ppm) confirmed the possible sequence of C-F-F-A. The peaks of H1 of A (5.52 ppm) with H6 of B (3.68 ppm), H1 of B (4.20 ppm) with H6 of B (3.68 ppm) and D (3.68 ppm) indicated the possible linkage pattern of A-B-B-D. The linkage patterns of G-D and G-E were assigned by H1 of G (4.93 ppm) with H2 of D (4.20 ppm) and H6 of E (3.76 ppm). The related signals of H1 of D (5.16 ppm) and E (5.11 ppm) with H2 of E (4.13 ppm) confirmed the possible sequence of D-E-E. The 1H-13C HMBC spectrum of TP1A (Fig. S2d) further confirmed the sequence of different glycosidic residues. The H1 of C (5.18 ppm) correlated to C6 of F (70.6 ppm), and H1 of F (5.09 ppm) related to C6 of A (70.6 ppm) and F (70.6 ppm) indicated the presence of C-F-F-A. The signals from H1 of A (5.52 ppm) with C6 of B (70.6 ppm), H1 of B (5.20 ppm) with C6 of D (70.6 ppm), and H1 of D (5.16 ppm) with C2 of E (78.2 ppm) further confirmed the sequences of A-B, B-D, and D-E. The correlated signals for H1 of G (4.93 ppm) with C2 of D (88.3 ppm) and C6 of E (70.6 ppm) in the 1H-13C HMBC spectrum of TP1A (Fig. S2d) were not shown, but the signals of C1 of G (99.9 ppm) with H2 of D (4.20 ppm) and H6 of E (70.6 ppm) were found, so the sequences of G-D and G-E may be established. Combined of the data from NMR spectra and methylation analysis, TP1A was a complex heteropolysaccharide with branches. The possible structure of TP1A was shown in Fig. 3c, being n ≈ 15.
Polysaccharides with different structures had been isolated from fungi. The polysaccharides from Trichoderma and Hypocrea species were all complex heteropolysaccharides [26]. They had the backbone of [→6)-α-D-Manp(1→] with different branches substituted at C-2 or C-3 position [26]. The extracellular polysaccharides with the backbone of [→2)-α-D-Manp(1→] had been isolated from Aspergillus ochraceus [27] and Aspergillus terreus [6] with branches mainly substituted at C-6 position. Galactomannans with a mannan core had been isolated from Aspergillus wentii, Chaetosartorya chrysella, and Aspergillus fumigatus [23]. The polysaccharides from Aspergillus wentii and Chaetosartorya chrysella were [→3)-β-D-Galf(1→5)-β-D-Galf(1→]n→mannan core, and the polysaccharide from Aspergillus fumigatus was [→5)-β-D-Galf(1→]n→mannan core. The mannan core was (1→6)-α-mannan by chains from 1 to 7 residues of (1→6) linked α-mannopyranoses substituted at C-2 position [23]. A disaccharide repeating unit consisted of [→2)-β-D-Galf(1→6)-α-D-Glcp(1→] was isolated from the fungus Fusarium oxysporum Y24-2 [28]. The glucan of [→6)-α-D-Glcp(1→], [→3)-α-D-Glcp(1→], and [→3)-β-D-Glcp(1→] had been isolated from the fungi Aspergillus versicolor [29], Aspergillus wentii [30], and Thelebolus [31], respectively. The polysaccharide from Ganoderma lucidum has the main chain of [→4)-α-D-Galp(1→] with [α-L-Araf(1→], [α-L-Rhap(1→], [→6)-α-D-Manp(1→], and [→4)-β-D-Glcp(1→] as branches [32]. TP1A was a heteropolysaccharide with the structure different from the fungi polysaccharides previously reported. The fact that the extracellular polysaccharides could be contaminated by yeast extract and potato extract cannot be ruled out. But by comparison with early reported cases in the literatures, TP1A was different from the polysaccharide from yeast extract in monosaccharide composition, molar mass, linkage pattern, and sequence [27, 33, 34]. The starch from potato was a glucan composed of 1,4-Glcp or 1,4-Glcp with 1,6-Glcp branches and had a high average molar mass. TP1A was different from the starch from potato in monosaccharide composition, molecular weight, and linkage pattern. TP1A was a polysaccharide with unique structure to be worth being further studied.
Antitumor Activity
In this study, cancer cells HeLa and MCF-7 were incubated with different concentrations of TP1A (25, 50, 100, 200, 400 μg/mL) and determined by MTT method in vitro. The antitumor effect of TP1A against HeLa and MCF-7 cells was shown in Fig. 4. TP1A exhibited an obvious inhibition effect on HeLa cells. When the concentration of TP1A was 400 μg/mL, the inhibition rate was about 50.5% on HeLa cells for 48 h. No obvious change of the inhibition rate was observed on MCF-7 cells with the increasing concentration of TP1A. The inhibition rate was below 20% at the high concentration of 400 μg/mL on MCF-7 cells. As shown in Fig. 4, the cell viability decreased after treatment with higher concentration of TP1A. But with the prolongation of incubation time, the cytotoxicity of TP1A on cancer cells was slightly changed. The results indicated that the antitumor effect of TP1A on HeLa and MCF-7 cells was in a dose-dependent but not time-dependent manner. The in vitro antitumor activity of TP1A was weaker than the polysaccharides from Aspergillus fumigatus [35], Aspergillus fumigatus AF1 [36], and Thelebolus [31], but stronger than the polysaccharides from Longan [22], Mosla chinensis Maxim. cv. Jiangxiangru [37], Portulaca oleracea L. [38], Taxus chinensis var. Mairei [39], Phynchosia minima [40], and Pseudoaltermonas sp. [41]. For example, after incubated with the extracellular polysaccharide from Aspergillus fumigatus [35] and Aspergillus fumigatus AF1 [36] for 24 h, the inhibition rates on S180, HepG2, and MCF-7 cells were nearly 100% at 25 μg/mL. The antiproliferative effect on HepG2 cells was about 15% at 500 μg/mL for the glucan from Longan, and the inhibition rate was about 6% on MCF-7 cells at 250 μg/mL in vitro [22]. The pectic polysaccharide from Mosla chinensis Maxim. cv. Jiangxiangru could inhibit K562 cells for 30% at 200 μg/mL in vitro [37]. The polysaccharide from Portulaca oleracea L. could inhibit HeLa cells about 51.8% at 500 μg/mL for 48 h in vitro [38]. The alkali-soluble polysaccharide from Taxus chinensis var. Mairei had an antiproliferative effect on SGC-7901 cells about 36.53% at 300 μg/mL [39]. Three acidic polysaccharides from Phynchosia minima exhibited anticancer activity against A549 cells under 25% and against HepG2 cells under 40% for 24 h at 2.0 mg/mL in vitro [40]. The exopolysaccharide from Antarctic bacterium Pseudoaltermonas sp. S-5 could inhibit K562 cells about 36.0% at 600 μg/mL for 48 h [41].

Effects of TP1A at different concentration on HeLa and MCF-7 cells for 24 and 48 h by MTT assay in vitro. a HeLa cells. b MCF-7 cells
To investigate the possible mechanism of the antitumor effect of TP1A, the apoptosis of HeLa cells was detected by flow cytometry with Annexin V-PI double staining. As can be seen from Fig. 5a, the apoptosis of the untreated cells in the control group was 2.4%, while the apoptosis of cells treated with 100, 200, 400 μg/mL of TP1A was 8.6, 10.3, and 20.3%, respectively. The apoptotic cells were significantly increased in the TP1A treated groups compared to the control group. Cell cycle arrest at different phases (G0/G1, S, G2/M) could also induce cells apoptosis. To confirm the possible antitumor mechanism of TP1A treated cells, cell cycle was detected by flow cytometry. As shown in Fig. 5b, when the concentration of TP1A increased, percentages of cells in S phase decreased with a parallel increase in G2/M phase. Thus, TP1A could arrest HeLa cells in G2/M phase. G2/M checkpoint is an important cell cycle checkpoint. Cells that have a defective G2/M checkpoint enter mitosis before repairing their DNA and lead to death after cell division. The results demonstrated that TP1A could inhibit the cell viability of HeLa cells via apoptosis and cell cycle arrest. The exopolysaccharide (EPS) isolated from the fermentation liquor of Trichoderma pseudokoningii could induce apoptosis of K562 and MCF-7 cells through an intrinsic mitochondrial pathway [42, 43]. EPS could also arrest MCF-7 cells in S phase [43].

a TP1A induces HeLa cells apoptosis. b Induction of cell cycle arrest at G2/M phase in HeLa cells by TP1A. Data are shown as means ± SD (n = 3)
Conclusion
The extracellular polysaccharide TP1A was isolated from the fungus Trichoderma sp. KK19L1. TP1A was a complex heteropolysaccharide with the average molar mass about 40.0 kDa. TP1A possessed the antitumor activity on HeLa and MCF-7 cells; it could arrest HeLa cells in G2/M phase and induce HeLa cell apoptosis. Further work is needed to reveal the detailed structure and the mechanism of TP1A on cancer cells. Generally, Trichoderma species are a good resource of polysaccharides with new structures and antitumor effect. Microorganism could be an economical alternative to plants and animals to produce novel secondary metabolites.
References
Cai, S. X., Luan, Y. P., Kong, X. L., Zhu, T. J., Gu, Q. Q., & Li, D. H. (2013). Isolation and photoinduced conversion of 6-epi-stephacidins from Aspergillus taichungensis. Organic Letters, 15, 2168–2171.
Takagi, M., Cabrera-Crespo, J., Baruque-Ramos, J., Zangirolami, T. C., Raw, I., & Tanizaki, M. M. (2003). Characterization of polysaccharide production of Haemophilus influenza type b and its relationship to bacterial cell growth. Applied Biochemistry and Biotechnology, 110, 91–100.
Cao, X. Y., Liu, R. H., Liu, J. L., Hou, Y. P., Yang, W., Zeng, M., & Yang, C. (2013). A novel polysaccharide from Lentinus edodes mycelia exhibits potential antitumor activity on laryngeal squamous cancer cell line Hep-2. Applied Biochemistry and Biotechnology, 171, 1444–1453.
Vasanthakumari, D. S., Harikumar, S., Beena, D. J., Pandey, A., & Nampoothiri, K. M. (2015). Physicochemical characterization of an exopolysaccharide produced by a newly isolated Weissella cibaria. Applied Biochemistry and Biotechnology, 176, 440–453.
Li, L. Y., Li, D. H., Luan, Y. P., Gu, Q. Q., & Zhu, T. J. (2012). Cytotoxic metabolites from the antarctic psychrophilic fungus Oidiodendron truncatum. Journal of Natural Products, 75, 920–927.
Wang, C. Y., Mao, W. J., Chen, Z. Q., Zhu, W. M., Chen, Y. L., Zhao, C. Q., Li, N., Yan, M. X., Liu, X., & Guo, T. T. (2013). Purification, structural characterization and antioxidant property of an extracellular polysaccharide from Aspergillus terreus. Process Biochemistry, 48, 1395–1401.
Sheng, L., Chen, J. P., Li, J., & Zhang, W. Y. (2011). An exopolysaccharide from cultivated Cordyceps sinensis and its effects on cytokine expressions of immunocytes. Applied Biochemistry and Biotechnology, 163, 669–678.
Druzhinina, I. S., Kopchinskiy, A. G., & Kubicek, C. P. (2006). The first 100 Trichoderma species characterized by molecular data. Mycoscience, 47, 55–64.
Azin, M., Moravej, R., & Zareh, D. (2007). Production of xylanase by Trichoderma longibrachiatum on a mixture of wheat bran and wheat straw: optimization of culture condition by taguchi method. Enzyme and Microbial Technology, 40, 801–805.
Felse, P. A., & Panda, T. (1999). Self-directing optimization of parameters for extracellular chitinase production by Trichoderma harzianum in batch mode. Process Biochemistry, 34, 563–566.
Martinez, D., Berka, R. M., Henrissat, B., et al. (2008). Genome sequencing and analysis of the biomass-degrading fungus Trichoderma reesei (syn. Hypocrea jecorina). Nature Biotechnology, 26, 553–560.
Messner, R., Hagspiel, K., & Kubicek, C. P. (1990). Isolation of a β-glucosidase binding and activating polysaccharide from cell walls of Trichoderma reesei. Archives of Microbiology, 154, 150–155.
Rath, J., Messner, R., Kosma, P., Altmann, F., März, L., & Kubicek, C. P. (1995). The α-D-mannan core of a complex cell-wall heteroglycan of Trichoderma reesei is responsible for β-glucosidase activation. Archives of Microbiology, 164, 414–419.
Dubois, M., Gilles, K. A., Hamilton, J. K., Rebers, P. A., & Smith, F. (1956). Colorimetric method for determination of sugars and related substances. Analytical Chemistry, 28, 350–356.
Bradford, M. M. (1976). A rapid and sensitive method for the quantization of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry, 72, 248–254.
Liu, X., Zhang, M., Guo, K., Jia, A., Shi, Y., Gao, G., Sun, Z., & Liu, C. (2015). Cellulose-assisted extraction, characterization, and bioactivity of polysaccharide from Polygonatum odoratum. International Journal of Biological Macromolecules, 75, 258–265.
Hakomori, S. (1964). A rapid permethylation of glycolipid, and polysaccharide catalyzed by methylsulfinyl carbanion in dimethyl sulfoxide. Journal of Biochemistry, 55, 205–208.
Mosman, T. (1983). Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. Journal of Immunological Methods, 65, 55–63.
Ahrazem, O., Prieto, A., Leal, J. A., Giménez-Abián, M. I., Jiménez-Barbero, J., & Bernabé, M. (2007). Fungal cell wall polysaccharides isolated from Discula destructiva spp. Carbohydrate Research, 342, 1138–1143.
Bernabé, M., Salvachúa, D., Jiménez-Barbero, J., Leal, J. A., & Prieto, A. (2011). Structures of wall heterogalactomannans isolated from three genera of entomopathogenic fungi. Fungal Biology, 115, 862–870.
Niu, Y. G., Yan, W., Lv, J. L., Yao, W. B., & Yu, L. L. (2013). Characterization of a novel polysaccharide from tetraploid Gynostemma pentaphyllum Makino. Journal of Agricultural and Food Chemistry, 61, 4882–4889.
Zhu, Q. Q., Jiang, Y. M., Lin, S., Wen, L. R., Wu, D., Zhao, M. M., Chen, F., Jia, Y. X., & Yang, B. (2013). Structural identification of (1→6)-α-D-glucan, a key responsible for the health benefits of Longan, and evaluation of antitumor activity. Biomacromolecules, 14, 1999–2003.
Gόmez-Miranda, B., Prieto, A., Leal, A. P., Ahrazem, O., Jiménez-Barbero, J., & Bernabé, M. (2004). Differences among the cell wall galactomannans from Aspergillus wentii and Chaetosartorya chrysella and that of Aspergillus fumigatus. Glycoconjugate Journal, 20, 239–246.
Shashkov, A. S., Potekhina, N. V., Kachala, V. V., Senchenkova, S. N., Dorofeeva, L. V., & Evtushenko, L. I. (2012). A novel galactofuranan from the cell wall of Arthrobacter sp. VKM Ac-2576. Carbohydrate Research, 352, 215–218.
Svensson, M. V., Zhang, X., Huttunen, E., & Widmalm, G. (2011). Structural studies of the capsular polysaccharide produced by Leuconostoc mesenteroides ssp. cremoris PIA2. Biomacromolecules, 12, 2496–2501.
Prieto, A., Leal, J. A., Poveda, A., Jiménez-Barbero, J., Gómez-Miranda, B., Domenech, J., Ahrazem, O., & Bernabé, M. (1997). Structure of complex cell wall polysaccharides isolated from Trichoderma and Hypocrea species. Carbohydrate Research, 304, 281–291.
Guo, S. D., Mao, W. J., Yan, M. X., Zhao, C. Q., Li, N., Shan, J. M., Lin, C., Liu, X., Guo, T., Guo, T. T., & Wang, S. Y. (2014). Galactomanan with novel structure produced by the coral endophytic fungus Aspergillus ochraceus. Carbohydrate Polymers, 105, 325–333.
Guo, S. D., Mao, W. J., Li, Y. L., Tian, J. H., & Xu, J. (2013). Structural elucidation of the exopolysaccharide produced by fungus Fusarium oxysporum. Carbohydrate Research, 365, 9–13.
Chen, Y., Mao, W. J., Gao, Y., Teng, X. C., Zhu, W. M., Chen, Y. L., Zhao, C. Q., Li, N., Wang, C. Y., Yan, M. X., Shan, J. M., Lin, C., & Guo, T. (2013). Structural elucidation of an extracellular polysaccharide produced by the marine fungus Aspergillus versicolor. Carbohydrate Polymers, 93, 478–483.
Choma, A., Wiater, A., Komaniecka, I., Paduch, R., Pleszczyńska, M., & Szczodrak, J. (2013). Chemical characterization of a water insoluble (1→3)-α-D-glucan from an alkaline extraxt of Aspergillus wentii. Carbohydrate Polymers, 91, 603–608.
Mukhopadhyay, S. K., Chatterjee, S., Gauri, S. S., Das, S. S., Mishra, A., Patra, M., Ghosh, A. K., Das, A. K., Singh, S. M., & Dey, S. (2014). Isolation and characterizaion of extracellular polysaccharide Thelebolan produced by a newly isolated psychrophilic Antarctic fungus Thelebolus. Carbohydrate Polymers, 104, 204–212.
Li, Y. Q., Fang, L., & Zhang, K. C. (2007). Structure and bioactivities of a galactose rich extracellular polysaccharide from submergedly cultured Ganoderma lucidum. Carbohydrate Polymers, 68, 323–328.
Kath, F., & Kulicke, W. (1999). Polymer analytical characterization of glucan and mannan from yeast Saccharomyces cerevisiar. Die Angewandtr Makromolekulare Chemie, 268, 69–80.
Komura, D. L., Ruthes, A. C., Carbonero, E. R., Alquini, G., Rosa, M. C. C. R., Sassaki, G. L., & Iacomini, M. (2010). The origin of mannans found in submerged culture of basidiomycetes. Carbohydrate Polymers, 79, 1052–1056.
Jin, X. C., & Zhao, S. J. (2014). Extraction optimization and bioactivities of an extracellular polysaccharide produced by Aspergillus fumigatus. International Journal of Biological Macromolecules, 68, 13–17.
Jin, X. C., & Ning, Y. (2013). Extraction optimization and bioactivity of polysaccharides from Aspergillus fumigatus AF1. Carbohydrate Polymers, 96, 411–416.
Li, J. E., Cui, S. W., Nie, S. P., & Xie, M. Y. (2014). Structure and biological activities of a pectic polysaccharide from Mosla chinensis Maxim. cv. Jiangxiangru. Carbohydrate Polymers, 105, 276–284.
Zhao, R., Gao, X., Cai, Y. P., Shao, X. Y., Jia, G. Y., Huang, Y. L., Qin, X. G., Wang, J. W., & Zheng, X. L. (2013). Antitumor activity of Portulaca oleracea L. polysaccharides against cervical carcinoma in vitro and in vivo. Carbohydrate Polymers, 96, 376–383.
Wu, M. B., Zhang, F. F., Yu, Z. P., Lin, J. P., & Yang, L. R. (2015). Chemical characterization and in vitro antitumor activity of a single-component polysaccharide from Taxus chinensis var. Mairel. Carbohydrate Polymers, 133, 294–301.
Jia, X. J., Zhang, C., Qiu, J. F., Wang, L. L., Bao, J. L., Wang, K., Zhang, Y. L., Chen, M. W., Su, H. X., Han, J. P., & He, C. W. (2015). Purification, structural characterization and anticancer activity of the novel polysaccharides from Rhynchosia minima root. Carbohydrate Polymers, 132, 67–71.
Chen, G. C., Qian, W., Li, J., Xu, Y. H., & Chen, K. S. (2015). Exopolysaccharide of Antarctic bacterium Pseudoaltermonas sp. S-5 induces apoptosis in K562 cells. Carbohydrate Polymers, 121, 107–114.
Huang, T. T., Lin, J., Cao, J. F., Zhang, P. Y., Bai, Y. G., Chen, G. C., & Chen, K. S. (2012). An exopolysaccharide from Trichoderma pseudokoningii and its apoptotic activity on human leukemia K562 cells. Carbohydrate Polymers, 89, 701–708.
Wang, G. D., Liu, C. Y., Liu, J., Liu, B., Li, P., Qin, G. Z., Xu, Y. H., Chen, K., Liu, H. X., & Chen, K. S. (2016). Exopolysaccharide from Trichoderma pseudokoningii induces the apoptosis of MCF-7 cells through an intrinsic mitochondrial pathway. Carbohydrate Polymers, 136, 1065–1073.
Acknowledgements
This work was supported by financial support from Hebei University, Natural Science Foundation of Hebei province in China (B2015201069), Youth Fund Project of Hebei Education Department (QN2014131), and Bureau of Baoding city science and technology project (14ZF083).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Li, H., Yu, H. & Zhu, H. Structure Studies of the Extracellular Polysaccharide from Trichoderma sp. KK19L1 and Its Antitumor Effect via Cell Cycle Arrest and Apoptosis. Appl Biochem Biotechnol 182, 128–141 (2017). https://doi.org/10.1007/s12010-016-2315-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-016-2315-1