
Abstract
Pholiota adiposa is an edible chestnut mushroom with many health benefits, such as antioxidant and anticancer activity. In this paper, polysaccharides were extracted from Pholidota adiposa using an acid extraction process. The crude polysaccharide was purified using DEAE-cellulose chromatography, and two polysaccharide fractions of SPAP2-1 and SPAP2-2 were obtained. The structure was characterized using UV, GPC, GC, FT-IR, methylation, and NMR analysis. Monosaccharide component analysis indicated that SPAP2-1 (19 kDa) and SPAP2-2 (20 kDa) contained mannose, glucose, and galactose with different molecular ratios. Their antitumor effects were investigated using the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl tetrazolium (MTT) assay, Annexin V-fluorescein isothiocyanate (FITC), propidium iodide (PI) staining, and flow cytometry. By analyzing the changes in the cells, SPAP2-1 caused damage and changed the proliferation rate of HeLa cells. SPAP2-1 showed strong interference to the cell cycle of HeLa cells and induced cell apoptosis. Overall, these results suggested that polysaccharides from Pholiota adiposa, especially SPAP2-1, may have the potential to be used as a tumor cell inhibitor, which needs further study.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pholiota adiposa, an edible chestnut mushroom, is cultivated in Japan, China, Europe, and North America. It is a good source of trace minerals and contains free amino acids and vitamins[1, 2]. Cultivation parameters, such as optimal temperature, pH, and cultivation media formula can affect the nutrient composition of P. adiposa [3]. The components isolated from P. adiposa have been reported to be bioactive with multiple health-related functions. The stigmasterol, pentapeptide, adenosine, lectin, water extract, and methanol extract obtained from P. adiposa were reported to have anti-tumor, antioxidant, anti-hypertension, anti-microbial, anti-proliferative, anti-viral, and immunomodulatory activity [1, 4,5,6,7,8,9,10,11].
Polysaccharides are biological macromolecules composed of monosaccharides linked together through glycosidic bonds and widely distributed in animals, plants, and microorganisms. In recent years, much attention has focused on polysaccharides due to their various potential bioactivities such as anti-tumor, antioxidant, anti-inflammatory, anti-microbial, anti-viral, and immunomodulatory activity [12,13,14,15]. The antitumor activity of P. adiposa polysaccharides (PAP) was investigated and showed a prominent effect in counteracting Lewis lung cancer in mice. The investigations have demonstrated that PAP can be an effective antioxidant compound in scavenging oxyradicals, superoxide ions, and 1,1-diphenyl-2- picrylhydrazyl (DPPH) radicals [5, 16].
In our previous work, the results suggested that PAP can inhibit the proliferation of HeLa, Caco2, and HepG2 cells, especially the acid-extracted PAP (SPAP2) showed 91% inhibition of HeLa cells [17]. However, the structural properties have not been reported yet. In this study, the extraction, purification, and structural characterization of the acid-extracted PAP from the fruit body of P. adiposa were investigated. The anti-tumor activities of PAP were explored by 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl tetrazolium (MTT) assay, Annexin V-fluorescein isothiocyanate (FITC), propidium iodide (PI) staining, and flow cytometry.
Materials and methods
Materials
Dried fruit bodies of P. adiposa were grown and provided by the Forest Protection Laboratory in the College of Forestry, Northeast Forestry University (Harbin, China). P. adiposa spawn was cultured in sterilized substrates in polythene bags and then incubated at 30 °C for three months to grow into a fruiting body which was dehydrated using an air-drying oven at 55 °C for two days. HeLa cells (Cat. No. D611002-0001) were purchased from Sangon Biotechnology Co. (Shanghai, China).
Pyridine, acetic anhydride, DEAE-cellulose, Sepharose CL-6B, standard monosaccharides of L-rhamnose (RHA), D-fucose (Fuc), D-arabinose (Ara), D-xylose (Xyl), D-mannose (Man), D-glucose (D-Glc), D-galactose (Gal), and D-inositol (Ino), standard dextrans of known MW (5, 12, 25, 50, 80, 150, 410, 670 kDa) according to the manufacturer were purchased from Sinopharm Chemical Reagent Co. (Shanghai, China). Formic acid, acetic acid, methanol, absolute ethanol, and sodium hydroxide were purchased from Beijing Chemical Works Co. (Beijing, China). Dulbecco’s modified essential medium (DMEM), trypsin (bovine, ~ 10,000 BAEE units/mg protein), trifluoroacetic acid (TFA), dimethyl sulfoxide (DMSO), and hydroxylamine hydrochloride were purchased from Sigma-Aldrich (St. Louis, MO, USA). Sodium phosphate-buffered saline (PBS), fetal bovine serum (FBS), penicillin, streptomycin, and lysine amide were purchased from Gibico (Grand Island, NY, USA). Fluorouracil (5-Fu) was purchased from Lima Biotechnology Co. (Shanghai, China). Annexin V-FITC/PI staining kit was purchased from Sangong Biotechnology Co. (Shanghai, China). All other chemicals and reagents were analytical grade.
Isolation and purification of the crude polysaccharides
The dry fruit bodies of P. adiposa were ground into powder (60 mesh sieved) using a high-speed disintegrator (SL-250ABS, Yunda Co., Yongkang, China). The powder was soaked in petroleum ether (1:4, w/v) for 24 h to remove lipid. To remove the petroleum ether, the mixture was rotary evaporated under a vacuum at 45 °C (RE-5205, Biobase Co., Ji’nan, Shandong, China) before air-drying. The dry mixture was then added to 0.1 mol/L HCl (1:30, w/v) and extracted at 45 °C for 4 h (2×) using a water bath. Extracts were combined and the pH adjusted to 7.0 (Mettler-Toledo Co., Greifensee, Switzerland) and concentrated at 45 °C using the evaporator to approximately 1/3 of the original volume. The concentrated extracts were dialyzed using Spectra/Pro 3 dialysis tubing (Repligen Co., Shanghai, China, with a nominal cut-off of 3.5 KDa according to the manufacturer) against distilled water for 48 h. Then four volumes of 95% (v/v) ethanol were added to the dialysate, and the mixture was allowed to stand overnight at 4 °C. After 10 min centrifugation at 10,000 rpm (25 °C; BKC-VH20R, Biobase Co.), filtration (Whatman # 1 filter paper; Starlab Scientific Co., Xi’an, Shaanxi, China) and lyophilization (Bilang Instrument Co., Shanghai, China), crude SPAP1 was obtained.
The polysaccharide content of Pholiota adiposa was determined by the phenol-sulfuric acid method. Using glucose as the standard, draw the content curve. The polysaccharide content was calculated by measuring the absorbance value of the sample and then substituting it into the standard curve.
where C represents the mass concentration of polysaccharide of Pholiota adiposa calculated according to the standard curve, %; V is the total volume of the polysaccharide extract, mL; N is the final dilution ratio of the solution; m is the total polysaccharide content of Pholiota adiposa, g.
The crude SPAP1 powder was dissolved and then deproteinized using the Sevag method four times[18]. Briefly, 17 mL of Sevag reagent (Vchloroform: V1-butanol = 4:1) was added to 50 mL of SPAP1 solution (10.0 mg/mL), then left for 30 min after vortex treatment. The supernatant was removed and the precipitate was discolored for 3 h at 37 °C using 5% H2O2 (v/v). The deproteinized and discolored SPAP1, termed SPAP2, was obtained after dialysis, alcohol precipitation, and lyophilization previously described for SPAP1 extraction. The SPAP2 was dissolved in distilled water (10.0 mg/mL), and centrifuged (10,000 rpm, 25 °C) for 10 min. The supernatant was obtained after suction filtration (Starlab Scientific Co.) using Whatman # 1 filter paper. Then 50 mL of SPAP2 solution was applied to a DEAE-cellulose (Sigma-Aldrich) column (1.6 × 10 cm) and eluted with distilled water, followed by gradient elution with 1 and 2 mol/L NaCl and collected in 10 mL tubes (60 tubes for each elution solution) to obtain the elution curves. The same fractions were combined and concentrated using rotary evaporation and dialyzed against distilled water for 48 h. Two polysaccharides, named SPAP2-1 and SPAP2-2, were obtained after lyophilizing the dialysate.
Structure characterization of SPAP2
UV and MW analysis
Proteins and nucleic acids were detected using an ultraviolet spectrophotometer (model 752, Shanghai Optical Instrument Factory Co., Shanghai, China) as described by Yuan et al. [19]. Briefly, two fractions were dissolved and diluted to 2 mg/mL respectively, and they were scanned from 200 to 1200 nm.
The MW of SPAP2-1 and − 2 were determined using gel permeation chromatography (GPC) using a Shimadzu LC-10 A high-performance liquid chromatography (HPLC) system (Shimadzu Corp., Tokyo, Japan) fitted with a Sepharose CL-6B column (1.5 × 100 cm) (Sigma-Aldrich) and a differential refractive index detector (RID) (Shimadzu). The sample was dissolved in ultrapure water (0.2%, w/v) and centrifuged at 10,000 rpm, 25 °C, for 5 min, the supernatant (20 µL) was then applied to the column at 0.6 mL/min at 35 °C (column), 7 MPa. The running time was 30 min, and 0.15 mol/L NaCl solution was used as the eluent at 0.15 mL/min. The retention time was obtained using this method. Dextrans of different MW (5, 12, 25, 50, 80, 150, 410, 670 kDa) according to the manufacturer (Sinopharm Chemical Reagent Co.) were used as standards for MW measurements.
Monosaccharide composition
Monosaccharide composition was determined using gas chromatography (GC) using the method of Yuan et al. [19] with slight modifications. L-rhamnose, D-fucose, D-arabinose, D-xylose, D-mannose, D-glucose, and D-galactose (Sigma-Aldrich) were the monosaccharide standards. The polysaccharide samples (5 mg) were degraded with 4 mL TFA (2 mol/L) at 120 °C for 3 h, then extra TFA was removed by evaporating three times after adding methanol. Hydroxylamine hydrochloride (10 mg) and 0.5 mL of pyridine were added into the degraded product with 7 mg inositol used as the internal reference, mixed by vortex, and then incubated at 90 °C in a water bath for 30 min. After incubation, the mixture was cooled to room temperature and acetylated using 0.5 mL of acetic anhydride in a water bath at 90 °C for 30 min. All standard sugars were converted to their acetylated derivatives using the above method.
The acetylated mixture was analyzed using a GC system (Agilent 6890 N, Wilmington, DE, USA) equipped with a fused silica capillary column (RTX-1701, 0.25 μm×30.0 m, Supelco, Bellefonte, PA, USA) and a flame ionization detector (FID) (Agilent), with nitrogen (99.9% of purity) as the carrier gas. The temperature program was: 180 to 220 °C at 5 °C/min, holding for 5 min, and then to 280 °C at 10 °C/min holding for 20 min. The monosaccharide composition was determined based on the retention time of the monosaccharide standards. The area normalization method was used to calculate the molar proportion of monosaccharides in SPAP2-1 and 2.
Methylation analysis
Polysaccharides were methylated by the modified method of Ciucanu (1984). Dried polysaccharide (5 mg) was dissolved in a vial with 0.5 mL of anhydrous DMSO (99.9% purity) at room temperature, followed by the addition of 0.5 mL of NaOH-DMSO suspension. A total of 1 mL of methyl iodide was added dropwise to the mixture and the mixture was stirred in an ice bath for 30 min. The resulting mixture was dialyzed and the retentate was subjected to hydrophilic treatment. The methylation step was repeated twice before washing three times with water and dichloromethane (1:1, v/v). The dichloromethane was removed by vacuum distillation and the permethylated polysaccharide was hydrolyzed and converted to partially methylated aldehyde condensate (PMAA) and analyzed by an Agilent Technologies 7890B GC and 5977B MSD system equipped with an Agilent DB-35MS capillary column (Wilmington, DE, USA). The temperature program was 140 °C (2 min) to 170 °C (3 min) at 5 °C/min, to 180 °C (5 min) at 1 °C/min, to 220 °C (1 min) at 3 °C/min and to 295 °C (3 min) at 20 °C/min. Gas chromatography uses helium as a carrier.
FT-IR
The freeze-dried sample was ground and mixed with spectroscopic grade KBr powder. The mixture was pressed into a 1 mm pellet for FT-IR analysis with an Affinity-1 IR spectrophotometer (Brucker Technology Co., Beijing, China) within 4000–400 cm− 1 at a resolution of 4 cm− 1 [20, 21].
NMR spectroscopy analysis
The sample (about 50 mg) was dissolved in 1 mL D2O in a NMR tube. 1 H and 13 C NMR spectra were recorded at 600 and 150 MHz, respectively, on a Bruker NMR spectrometer (AVANCE III HD 600, Bruker, Germany) at 25 °C. Two-dimensional spectra (heteronuclear single quantum coherence, HSQC, and heteronuclear multiple bond correlation, HMBC) were recorded using the standard Bruker pulse procedures.
Cell inhibitory assay
The inhibitory effects of SPAP2 on the tumor cell were determined using the colorimetric 3-(4, 5-dimethyl-2-thiazolyl)-2, 5-diphenyltetrazolium bromide (MTT) assay using the method of Li et al[22]. with slight modifications. HeLa cells were cultured with DMEM containing 10% FBS, penicillin (100 U/mL), and streptomycin (100 mg/mL) in a 37 °C, 5% CO2 humidified incubator [23, 24].
Cells (100 uL/well) in logarithmic phase were treated with 0.25% trypsin solution at 5 × 104 cells/mL suspension in 96-well plates. Three groups were set as blank (DMEM only), control (DMEM and cells), and SPAP2 (DMEM, SPAP2 solution, and cells). SPAP2 solutions were added to final concentrations of 0.25, 0.5, 0.75, 1.0, 2.0, 3.0, 4.0, 5.0, and 6.0 mg/mL. Six repeats were conducted for each group. All groups were incubated for 24, 48, and 72 h.
After incubation, 20 µL of MTT was added to each well, and groups were incubated for 4 more h. After the second incubation, cells were centrifuged (1,000 rpm) for 3 min at 4 °C (TGL-16 K, Xi’an Yima Opto-electrical Technology Co., Xi’an, Shanxi, China) and precipitates were collected followed by the addition of 150 µL of DMSO. The absorbance of each well was measured at 490 nm using an MR-96 A microplate reader (Mindray, Shenzhen, Guangdong, China). The proliferation rate and inhibition rate were calculated using Eqs. (1) and (2), and they were used to determine the IC50 concentration for further research.
Cell cycle distribution assay
HeLa cell cycle distribution was determined using propidium iodide (PI) staining and flow cytometry as previously described [25]. The control group (HeLa cells) and the SPAP2 group (SPAP2 treated HeLa cells) were used. Cells in the logarithmic phase were treated using the 0.25% trypsin solution at a density of 1–5 × 106 cell/mL and pelleted by centrifugation at 1,000 rpm for 3 min at 4 °C (Xi’an Yima centrifuge), washed twice in pre-cooled PBS (pH 7.4), and centrifuged again. Cells were then fixed in a dilution of pre-cooled PBS and ethanol (70%, v/v) at 4 °C for 2 h, the cell pellet was washed once in pre-cooled PBS and re-centrifuged. The pellet was stained using 100 µL of 1 mg/mL propidium iodide solution and incubated at 4 °C for 30 min in the dark. Then 2 µL of Triton X-100 was added at the 25th min to assist with staining. The stained cells were centrifuged at 1,000 rpm for 3 min at 4 °C and re-suspended in 1 mL of pre-cooled PBS for flow cytometric analysis using FACSAria flow cytometry (Becton- Dickinson, Mansfield, MA, USA) and Modfit software (Version 3.2, Bio-Rad, Hercules, CA, USA).
Assay of apoptosis induced by purified SPAP2
Control (HeLa cells), SPAP2 (SPAP2 treated HeLa cells), and the positive control (5-Fu group, 0.15 mg/mL fluorouracil treated HeLa cells) were used to study HeLa cell apoptosis induced using purified SPAP2. Cells in the logarithmic phase were treated with 0.25% trypsin solution (without EDTA) after reaching 1–5 × 105 cells/mL and pelleted by centrifugation at 1,000 rpm for 3 min at 4 °C, washed twice in pre-cooled PBS (pH 7.4), centrifuged again to collect cells. Then 100 µL of Binding Buffer was added before straining with 5 µL of Annexin V-FITC (Sangon Biotech) and 5 µL of propidium iodide solution (Sangon Biotech) in the dark for 10 min. Following the staining, another 400 µL of Binding Buffer has added to re-suspend the cells. The proportions of apoptotic cells were detected using flow cytometry, and the results were analyzed using the Modfit software.
Statistical analysis
Cell inhibition assays were carried out with 6 repeats. Cell cycle distribution and apoptosis assays were carried out in triplicate. The results were expressed as mean ± SD, and P < 0.05 was considered statistically significant. The data were analyzed using the T-test by SPSS 13 software (SPSS Inc., Chicago, IL, USA).
Results and discussion
Isolation and purification of polysaccharides
After dialysis and ethanol precipitation, the yield of the crude polysaccharide SPAP1 was 9.26% ± 0.81%. SPAP1 was deproteinized and discolored to produce SPAP2 (yield of 6.61% ± 0.56%), which was further fractionated on a DEAE-cellulose column and target fractions SPAP2-1 (yield of 62.41% ± 0.43%) and SPAP2-2 (yield of 3.82% ± 0.64%) was obtained.
MW and monosaccharide composition of SPAP2-1 and − 2
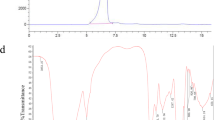
After purification with a DEAE-Sepharose fast flow column (Fig. 1 A), two major fractional peaks, eluted with ultrapure water and 0.15 M NaCl, were obtained.

Elution curve (A) and UV spectra (B) of SPAP2-1 and -2
In this study, we mainly focused on the SPAP2-1 fraction and SPAP2-2 fraction. Ultraviolet spectra (Fig. 1B) of these two fractions showed no absorbance at 260 or 280 nm, suggesting the absence of proteins and nucleic acids[26]. As shown in Fig. 1 A, a single absorption peak was observed and the average molecular mass of SPAP2-1 was estimated to be 19 kDa according to the dextran standard curve. Since both SPAP2-1 and SPAP2-2 were eluted as a single and symmetrical peak, this indicated that both SPAP2-1 and SPAP2-2 may be a homogeneous polysaccharide. The molecular weight of a polymer and its retention time (or efflux volume) is inversely proportional, the smaller the molecular weight, the later the peak emergence time.
The monosaccharide compositions of SPAP2-1 and -2 using GC are shown in Fig. 2; Table 1. It suggested that both fractions were composed of mannose, glucose, and galactose, and glucose was the predominant monosaccharide in both fractions (especially in SPAP2-2), which was similar to the findings of [27].

Gas chromatogram of SPAP2-1 and -2. (A) A mixture of standard monosaccharides, (B) the monosaccharide composition of SPAP2-1, and (C) the monosaccharide composition of SPAP2-2
Methylation and GC–MS analysis
PAP2-1 and -2 were methylated using the modified method of Ciucanu and the obtained PMAA were analyzed by GC-MS. The related linkage patterns and the molar ratio of the glycoside residues of SPAP2-1 and -2 were summarized in Table 2. The GC-MS separation chromatograms of SPAP2-1 and SPAP2-2 are shown in Fig. 3.

Total ion chromatogram (TIC) of SPAP2-1 and -2
The results of the methylation analysis showed that both fractions mainly consisted of 1,4-Glcpand 1,3,4-Glcp, and terminal Glcp and terminal mannose were also presented. However, the proportions of other glycosidic linkages in SPAP2-1 and -2 showed a significant difference. In both polysaccharides, glucose was also linked by a 1,3- and 1,3,6- linked glycosidic bond. Mannose was also linked by (1→3)glycosidic bond. Galactose was linked by 1,6- and 1,2,6- linked glycosidic bonds. These results suggested that SPAP2-1 and -2 were heteroglycans with multiple linkages in the backbone and had glucose and galactose as the branching points with glucose and mannose as the terminal sugars.
FT-IR spectrum
The FT-IR spectrum of SPAP2-1 and -2 are presented in Fig. 4. Both fractions showed strong absorption peaks near 3411 cm− 1, which was attributed to the stretching vibrations of O-H. The weak absorption bands at 2927/2929 cm− 1 are related to the C-H stretching vibrations of free sugars, which are characteristic groups of carbohydrates [28]. There were no absorption peaks in both fractions in the range of 1700–1750 cm− 1 or around 1590 cm− 1, indicating the absence of uronic acid [29]. Furthermore, it was reported that the band at 1637 cm− 1 was related to the bound water and that the 1417 cm− 1 band represented C-H bending vibrations in SPAP2-1 and -2 molecules [30, 31].

FT-IR spectra of SPAP2-1 and − 2
Each fraction had a specific absorption in the 1200 − 1000 cm-1 region, which is dominated by ring vibrations overlapped with stretching vibrations of C-OH side groups and with the C-O-C glycosidic bond vibration [32]. The absorption peaks at 1153/1079/1024 cm-1 were typical for a pyranose ring [33]. In particular, the 1079 cm-1 peak was characteristic of β-type glycosidic linkages[28]. The peaks at 850 and 575 cm-1 were characteristic of α-type glycosidic linkages and mannose, respectively[34, 35]. The results suggested that SPAP2-1 and - 2 had typical functional groups, bonds, and linkages of polysaccharides.
NMR spectrum
The 1D and 2D NMR spectra of the SPAP2-1 and -2 fractions were analyzed. Based on monosaccharide composition analysis, FT-IR analysis, methylation analysis, and NMR analysis, the major glycosyl residues were assigned. The 1H, 13C, HSQC, HMBC, and COSY spectra of SPAP2-1 were shown in Fig. 5.

NMR spectra of SPAP2-1 in D2O. 1H and 13C NMR spectra (A). HSQC (B), HMBC (C), and COSY (D) NMR spectra of SPAP2-1
The 600 MHz 1H NMR spectrum (Fig. 5 A) of SPAP2-1 showed three anomeric proton signals, indicating three residues. The strong δ5.39 ppm signal was assigned to 1,4-α-D-Glcp (A), two medium δ5.13 and 5.00 ppm signals were assigned to terminal-β-D-Glcp (B) and 1,3,4-α-D-Glcp (C), respectively [36,37,38]. In 151 MHz 13 C NMR spectrum (Fig. 5 A) of SPAP2-1, only two anomeric signals appeared at δ101.74, 100.05 ppm, which were assigned to C-1 of 1,3-β-D-Glcp (D) and 1,4-α-D-Glcp (A), respectively[39]. Accordingly, the cross-peaks at δ5.39/100.05, 5.13/98.70, 5.00/98.15, and 4.89/101.74 ppm in HSQC (Fig. 5B) confirmed these 1 H and 13 C NMR results. The signals at δ61.22 and 77.64 ppm belonged to C-6 of 1,4-α-D-Glcp (A) and terminal-β-D-Glcp (B), respectively. The strong signals at δ73.10, 73.49, and 71.74 ppm were assigned to C-5, C-4, C-3, and C-2 of 1,4-α-D-Glcp (A), respectively [40, 41]. Resonances at δ70.57, 68.69, and 66.98 ppm were assigned to C-5, C-3, and C-2 of terminal-β-D-Glcp (B), respectively. As for SPAP2-2 (data not shown), only one significant anomeric proton signal showed at δ5.38 ppm in the 1 H spectrum, which was assigned to 1,4-α-D-Glcp (A). And the resonance appeared at δ 99.92 ppm in the 13 C spectrum belonging to C-1 of 1,4-α-D-Glcp (A). The other chemical shifts failed to assign due to the poor quality of the 2D spectra. Both fractions showed no shifts around 170 ppm in the C NMR spectrum, indicating the absence of uronic acid, which confirmed the FT-IR results. FT-IR analysis showed that SPAP2-1 is a typical α-pyran type polysaccharide. 1 H-NMR and 13 C-NMR analysis demonstrated that the sugar chain structure of SPAP2-1 is [α-Rha (1→3)-]n, and GC-MS analysis showed that rhamnose is located at the non-reducing end of the sugar chain, while glucose is located at the non-reducing end or in the sugar chain in 1,2,6- and 1,3,6-linked forms. Our study clearly illuminates the primary structure of SPAP2-1, but the 3D structure of SPAP2-1and its structure-function relationship is a future challenge.
Cell inhibitory assay of SPAP2-1 and − 2
The antitumor activities of SPAP2-1 and − 2 had determined by calculating the inhibition rate of HeLa cells in vitro. In Fig. 6, the antiproliferative effects of SPAP2-1 and − 2 on HeLa cells were concentration- and time-dependent. As for concentration, the inhibition rate of both fractions increased significantly to 0.25-4 mg/mL, and this trend slowed down to 4.0–6.0 mg/mL. Moreover, polysaccharides from fungus, Phellinus gilvus, inhibited the proliferation of HeLa and SGC-7901 cells in a dose-dependent manner and such inhibitory effects were efficient at low concentrations.

The effect of SPAP2-1 and SPAP2-2 on cell proliferation activity of Hela cells. Hela cells were treated with SPAP2-1 at different concentration gradients and at different times (A). Hela cells were treated with SPAP2-2 at different concentration gradients and at different times (B). The error bars represent standard deviations of means from three repeat experiments.
Meanwhile, in the aspect of time, the 48 h samples showed stronger inhibitive effects than 24 and 72 h. SPAP2-1 showed higher antiproliferative activities against HeLa cell (P < 0.05) with an IC50 concentration of 0.25 mg/mL, which is significantly lower than that of SPSP2-2 (5.5 mg/mL).
Effects of SPAP2-1 on HeLa cell cycle distribution
The effect of SPAP2-1 on HeLa cell cycle distribution is shown in Fig. 7 A. Compared with the control group, the ratio of HeLa cells in the G1 phase grew significantly (P < 0.05). Consequently, cell numbers in the S and G2/M phases decreased. The percentage of HeLa cells in the S phase decreased significantly after treatment. The proportion of SPAP2-1 treated HeLa cells in the G2/M phase was reduced. SPAP2-1 treated HeLa cells increased the population in the G1 phase, hinting that SPAP2-1 might cause G1 phase arrest, thus interfering with the cell cycle in HeLa cells.

Cell cycle distribution and apoptosis induced by SPAP2-1. (A) Effects of SPAP2-1 on cell cycle distribution in HeLa cells, (B) Apoptosis in HeLa cells (control group), (C) Apoptosis induction in SPAP2-1 treated HeLa cells (SPAP2-1 group), and (D) Apoptosis induction in 5-Fu-treated HeLa cells (positive control group)
SPAP2-1 induced HeLa cell apoptosis
To determine the putative mechanism for the SPAP2-1-potential interference with the cell cycle the flow cytometry analysis was done and the results have shown in Fig. 7. In the control group (Fig. 7B), intact HeLa cells accounted for the majority, while necrotic cells, and early and late apoptotic cells were lower. However, the ratios of apoptotic cells in SPAP2-1 and positive control 5-Fu groups were significantly higher (Fig. 7 C and D) than in the control. Furthermore, the necrotic cell increased for the SPAP2-1 group and even more for the 5-Fu group.
Our results also demonstrated that the proportions of each phase had rearranged after SPAP2-1 and 5-Fu treatment. In particular, the apoptotic cell population increased, which indicated that SPAP2-1 played a role in HeLa cell apoptosis induction but less than that of 5-Fu. Research has proved that polysaccharides inhibited the proliferation of cancer cells by inducing apoptosis and G2 /M phase arrest [42,43,44]. Bae et al. also supported this mechanism, which reported that polysaccharides inhibited the proliferation of melanoma cells by leading cells to apoptosis [45]. Zhang et al. reported that polysaccharides from Lycium barbarum lead to significant accumulation of cells in the S phase [46]. Macrophages play a central role in initiating the innate immune response, which leads to the activation of the adaptive response in a later period [47].
Conclusions
This study identified two acid-extracted P. adiposa polysaccharides (SPAP2-1 and -2) that showed antitumor activity. The MW of SPAP2-1 and 2 were 19 and 20 kDa, respectively. Both fractions consisted of mannose, glucose, and galactose. The in vitro cell inhibitory assay suggested that SPAP2-1 had a stronger inhibitory effect on cell development with HeLa cells than SPAP2-2. Further analysis proved that SPAP2-1 had an impact on cell cycle distribution and apoptosis, and consequently inhibited tumor cell development. Therefore, the polysaccharide from P. adiposa should be explored as a potential inhibitor of tumor cell development.
References
Zou, Y., Du, F., Hu, Q., Wang, H.: The structural characterization of a polysaccharide exhibiting antitumor effect from Pholiota adiposa mycelia.Scientific Reports. 9. (2019)
Lee, J.W., Park, M.S., Park, J.-H., Cho, Y., Kim, C., Kim, C.S., Jo, J.W., Lim, Y.W.: Taxonomic Study of the Genus Pholiota (Strophariaceae, Basidiomycota) in Korea. Mycobiology. 48, 476–483 (2020)
Jatuwong, K., Kumla, J., Suwannarach, N., Matsui, K., Lumyong, S.: Bioprocessing of Agricultural Residues as Substrates and Optimal Conditions for Phytase Production of Chestnut Mushroom, Pholiota adiposa, in Solid State Fermentation.Journal of Fungi.6 (2020)
Wang, C.R., Qiao, W.T., Zhang, Y.N., Liu, F.: Effects of Adenosine Extract from Pholiota adiposa (Fr.) Quel on mRNA Expressions of Superoxide Dismutase and Immunomodulatory Cytokines. Molecules. 18, 1775–1782 (2013)
Deng, P., Zhang, G., Zhou, B., Lin, R., Jia, L., Fan, K., Liu, X., Wang, G., Wang, L., Zhang, J.: Extraction and in vitro antioxidant activity of intracellular polysaccharide by Pholiota adiposa SX-02. J. Bioscience Bioeng. 111, 50–54 (2011)
Dulger, B.: Antimicrobial activity of the macrofungus Pholiota adiposa. Fitoterapia. 75, 395–397 (2004)
Chul, K.K., Hyoung, L.D., Hoyu, K.J., Yu, H.E., Sik, P.G., Soo, L.J.: Production and characterization of antihypertensive angiotensin I-converting enzyme inhibitor from Pholiota adiposa. J. Microbiol. Biotechnol. 16, 757–763 (2006)
Muk, C.H., Min, L.Y., Hyoung, L.D., Kyung, C.H., Soo, L.J.: Effect of a Pholiota adiposa Extract on Fat Mass in Hyperlipidemic Mice.Mycobiology.34 (2006)
Zhang, G.Q., Sun, J., Wang, H.X., Ng, T.B.: A novel lectin with antiproliferative activity from the medicinal mushroom Pholiota adiposa. Acta Biochim. Pol. 56, 415–421 (2009)
Yu, H.E., Lee, D.H., Seo, G.S., Cho, S.M., Lee, J.S.: Characterization of a novel beta-hydroxy-beta-methyl glutaryl coenzyme a reductase-inhibitor from the mushroom, Pholiota adiposa. Biotechnol. Bioprocess Eng. 12, 618–624 (2007)
Wang, C.R., Zhou, R., Ng, T.B., Wong, J.H., Qiao, W.T., Liu, F.: First report on isolation of methyl gallate with antioxidant, anti-HIV-1 and HIV-1 enzyme inhibitory activities from a mushroom (Pholiota adiposa). Environ. Toxicol. Pharmacol. 37, 626–637 (2014)
Yi, Y., Xu, W., Wang, H.X., Huang, F., Wang, L.M.: Natural polysaccharides experience physiochemical and functional changes during preparation: A review.Carbohydrate Polymers.234(2020)
Liu, Z.H., Niu, F.J., Xie, Y.X., Xie, S.M., Liu, Y.N., Yang, Y.Y., Zhou, C.Z., Wan, X.H.: A review: Natural polysaccharides from medicinal plants and microorganisms and their anti-herpetic mechanism.Biomedicine & Pharmacotherapy.129(2020)
Hou, C., Chen, L., Yang, L., Ji, X.: An insight into anti-inflammatory effects of natural polysaccharides. Int. J. Biol. Macromol. 153, 248–255 (2020)
Mohan, K., Muralisankar, T., Uthayakumar, V., Chandirasekar, R., Revathi, N., Ganesan, A.R., Velmurugan, K., Sathishkumar, P., Jayakumar, R., Seedevi, P.: Trends in the extraction, purification, characterisation and biological activities of polysaccharides from tropical and sub-tropical fruits - A comprehensive review.Carbohydrate Polymers.238 (2020)
Yiemei, J.I., Qingxiu, H.U., Chunyu, G., Guang, G.U.O., Rongshu, A.N.: Antioxidation Study of Extra-polysaccharides Extracted from Pholiota adiposa(Fr.). Quel Biotechnol. 17, 29–31 (2007)
Zhou, C., Sun, Y., Li, D.H., Gu, J.L., Gong, J.H.: Inhibitory Effect of Polysaccharides from Pholiota adiposa on Tumor Cell Proliferation. Mod. Food Sci. Technol. 33, 56–62 (2017)
Zhao, J.-L., Zhang, M., Zhou, H.-L.: Microwave-Assisted Extraction, Purification, Partial Characterization, and Bioactivity of Polysaccharides from Panax ginseng.Molecules.24 (2019)
Yuan, S., Xu, C.-Y., Xia, J., Feng, Y.-N., Zhang, X.-F., Yan, Y.-Y.: Extraction of polysaccharides from Codonopsis pilosula by fermentation with response surface methodology. Food Sci. Nutr. 8, 6660–6669 (2020)
Yuan, J., Yan, X., Chen, X., Jiang, X., Ye, K., Xiong, Q., Kong, J., Huang, Y., Jiang, C., Xu, T., Xie, G.: A mild and efficient extraction method for polysaccharides from Sinonovacula constricta and study of their structural characteristic and antioxidant activities. Int. J. Biol. Macromol. 143, 913–921 (2020)
Wang, Y., Wang, C., Guo, M.: Effects of Ultrasound Treatment on Extraction and Rheological Properties of Polysaccharides from Auricularia Cornea var.Li. Molecules.24 (2019)
Li, Y., Qin, G., Cheng, C., Yuan, B., Huang, D., Cheng, S., Cao, C., Chen, G.: Purification, characterization and anti-tumor activities of polysaccharides from Ecklonia kurome obtained by three different extraction methods. Int. J. Biol. Macromol. 150, 1000–1010 (2020)
Xu, J., Tan, Z.-C., Shen, Z.-Y., Shen, X.-J., Tang, S.-M.: Cordyceps cicadae polysaccharides inhibit human cervical cancer hela cells proliferation via apoptosis and cell cycle arrest.Food and Chemical Toxicology.148 (2021)
Yang, J., Chanok, L., Heekyu, K., Kwon, Y.S., Kim, M.J.: Biological Activities and Phenolic Compound Content of Astilboides tabularis (Hemsl.) Engler Extracts. Curr. Pharm. Biotechnol. 21, 1070–1078 (2020)
Chai, Y., Zhao, M.: Purification, characterization and anti-proliferation activities of polysaccharides extracted from Viscum coloratum (Kom.) Nakai. Carbohydr. Polym. 149, 121–130 (2016)
El-Naggar, N.E.-A., Hussein, M.H., Shaaban-Dessuuki, S.A., Dalal, S.R.: Production, extraction and characterization of Chlorella vulgaris soluble polysaccharides and their applications in AgNPs biosynthesis and biostimulation of plant growth.Scientific Reports.10 (2020)
Wang, Z., Luo, D., Liang, Z.: Structure of polysaccharides from the fruiting body of Hericium erinaceus Pers.Carbohydrate Polymers.57(2004)
Kozarski, M., Klaus, A., Niksic, M., Jakovljevic, D., Helsper, J.P.F.G., Van Griensven, L.J.L.D.: Antioxidative and immunomodulating activities of polysaccharide extracts of the medicinal mushrooms Agaricus bisporus, Agaricus brasiliensis, Ganoderma lucidum and Phellinus linteus. Food Chem. 129, 1667–1675 (2011)
Zeng, W.-C., Zhang, Z., Gao, H., Jia, L.-R., Chen, W.-Y.: Characterization of antioxidant polysaccharides from Auricularia auricular using microwave-assisted extraction.Carbohydrate Polymers.89 (2012)
Zhao, G.H., Kan, J.Q., Li, Z.X., Chen, Z.D.: Structural features and immunological activity of a polysaccharide from Dioscorea opposita Thunb roots. Carbohydr. Polym. 61, 125–131 (2005)
Parker, F.S.: Application of IR Sepectroscopy in Biochemistry, Biology, and Medicine. Plenum, New York (1971)
Zhu, Y., Li, Q., Mao, G., Zou, Y., Feng, W., Zheng, D., Wang, W., Zhou, L., Zhang, T., Yang, J., Yang, L., Wu, X.: Optimization of enzyme-assisted extraction and characterization of polysaccharides from Hericium erinaceus. Carbohydr. Polym. 101, 606–613 (2014)
Yang, W., Pei, F., Shi, Y., Zhao, L., Fang, Y., Hu, Q.: Purification, characterization and anti-proliferation activity of polysaccharides from Flammulina velutipes. Carbohydr. Polym. 88, 474–480 (2012)
Guo, L., Xie, J., Ruan, Y., Zhou, L., Zhu, H., Yun, X., Jiang, Y., Lue, L., Chen, K., Min, Z., Wen, Y., Gu, J.: Characterization and immunostimulatory activity of a polysaccharide from the spores of Ganoderma lucidum. Int. Immunopharmacol. 9, 1175–1182 (2009)
Khaskheli, S.G., Zheng, W., Sheikh, S.A., Khaskheli, A.A., Liu, Y., Soomro, A.H., Feng, X., Sauer, M.B., Wang, Y.F., Huang, W.: Characterization of Auricularia auricula polysaccharides and its antioxidant properties in fresh and pickled product. Int. J. Biol. Macromol. 81, 387–395 (2015)
Li, S.-G., Wang, D.-G., Tian, W., Wang, X.-X., Zhao, J.-X., Liu, Z., Chen, R.: Characterization and anti-tumor activity of a polysaccharide from Hedysarum polybotrys Hand.-Mazz. Carbohydr. Polym. 73, 344–350 (2008)
Li, B., Dobruchowska, J.M., Gerwig, G.J., Dijkhuizen, L., Kamerling, J.P.: Structural investigation of water-soluble polysaccharides extracted from the fruit bodies of Coprinus comatus. Carbohydr. Polym. 91, 314–321 (2013)
Zhang, H., Li, W.-J., Nie, S.P., Chen, Y., Wang, Y.X., Xie, M.Y.: Structural characterisation of a novel bioactive polysaccharide from Ganoderma atrum. Carbohydr. Polym. 88, 1047–1054 (2012)
Li, S.-G., Zhang, Y.Q.: Characterization and renal protective effect of a polysaccharide from Astragalus membranaceus. Carbohydr. Polym. 78, 343–348 (2009)
Gonzaga, M.L.C., Ricardo, N., Heatley, F., Soares, S.D.: Isolation and characterization of polysaccharides from Agaricus blazei Murill. Carbohydr. Polym. 60, 43–49 (2005)
Kawagishi, H.: Formolysis of a potent antitumor (1 → 6)-β-d-glucan-protein complex from Agaricus blazei fruiting bodies and antitumor activity of the resulting products. Carbohydr. Polym. 12, 393–403 (1990)
Liu, C., Sun, Y., Mao, Q., Guo, X., Li, P., Liu, Y., Xu, N.: Characteristics and Antitumor Activity of Morchella esculenta Polysaccharide Extracted by Pulsed Electric Field.International Journal of Molecular Sciences.17 (2016)
Mei, Y., Zhu, H., Hu, Q., Liu, Y., Zhao, S., Peng, N., Liang, Y.: A novel polysaccharide from mycelia of cultured Phellinus linteus displays antitumor activity through apoptosis. Carbohydr. Polym. 124, 90–97 (2015)
Ren, D., Jiao, Y., Yang, X., Yuan, L., Guo, J., Zhao, Y.: Antioxidant and antitumor effects of polysaccharides from the fungus Pleurotus abalonus. Chemico-Biol. Interact. 237, 166–174 (2015)
Bae, J.S., Jang, K.H., Yim, H., Jin, H.K.: Polysaccharides isolated from Phellinus gilvus inhibit.melanoma growth in mice. Cancer Lett. 218, 43–52 (2005)
Zhang, M., Chen, H., Jin, H., Li, Z., Zhu, C., Zhang, S.: Effect of lycium barbarum polysaccharide on human hepatoma qgy7703 cells: inhibition of proliferation and induction of apoptosis. Life Sci. 76, 2115–2124 (2005)
Zirk, N.M., Hashmi, S.F., Ziegler, H.K.: The polysaccharide portion of lipopolysaccharide regulates antigen-specific t-cell activation via effects on macrophage-mediated antigen processing. Infect. Immun. 6, 319–326 (1999)
Acknowledgements
This work was supported by the Hundred Million Science and Technology Major Special Project of Heilongjiang Province (2020ZX07B01-3), Fundamental Research Funds for the Central Universities [2572019BA04], Harbin Science and Technology Bureau Science and Technology Innovation Talent Research Fund [2017RAQXJ091], the Natural Science Foundation of Heilongjiang Province [C2015062], and the China Scholarship Commission.
Author information
Authors and Affiliations
Contributions
Dehai Li designed the experiments; Jiao Zhou supervised the research and drafted the manuscript. Jinhua Gong experiment performance and data analysis; Cong Zhou performed the extraction and purification of polysaccharides. Joe M. carried out the chemical and statistical analyses. Changyan Sun and Yangyang Chai revised the manuscript. All authors read and approved the final article.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhou, J., Gong, J., Chai, Y. et al. Structural analysis and in vitro antitumor effect of polysaccharides from Pholiota adiposa. Glycoconj J 39, 513–523 (2022). https://doi.org/10.1007/s10719-022-10065-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10719-022-10065-9