
Abstract
Adenosine monophosphate-activated protein kinase (AMPK) is viewed as a privileged therapeutic target for several diseases such as cancer, diabetes, inflammation, obesity, etc. In addition, AMPK has entered the limelight of current drug discovery with its evolution as a key metabolic regulator. AMPK also plays a key role in the maintenance of cellular energy homeostasis. Structurally, AMPK is a heterotrimeric protein, which consists of three protein subunits (α, β, and γ). The crystal structure of AMPK was solved, and several computational studies including homology modeling, molecular docking, molecular dynamics, and QSAR have been reported in order to explore the structure and function of this diverse therapeutic target. In this review, we present a comprehensive up-to-date overview on the computational and molecular modeling approaches that have been carried out on AMPK in order to understand its structure, function, dynamics, and its drug binding landscape. Information provided in this review would be of great interest to a wide pool of researchers involved in the design of new molecules against various diseases where AMPK plays a predominant role.

ᅟ
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
AMPK
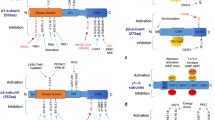
5′-Adenosine monophosphate-activated protein kinase (AMPK) is a serine/threonine-specific protein kinases (EC 2.7.11.11) [1–3]. It was discovered while referring to metabolic activation of acetyl-CoA carboxylase (ACC) and HMG-CoA reductase (HMGR) by phosphorylation. AMPK controls energy metabolism and appetite. Structurally, AMPK is composed of (i) a α catalytic subunit (ii) a scaffolding β subunit, and (iii) a nucleotide-binding γ subunit (Fig. 1) [4]. AMPK activates a wide range of proteins containing evolutionarily conserved sequences. It is expressed highly in the liver, brain, and skeletal muscle. AMPK stimulates the transport of glucose and oxidation of fatty acids in skeletal muscle, and it also decreases glucose output and increases fatty acid oxidation in the liver. AMPK is regulated by AMP/ATP ratio and upstream kinases [1–3].

Molecular assembly of AMPK: Catalytic α subunit (blue), β subunit (green), and γ subunit (orange red) with a glycogen and nucleotide-binding domain and b Glycogen and inhibitor binding domain
AMPK Activators
Activating Molecules
Several efforts have been made to discover small molecules acting as AMPK activators [5–10]. 5-Aminoimidazole-4-carboxamide-1-β-d-ribonucleoside (AICAR) is currently employed as the reference AMPK activator for biochemical studies. Recently, the development of small molecules as AMPK modulators was studied by emphasizing their pharmacological significance [11]. The activation of AMPK also poses a relook at the pharmacology of existing drugs that alter the biochemical pathways. Salicylate has been recently reported as a direct activator of AMPK [12]. Indirect activation of AMPK by the existing antidiabetic drug (metformin) [1.1] has been proposed and its new application for the treatment of cancer is being clinically investigated [13]. Pharmacology of a number of drugs and natural products, including resveratrol [1.2], berberine [1.3], rosiglitazone [1.4], α-lipoic acid [1.5], bortezomib [1.6], cilostazol [1.7], estrogen [1.8], 3,3′-diindolylmethane (DIM) [1.9], genistein [1.10], lycopene [1.11], ketamine [1.12], pitavastatin [1.13], olanzapine [1.14], and atorvastatin [1.15] are also postulated to act by indirect AMPK activation (Fig. 2) [7, 10, 13–26].

These kind of reports may support the claims for considering AMPK as obligatory screening target for predicting the side effects of drugs. Though the current drug design strategy seeks AMPK activation, a contradictory approach may be required at some instances when a tumor adopts activated AMPK consequences for its growth under stress [26]. In a recent study, it was proposed that inhibition of AMPK activity may provide a therapeutic strategy for amyotrophic lateral sclerosis (ALS) [27].
Activation Mechanism
AMPK is activated by phosphorylation of the Thr172 moiety by various upstream kinases like liver kinase B1 (LKB1), Ca(2+)/CaM-dependent protein kinase β (CaMKKβ) and transforming growth factor beta-activated kinase 1 (TAK1) (Fig. 3) [28–31]. A 15-mer peptide sequence is identified to be present in the kinase domain as a substrate recognition motif [Φ(X,b)XXS/TXXXΦ], where Φ is a hydrophobic residue and b is a basic residue. Recently the motif has been revised with six additional residues [32]. Phosphorylation of the catalytic domain at the α subunit is induced by conformational changes in the protein brought about by AMP/ADP binding to cystathionine-β-synthase sequences (CBS) located in the N-terminus of γ subunit [28, 33–37]. ATP binding to the CBS domain will antagonize this process via dephosphorylation by protein phosphatases (Fig. 3) [38]. These distinct allosteric modulations in the complex is induced by the binding of adenosine nucleotides, which paved the way for identifying AMPK as plausible target for synthetic activation. Mutational studies in the γ subunit further supported its role in activation of the AMPK complex. The most interesting finding is that the γ2 isomeric complex is more dependent on AMP in comparison with the γ3 isoform to produce active complex conformation [34]. The β subunit in the AMPK complex acts as a basement and contains recognition sequences in the C-terminus to hold α & β subunits [39, 40], whereas, the N-terminus of the β subunit contains a carbohydrate binding module CBD (also referred as glycogen binding domain) for glycogen binding and a myristoylation site for reversible membrane binding [41–44]. Membrane docking is found to be significant for controlling the activity of the AMPK complex. It is mentioned that point mutation at the myristoylation site produced a 4-fold increase in AMPK activity [44]. Synthetic activator binding studies at the interface of kinase-CBD domains explored a significant hydrophobic cavity (Fig. 4), which was postulated as binding site for natural ligand. However, no such natural substrate has been discovered so far [45].

Activation mechanism of AMPK

Hydrophobic part of leucine residue interacting with cyclodextrin (red) in β subunit of AMPK
AMPK Crystal Structures
Functional behavior of AMPK by an energy sensing mechanism allured the structural biologists to elucidate its structure. Several crystal structures of AMPK have been published and so far 53 structures have been reported in Protein Data Bank (PDB) (Table 1). Structural studies indicate that AMPK is a heterotrimeric supramolecule [41, 67]. The protein is composed of three different subunits α, β, and γ [53, 68]. Two isoforms for α as well as β and four isoforms for γ (α1, α2, β1, β2, γ1, γ2, γ3, γ4) have been identified so far. Individual crystal structure for subunits α1 (4RED) [66], α2 (2H6D) [49], β1 (1ZOM) [46], β2 (2FI5) [47], and γ1 (2UV4) [55] were reported in PDB. Full-length ligand bound complex crystal structures are available for α2β1γ1 (4CFE), α1β2γ1 (2V8Q), and α1β1γ1 (4EAK) only, out of 12 possible forms. The α subunit of AMPK is relatively bigger than the others and includes a conventional protein kinase domain toward its N-terminal [68].
The low-resolution crystal structure of human α1β2γ1 holo-AMPK in complex with AMP and cyclodextrin has been reported [66]. The study describes AMP-induced compact and ATP-induced extended conformational changes. The conformational changes suggest ATP as a competitor for AMP as well as an allosteric modulator [66]. In addition, the structure of the kinase domain from the α2 subunit of AMPK was also solved [49]. The study suggested the changes in the ATP site and catalytic loop which are involved in AMPK regulation. The catalytic loop was found to be stabilized by the conserved residues of Ser/Thr protein kinase [49]. The crystal structure of cystathionine β-synthase from γ subunit of AMPK in complex with AMP and AICAR was published [55]. The structure showed a fold for CBS domain pairs. The reported structure suggested the formation of recognition motif from (311–317) for the ribose phosphate pathway [55].
AMPK as a Drug Target
AMPK is primarily involved in the elevation of cellular ATP levels on demand [69]. Regulatory functioning of AMPK with respect to the cell requirements for ATP makes this protein a potential resource for understanding the cellular energy balance mechanism. AMPK is activated when cells demand more energy to perform. Furthermore, it is also activated when the concentration of AMP/ADP is more than that of ATP [69]. The feedback regulation of cellular ATP levels during metabolic stress or increased ATP consumption (muscle contraction) is achieved by the inhibition of ATP-consuming anabolic pathways (protein, fatty acid, and glycogen synthesis) and activation of energy-producing catabolic pathways. Current research on AMPK claims that deregulation of energy levels often occurring during metabolic syndromes can be corrected with the aid of AMPK activation. Several studies in this context are carried out and the results support the role of AMPK as an emerging target for the treatment of metabolic disorders like diabetes, inflammation, obesity, and cancer (Table 2) [79–85].
Currently, AMPK is being evaluated as a potential target for the treatment of diabetes, cancer, inflammation, cardiac dysfunction, neurodegenerative disorders like ALS, obesity, and fatty liver fibrosis [10, 70–78]. For the development of specific agents and to avoid unwanted effects, more details on tissue distribution of AMPK isoforms and their binding affinities are required. Even more studies are required to understand the role of GBD in AMPK function, role of the β subunit myristoylation on phosphorylation, and elucidating the mechanism of allosteric activation by AMP/ADP. Identification of AMPK phosphatases and their physiological role is also need to be addressed. Overall, there is a demand for in-depth studies on supramolecular behavior of AMPK substrate, AMPK activator, and AMPK inhibitor. A few articles have been published to understand the structure and pharmacological status of AMPK. The current review is focused on computational studies conducted so far to understand the structure and function of AMPK. Further, the review briefly discusses the molecular modeling tools to comprehend the molecular pharmacology of AMPK for rational drug discovery.
Molecular Modeling Studies on AMPK
Computational Approaches Aimed at Understanding the Three-Dimensional Structural and Functional Features of AMPK
Several computational approaches have been reported to explore the three-dimensional structural and functional features of AMPK (Table 3). These include homology modeling [87], molecular docking [97–100], molecular dynamics [97, 98, 101], virtual screening [101], quantitative structure activity relationships [95], etc. These studies assisted in the identification of novel hits against various diseases where AMPK was found to be the critical target.
Homology Modeling
Homology modeling is used to identify the preferred conformation of target protein from known structures. The study is usually employed in the absence of X-ray crystal structure of target protein [102]. The homology model for human AMPK has been reported [86]. The crystal structure of yeast AMPK (PDB: 2OOX) was employed as a suitable template along with the sequences obtained from Swiss Prot (accession nos. Q13131, Q9Y478, and P54619) for homology modeling using Discovery Studio (www.accelrys.com). The model was validated by computing the profile 3D score for each of the residues of sequence. The model was further validated by molecular docking approaches. The profile 3D score measures the compatibility of the 3D structure of the protein with the sequence. The study identified the binding site of human AMPK based on the active site residues (Thr200, Ala205, Val225, Ser226, Ala227, Arg299, Ile312, Ser314, Ser316, and Asp317) of yeast AMPK [86].
Recently, rat AMPK was also employed to build a homology model for AMPK (Uniprot Knowledgebase: Q13131, human) (PDB: 2Y94) [25]. The identity between these two structures was found to be 89.4 % and similarity was found to be (89.6 %). The Ramachandran plot [103] showed 91.4 % of residues of the homology model in the favored region, 5.9 % residues in the allowed region, and 2.4 % residues in the disallowed region. The results of the Ramachandran plot indicated the reliability of the homology [103]. Further, the homology model was employed to conduct molecular dynamics and virtual screening studies. The study was conducted to identify the AMPK activators [25].
Homology modeling and in silico mutagenesis experiments were conducted to generate a structural model for cystic fibrosis transmembrane conductance regulator AMP-activated protein kinase (CFTR-AMPK) and to determine their interactions [87]. The sequence of AMPK (α1, id: Q13131; β1, id: Q9Y478; γ2, id: Q9UGJ0) was obtained from SwissProt/Trembl database. BLAST and HHPRED programs were used to identify the suitable template [87]. The crystal structures of mouse cAMP-dependent protein kinase (PDB: 1JBP:E), yeast proteins (PDB: 2OOX:B), and rat 5′-AMP-activated protein kinase were used as the templates. The homology model was energetically minimized using AMBER7 99 force field and simplex initial optimization by Powel method implemented in SYBYL8.0 [104, 105]. The study revealed the possibility of conformational changes while AMPK binds with CFTR [87]. A different study reported the homology model from the human protein sequence of AMPK α1 [91]. MOE-Align module was used to perform chain alignment. A three-dimensional model was constructed using MOE [106] to perform molecular docking and molecular dynamics studies [91].
Computational Approaches to Identify Ligand Binding Site and Binding Mode
Molecular Docking
Molecular Docking is a computational approach for identifying the binding pose between drug and target protein. The approach is one of the valuable types of molecular modeling to predict the strength of association of two molecules in the form of binding affinity. During the process of molecular docking, orientation and conformation of the ligands were determined by computing the binding affinity between two molecules (drug and enzyme/receptor) in the form of different scoring functions [107]. Molecular docking studies were reported on human AMPK using Ligandfit module [86]. Phenylamides, as the ligands, were docked at the binding site of AMPK. The derivatives of phenylamide were generated from de novo evolution. The program LUDI was employed to identify the suitable fragments that can be docked into AMPK. The “Rule of Five” [108] was employed to screen the derivatives for docking. The compounds with high affinity were identified based on docking scores and their key interaction with active site residues. AMP (5.1) and 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) (5.2) were used as a control for docking (Fig. 5). The approach explored pharmacophoric features of binding sites of AMPK. The study identified the anchoring residues (Ser226, Ala227, and Asp299) as hydrogen bond acceptor and (Ser316 and Thr200) as hydrogen bond donors (Table 4). The study suggested conformational changes before the binding of AMP [86].

Molecular docking using the atomic coordinates of AMPK (subunit α2) from a protein databank (PDB: 3AQV) was also reported [88]. The computational program AutoDock and FlexX implemented in SYBYL software were employed to conduct molecular docking [105, 109]. From the library of the database, 118 compounds were selected for in vitro studies since these compounds occupied the top 1,000 rank in molecular docking. The in vitro results were compared with the ranking of docking poses obtained from FlexX and AutoDock [105, 109]. AutoDock program was found to be slightly better in prediction. Ligand binding mode was investigated from the docking pose of AutoDock [109]. The residues (Ala43 and Lys45 in the N lobe, Tyr95 and Val96 in the hinge region, and Leu146, Ala156, and Met164 in the C lobe) were involved in several hydrogen bonding and van der Waals interactions (Table 4). The study also suggested the van der Waals interaction of ligand with Gly loop that may serve for the inhibition of AMPK [88].
The interaction between metformin (1.1) and AMPK was investigated based on molecular docking to identify the role AMPK in energy homeostasis [89]. The crystal structure of AMPK (PDB: 2Y94) was used for molecular docking. Docking was carried out using Internal Coordinate Mechanics (ICM) software (http://www.molsoft.com). Metformin occupied the active site of α-AMPK, β-AMPK, and γ-AMPK. The residues (Arg181, Thr251, and Thr271) in γ-AMPK were involved in strong interactions with metformin (Table 4). In addition, the study also explored the interaction of ZMP (analog of AMP) with AMPK. The results suggested the interaction of ZMP with Bateman domains in the γ subunit of AMPK. The study described the direct interaction of metformin at γ-subunit of AMPK [89].
A bis(4-hydroxy-3-methoxybenzylidene) cyclopentanone (CA) was docked into the active site of AMPK (PDB: 2OOX) for determining the role of AMPK in glucose and lipid metabolism [10]. The binding free energies were computed and interactive residues were analyzed using Ligplot [110]. Arg139, Lys214, Asn311, Asp328, Pro324, and Tyr342 were found to be active site residues involved in ligand binding (Table 4) [10, 90].
Molecular docking was performed on human AMPK α1 using Moloc force field, and the docking complex was energetically minimized using MAB force field [91]. The binding free energy was computed using a scoring function in the Moloc implementation. The study revealed the importance of Glu-96 and Lys-156 in autoinhibition (Table 4) [91].
Molecular Dynamics
Molecular dynamics is a simulation of atoms in molecular structure using force fields [111]. Dynamics can be used to determine the macroscopic thermodynamic properties of the system. The approach calculates the behavior of the system in time dependent manner [111]. Molecular dynamics studies were reported for AMPK using GROMACS (Groningen Machine for Chemical Simulations) [112] to identify small compounds as AMPK activators [25]. The hydrogen bonding interactions from the binding site residues (Gly19, Val90, Ser91, Glu94, Asn138, Asp151, Val18, Asp133, Lys135, and Asp151) with the ligands (Eugenyl beta-d-glucopyranoside, 6-O-cinnamoyl- d-glucopyranose, and AMP) were observed. Furthermore, the ligand pathway was also investigated. The simulation identified the different ligand pathways for Eugenyl beta-d-glucopyranoside, 6-O-cinnamoyl-d-glucopyranose, and AMP while binding with the AMPK protein. The data analysis based on RMSD, RMSF suggested the possible conformational changes of AMPK for different ligands [25].
The mechanism of autoinhibition was communicated from molecular dynamics simulation [92]. The dynamical nature of kinase domain between an inactive unphosphorylated open state and an active or semi-active phosphorylated closed state was studied in GROMACS [112]. The study reported the important role of autoinhibitory domain in regulating inter lobe conformational transition and functional rearrangements to inhibit AMPK. The results suggested the importance of mutations at L341D (mutant 2) and M316E (mutant 3), or elimination of AID altogether, in catalytic activity [92].
Computational study to design the peptide for the activation of AMPK was reported [93]. The peptide was designed from molecular dynamics approaches in GROMACS [112]. The designed peptide with the addition of the myristoyl (Myr) or acetyl (Ac) moiety activated AMPK in muscle cells. The activation mechanism results in reduction of adipose tissue weight, body weight, blood glucose level, insulin level and insulin resistance index [93].
Molecular dynamics study to refine the homology model of AMPK was reported [91]. The simulation conducted on homology model using AMBER force field for 5 ps [104]. The study helped to identify the active site of AMPK. Further, the study helped to perform a molecular docking of AMPK activators. Glu-96 and Lys-156 residues were found to reside autoinhibition domain and relieve the autoinhibition of AMPK [91].
Computational Approaches to Identify Potential Hits/Inhibitors Against AMPK
Virtual Screening
Virtual screening is a computational approach of database searching and was defined as automated evaluation of very large libraries of compounds using theoretical models. Pharmacophore models and rule based descriptors are generally employed for screening. Virtual screening was summarized to identify the potent ligands for AMPK based on (i) Rule of Five [108], and (ii) Molecular docking [86]. The molecule (5.3) was identified as a highly potent agonist to activate AMPK. The molecule (5.3) had the highest docking score (docking score 76.25; Eb: −215.99) as compare to AMP and AICAR. It formed eight hydrogen bonds with AMPK. Thr200, Ala205, Ser226, Ala227, Asp299, Ser314, Ser316, Asp317 were observed as the binding site residues for (5.3) (Fig. 5) [86].
Traditional Chinese Medicine (TCM) Database was screened to identify the potential candidates for activating AMPK [25]. The structure based virtual screening involves absorption, distribution, metabolism excretion and toxicity in association with molecular docking and molecular dynamics. In this study, AMP was used as a control and Eugenyl beta-d-glucopyranoside and 6-O-cinnamoyl-d-glucopyranose were screened as the potential compounds. These compounds were found to have good absorption profiles and formed a hydrogen bonding interactions with Asp151. The study also suggested the induction of conformational changes by these two molecules [25].
Structure based virtual screening was carried out to identify the potent inhibitors of AMPK2 [88]. Consensus scoring method associated with (i) molecular docking, and (ii) Rule of Five was employed in virtual screening. The virtual screening was carried out for the chemical database distributed by Interbioscreen (http://www.ibscreen.com) containing approximately 500,000 synthetic as well as natural products. The approach identified the seven AMPK2 inhibitors with micromolar inhibitory activity. Among the seven compounds, the compound (5.4) (Fig. 5) was found to be highly potent during in vitro AMPK2 kinase assay (IC50 = 1.4 μM). The study described the usefulness of consensus scoring function in virtual screening using modified scoring function [88].
The study on AMPK using mining frequent pattern indicated its impact on metabolic reactions toward energy demand and supply. The study has also been suggested to be useful in understanding the functions and metabolic pathways of AMPK [113]. In another study, phenylamide derivatives were described as a potent agonist for AMPK based on homology modeling and virtual screening. The interaction of the potent ligands with the anchoring residues was found to be same as observed in control [94].
Structure Activity Relationships
The approach relates the 2D/3D properties of the molecule against biological activities of target protein and helps to identify the factors that are involved in biological activity [114]. Structure activity relationship studies were reported to identify xylose derivatives to activate AMPK with improved pharmacokinetic properties [95]. The study identified the relationship of a hydroxyl group in xylose. Further, the study helped to identify a hydrophobic derivative of xylose (5.5) (Fig. 5). This was found to be highly potent in stimulating the rate of hexose transport by increasing the abundance of glucose transporter-4 in the plasma membrane of myotubes [95].
The highly potent multi-target berberine analogues were synthesized from the analysis of structure activity relationship [96]. Further, these compounds were evaluated on AMPK activation as well as up-regulatory low-density lipoprotein receptor (LDLR) gene expression. Out of 14 compounds, the compound (5.6) (Fig. 5) was found to be multi-target therapeutic agent and was suggested to be drug candidate for the treatment of metabolic syndrome [96].
Conclusions
AMPK is emerging as an indisputable target for correcting several metabolic syndromes that majorly include diabetes, obesity, and cancer. Both the academia and industry are extremely interested in understanding the biological consequences of AMPK activation to a deeper extent. The phenomenon of AMPK inactivation and its pharmacological significance is another exciting problem to be solved. Molecular modeling studies are facilitating their intuition by revealing the structural, functional, and conformational changes of AMPK while binding to its substrates. However, there are only limited number of computational studies in the literature. This review comprehends the structural and functional mechanism of AMPK from the computational studies. Further, this study may assist in the development of multi-therapeutic agents for the several human ailments by choosing AMPK as a therapeutic target.
References
Carlson, C. A., & Kim, K. H. (1973). Regulation of hepatic acetyl coenzyme A carboxylase by phosphorylation and dephosphorylation. Journal of Biological Chemistry, 248, 378–80.
Beg, Z. H., Allmann, D. W., & Gibson, D. M. (1973). Modulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity with cAMP. Biochemical and Biophysical Research Communications, 54, 1362–1369.
Carling, D. (2004). The AMP-activated protein kinase cascade—a unifying system for energy control. Trends in Biochemical Sciences, 29, 18–24.
Hardie, D. G., & Carling, D. (1997). The AMP-activated protein kinase—fuel gauge of the mammalian cell? European Journal of Biochemistry, 246, 259–273.
Tripodi, F., Pagliarin, R., Fumagalli, G., Bigi, A., Fusi, P., Orsini, F., Frattini, M., & Coccetti, P. (2012). Synthesis and biological evaluation of 1,4-diaryl-2-azetidinones as specific anticancer agents: activation of adenosine monophosphate activated protein kinase and induction of apoptosis. Journal of Medicinal Chemistry, 55, 2112–2124.
Zhang, W., Wu, R., Zhang, F., Xu, Y., Liu, B., Yang, Y., Zhou, H., Wang, L., Wan, K., Xiao, X., & Zhang, X. (2012). Thiazolidinediones improve hepatic fibrosis in rats with non-alcoholic steatohepatitis by activating the adenosine monophosphate-activated protein kinase signalling pathway. Clinical and Experimental Pharmacology and Physiology, 39, 1026–1033.
Guo, H., Zhao, H., Kanno, Y., Li, W., Mu, Y., Kuang, X., Inouye, Y., Koike, K., Jiang, H., & Bai, H. (2013). A dihydrochalcone and several homoisoflavonoids from Polygonatum odoratum are activators of adenosine monophosphate-activated protein kinase. Bioorganic and Medicinal Chemistry Letters, 23, 3137–3139.
Guh, J. H., Chang, W. L., Yang, J., Lee, S. L., Wei, S., Wang, D., Kulp, S. K., & Chen, C. S. (2010). Development of novel adenosine monophosphate-activated protein kinase activators. Journal of Medicinal Chemistry, 53, 2552–2561.
Bae, E. J., Yang, Y. M., Kim, J. W., & Kim, S. G. (2007). Identification of a novel class of dithiolethiones that prevent hepatic insulin resistance via the adenosine monophosphate-activated protein kinase-p70 ribosomal S6 kinase-1 pathway. Hepatology, 46, 730–739.
Gruzman, A., Babai, G., & Sasson, S. (2009). Adenosine monophosphate-activated protein kinase (AMPK) as a new target for antidiabetic drugs: a review on metabolic, pharmacological and chemical considerations. The Review of Diabetic Studies, 6, 13–36.
Rana, S., Blowers, E. C., & Natarajan, A. (2015). Small molecule adenosine 5′-monophosphate activated protein kinase (AMPK) modulators and human diseases. Journal of Medicinal Chemistry, 58, 2–29.
Hawley, S. A., Fullerton, M. D., Ross, F. A., Schertzer, J. D., Chevtzoff, C., Walker, K. J., Peggie, M. W., Zibrova, D., Green, K. A., Mustard, K. J., Kemp, B. E., Sakamoto, K., Steinberg, G. R., & Hardie, D. G. (2012). The ancient drug salicylate directly activates AMP-activated protein kinase. Science, 336, 918–922.
Chen, Z., Wang, L., & Chen, Y. (2013). Antitumor mechanism of metformin via adenosine monophosphate-activated protein kinase (AMPK) activation. Zhongguo Fei Ai Za Zhi, 16, 427–432.
Xu, Q., Hao, X., Yang, Q., & Si, L. (2009). Resveratrol prevents hyperglycemia-induced endothelial dysfunction via activation of adenosine monophosphate-activated protein kinase. Biochemical and Biophysical Research Communications, 388, 389–394.
Chang, W., Zhang, M., Li, J., Meng, Z., Wei, S., Du, H., Chen, L., & Hatch, G. M. (2013). Berberine improves insulin resistance in cardiomyocytes via activation of 5′-adenosine monophosphate-activated protein kinase. Metabolism, 62, 1159–1167.
Wang, Y., Li, X., Guo, Y., Chan, L., & Guan, X. (2010). Alpha-lipoic acid increases energy expenditure by enhancing adenosine monophosphate-activated protein kinase-peroxisome proliferator-activated receptor-gamma coactivator-1alpha signaling in the skeletal muscle of aged mice. Metabolism, 59, 967–976.
Tseng, S. Y., Chao, T. H., Li, Y. H., Liu, P. Y., Lee, C. H., Cho, C. L., Wu, H. L., & Chen, J. H. (2015). Cilostazol improves high glucose-induced impaired angiogenesis in human endothelial progenitor cells and vascular endothelial cells as well as enhances vasculoangiogenesis in hyperglycemic mice mediated by the adenosine monophosphate-activated protein kinase pathway. Journal of Vascular Surgery. doi:10.1016/j.jvs.2014.10.103.
Tamrakar, P., Ibrahim, B. A., Gujar, A. D., & Briski, K. P. (2015). Estrogen regulates energy metabolic pathway and upstream adenosine 5′-monophosphate-activated protein kinase and phosphatase enzyme expression in dorsal vagal complex metabolosensory neurons during glucostasis and hypoglycemia. Journal of Neuroscience Research, 93, 321–932.
Li, J., Li, J., Yue, Y., Hu, Y., Cheng, W., Liu, R., Pan, X., & Zhang, P. (2014). Genistein suppresses tumor necrosis factor alpha-induced inflammation via modulating reactive oxygen species/Akt/nuclear factor kappaB and adenosine monophosphate-activated protein kinase signal pathways in human synoviocyte MH7A cells. Drug Design, Development and Therapy, 8, 315–323.
Lin, H. Y., Huang, B. R., Yeh, W. L., Lee, C. H., Huang, S. S., Lai, C. H., Lin, H., & Lu, D. Y. (2014). Antineuroinflammatory effects of lycopene via activation of adenosine monophosphate-activated protein kinase-alpha1/heme oxygenase-1 pathways. Neurobiology of Aging, 35, 191–202.
Ohira, M., Endo, K., Saiki, A., Miyashita, Y., Terai, K., Murano, T., Watanabe, F., Tatsuno, I., & Shirai, K. (2012). Atorvastatin and pitavastatin enhance lipoprotein lipase production in L6 skeletal muscle cells through activation of adenosine monophosphate-activated protein kinase. Metabolism, 61, 1452–1460.
Zong, J., Deng, W., Zhou, H., Bian, Z. Y., Dai, J., Yuan, Y., Zhang, J. Y., Zhang, R., Zhang, Y., Wu, Q. Q., Guo, H. P., Li, H. L., & Tang, Q. Z. (2013). 3,3′-Diindolylmethane protects against cardiac hypertrophy via 5′-adenosine monophosphate-activated protein kinase-α2. PLoS One, 8, e53427.
Xu, S. X., Zhou, Z. Q., Li, X. M., Ji, M. H., Zhang, G. F., & Yang, J. J. (2013). The activation of adenosine monophosphate-activated protein kinase in rat hippocampus contributes to the rapid antidepressant effect of ketamine. Behavioural Brain Research, 253, 305–359.
Ikegami, M., Ikeda, H., Ohashi, T., Ohsawa, M., Ishikawa, Y., Kai, M., Kamei, A., & Kamei, J. (2013). Olanzapine increases hepatic glucose production through the activation of hypothalamic adenosine 5′-monophosphate-activated protein kinase. Diabetes, Obesity & Metabolism, 15, 1128–1135.
Tang, H. C., & Chen, C. Y. (2014). In silico design for adenosine monophosphate-activated protein kinase agonist from traditional chinese medicine for treatment of metabolic syndromes. Evidence-based Complementary and Alternative Medicine. doi:10.1155/2014/928589.
Chuang, H. C., Chou, C. C., SK., K., & Chen, C. S. (2014). AMPK as a potential anticancer target—friend or foe? Current Pharmaceutical Design, 2020, 2607–2618.
Zhao, Z., Sui, Y., Gao, W., Cai, B., & Fan, D. (2015). Effects of diet on adenosine monophosphate-activated protein kinase activity and disease progression in an amyotrophic lateral sclerosis model. Journal of International Medical Research, 43, 67–79.
Hawley, S. A., Davison, M., Woods, A., Davies, S. P., Beri, R. K., Carling, D., & Hardie, D. G. (1996). Characterization of the AMP-activated protein kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. Journal of Biological Chemistry, 271, 27879–27887.
Hong, S. P., Leiper, F. C., Woods, A., Carling, D., & Carlson, M. (2003). Activation of yeast Snf1 and mammalian AMP-activated protein kinase by upstream kinases. Proceedings of the National Academy of Sciences of the United States of America, 100, 8839–8843.
Hawley, S. A., Pan, D. A., Mustard, K. J., Ross, L., Bain, J., Edelman, A. M., Frenguelli, B. G., & Hardie, D. G. (2005). Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metabolism, 2, 9–19.
Hardie, D. G. (2004). The AMP-activated protein kinase pathway—new players upstream and downstream. Journal of Cell Science, 117, 5479–5487.
Scott, J. W., Norman, D. G., Hawley, S. A., Kontogiannis, L., & Hardie, D. G. (2002). Protein kinase substrate recognition studied using the recombinant catalytic domain of AMP-activated protein kinase and a model substrate. Journal of Molecular Biology, 2317, 2309–2323.
Davies, S. P., Helps, N. R., Cohen, P. T., & Hardie, D. G. (1995). 5′-AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP-activated protein kinase. Studies using bacterially expressed human protein phosphatase-2C alpha and native bovine protein phosphatase-2AC. FEBS Letters, 377, 421–425.
Cheung, P. C., Salt, I. P., Davies, S. P., Hardie, D. G., & Carling, D. (2000). Characterization of AMP-activated protein kinase gamma-subunit isoforms and their role in AMP binding. Biochemical Journal, 346, 659–669.
Sanders, M. J., Grondin, P. O., Hegarty, B. D., Snowden, M. A., & Carling, D. (2007). Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochemical Journal, 403, 139–148.
Hardie, D. G., Carling, D., & Gamblin, S. J. (2011). AMP-activated protein kinase: also regulated by ADP? Trends in Biochemical Sciences, 36, 470–477.
Xiao, B., Sanders, M. J., Underwood, E., Heath, R., Mayer, F. V., Carmena, D., Jing, C., Walker, P. A., Eccleston, J. F., Haire, L. F., Saiu, P., Howell, S. A., Aasland, R., Martin, S. R., Carling, D., & Gamblin, S. J. (2011). Structure of mammalian AMPK and its regulation by ADP. Nature, 472, 230–233.
Chen, L., Wang, J., Zhang, Y. Y., Yan, S. F., Neumann, D., Schlattner, U., Wang, Z. X., & Wu, J. W. (2012). AMP-activated protein kinase undergoes nucleotide-dependent conformational changes. Nature Structural and Molecular Biology, 19, 716–718.
Stapleton, D., Gao, G., Michell, B. J., Widmer, J., Mitchelhill, K., Teh, T., House, C. M., Witters, L. A., & Kemp, B. E. (1994). Mammalian 5′-AMP-activated protein kinase non-catalytic subunits are homologs of proteins that interact with yeast Snf1 protein kinase. Journal of Biological Chemistry, 269, 29343–29346.
Woods, A., Cheung, P. C., Smith, F. C., Davison, M. D., Scott, J., Beri, R. K., & Carling, D. (1996). Characterization of AMP-activated protein kinase beta and gamma subunits. Assembly of the heterotrimeric complex in vitro. Journal of Biological Chemistry, 271, 10282–10290.
Neumann, D., Woods, A., Carling, D., Wallimann, T., & Schlattner, U. (2003). Mammalian AMP-activated protein kinase: functional, heterotrimeric complexes by co-expression of subunits in Escherichia coli. Protein Expression and Purification, 30, 230–237.
Hudson, E. R. (2003). A novel domain in AMP-activated protein kinase causes glycogen storage bodies similar to those seen in hereditary cardiac arrhythmias. Current Biology, 13, 861–866.
Polekhina, G., Gupta, A., Michell, B. J., van Denderen, B., Murthy, S., Feil, S. C., Jennings, I. G., Campbell, D. J., Witters, L. A., Parker, M. W., Kemp, B. E., & Stapleton, D. (2003). AMPK beta subunit targets metabolic stress sensing to glycogen. Current Biology, 13, 867–871.
Oakhill, J. S., Chen, Z. P., Scott, J. W., Steel, R., Castelli, L. A., Ling, N., Macaulay, S. L., & Kemp, B. E. (2010). β-Subunit myristoylation is the gatekeeper for initiating metabolic stress sensing by AMP-activated protein kinase (AMPK). Proceedings of the National Academy of Sciences of the United States of America, 107, 19237–19241.
Xiao, B., Sanders, M. J., Carmena, D., Bright, N. J., Haire, L. F., Underwood, E., Patel, B. R., Heath, R. B., Walker, P. A., Hallen, S., Giordanetto, F., Martin, S. R., Carling, D., & Gamblin, S. J. (2013). Structural basis of AMPK regulation by small molecule activators. Nature Communications, 4, 3017.
Polekhina, G., Gupta, A., van Denderen, B. J., Feil, S. C., Kemp, B. E., Stapleton, D., & Parker, M. W. (2005). Structural basis for glycogen recognition by AMP-activated protein kinase. Structure, 13, 1453–1462.
Walker, J. R., Wybenga-Groot, L., Finerty, P. J., Newman, E., MacKenzie, F. M., Weigelt, J., Sundstrom, M., Arrowsmith, C., Edwards, A., Bochkarev, A., Dhe-Paganon, S. Structure of the glycogen-binding domain of the AMP-activated protein kinase beta2 subunit. Protein data bank. 10.2210/pdb2f15/pdb.
Nayak, V., Zhao, K., Wyce, A., Schwartz, M. F., Lo, W. S., Berger, S. L., & Marmorstein, R. (2006). Structure and dimerization of the kinase domain from yeast Snf1, a member of the Snf1/AMPK protein family. Structure, 14, 477–485.
Littler, D. R., Walker, J. R., Davis, T., Wybenga-Groot, L. E., Finerty, P. J., Newman, E., Mackenzie, F., & Dhe-Paganon, S. (2010). A conserved mechanism of autoinhibition for the AMPK kinase domain: ATP-binding site and catalytic loop refolding as a means of regulation. Acta Crystallographica Section F: Structural Biology and Crystallization Communications, 66, 143–151.
Xia, B., Hu, J. Solution structure of autoinhibitory domain of human AMP-activated protein kinase catalytic subunit. Protein data bank. 10.2210/pdb2ltu/pdb.
Koay, A., Petrie, E., Gorman, M., di Paolo, A., Mobbs, J., Parker, M., Stapleton, D., Gooley, P. Solution NMR structure of the apo-form of the beta2 carbohydrate module of AMP-activated protein kinase. doi:10.2210/pdb2lu3/pdb.
Townley, R., & Shapiro, L. (2007). Crystal structures of the adenylate sensor from fission yeast AMP-activated protein kinase. Science, 315, 1726–1729.
Amodeo, G. A., Rudolph, M. J., & Tong, L. (2007). Crystal structure of the heterotrimer core of Saccharomyces cerevisiae AMPK homologue SNF1. Nature, 449, 492–495.
Jin, X., Townley, R., & Shapiro, L. (2007). Structural insight into AMPK regulation: ADP comes into play. Structure, 15, 1285–1295.
Day, P., Sharff, A., Parra, L., Cleasby, A., Williams, M., Horer, S., Nar, H., Redemann, N., Tickle, I., & Yon, J. (2007). Structure of a CBS-domain pair from the regulatory 1 subunit of human AMPK in complex with AMP and ZMP. Acta Crystallographica Section D: Biological Crystallography, 63, 587–596.
Xiao, B., Heath, R., Saiu, P., Leiper, F. C., Leone, P., Jing, C., Walker, P. A., Haire, L., Eccleston, J. F., Davis, C. T., Martin, S. R., Carling, D., & Gamblin, S. J. (2007). Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature, 449, 496–500.
Handa, N., Takagi, T., Saijo, S., Kishishita, S., Takaya, D., Toyama, M., Terada, T., Shirouzu, M., Suzuki, A., Lee, S., Yamauchi, T., Okada-Iwabu, M., Iwabu, M., Kadowaki, T., Minokoshi, Y., & Yokoyama, S. (2011). Structural basis for compound C inhibition of the human AMP-activated protein kinase α2 subunit kinase domain. Acta Crystallographica. Section D, Biological Crystallography, 67, 480–487.
Chen, L., Jiao, Z. H., Zheng, L. S., Zhang, Y. Y., Xie, S. T., Wang, Z. X., & Wu, J. W. (2009). Structural insight into the autoinhibition mechanism of AMP-activated protein kinase. Nature, 459, 1146–1149.
Cho, Y. S., Lee, J. I., Shin, D., Kim, H. T., Jung, H. Y., Lee, T. G., Kang, L. W., Ahn, Y. J., Cho, H. S., & Heo, Y. S. (2010). Molecular mechanism for the regulation of human ACC2 through phosphorylation by AMPK. Biochemical and Biophysical Research Communications, 391, 187–92.
Gomez-Garcia, I., Oyenarte, I., & Martinez-Cruz, L. A. (2010). The crystal structure of protein MJ1225 from Methanocaldococcus jannaschii shows strong conservation of key structural features seen in the eukaryal gamma-AMPK. Journal of Molecular Biology, 399, 53–70.
Rudolph, M. J., Amodeo, G. A., & Tong, L. (2010). An inhibited conformation for the protein kinase domain of the Saccharomyces cerevisiae AMPK homolog Snf1. Acta Crystallographica Section F: Structural Biology and Crystallization Communications, 66, 999–1002.
Mayer, F. V., Heath, R., Underwood, E., Sanders, M. J., Carmena, D., McCartney, R. R., Leiper, F. C., Xiao, B., Jing, C., Walker, P. A., Haire, L. F., Ogrodowicz, R., Martin, S. R., Schmidt, M. C., Gamblin, S. J., & Carling, D. (2011). ADP regulates SNF1, the Saccharomyces cerevisiae homolog of AMP-activated protein kinase. Cell Metabolism, 14, 707–714.
Zhan, Y., Chen, Y., Zhang, Q., Zhuang, J., Tian, M., Chen, H., Zhang, L., Zhang, H., He, J., Wang, W., Wu, R., Wang, Y., Shi, C., Yang, K., Li, A., Xin, Y., Li, T. Y., Yang, J. Y., Zheng, Z., Yu, C., Lin, S., Chang, C., Huang, P., Lin, T., & Wu, Q. (2012). Crystal structure of human nur77 ligand-binding domain in complex with ethyl 2-[2,3,4 trimethoxy-6(1-octanoyl)phenyl]acetate. Nature Chemical Biology, 8, 897–904.
Chen, L., Xin, F. J., Wang, J., Hu, J., Zhang, Y. Y., Wan, S., Cao, L. S., Lu, C., Li, P., Yan, S. F., Neumann, D., Schlattner, U., Xia, B., Wang, Z. X., & Wu, J. W. (2013). Conserved regulatory elements in AMPK. Nature, 498, E8–10.
Calabrese, M. F., Rajamohan, F., Harris, M. S., Caspers, N. L., Magyar, R., Withka, J. M., Wang, H., Borzilleri, K. A., Sahasrabudhe, P. V., Hoth, L. R., Geoghegan, K. F., Han, S., Brown, J., Subashi, T. A., Reyes, A. R., Frisbie, R. K., Ward, J., Miller, R. A., Landro, J. A., Londregan, A. T., Carpino, P. A., Cabral, S., Smith, A. C., Conn, E. L., Cameron, K. O., Qiu, X., & Kurumbail, R. G. (2014). Structural basis for AMPK activation: natural and synthetic ligands regulate kinase activity from opposite poles by different molecular mechanisms. Structure, 22, 1161–1172.
Li, X., Wang, L., Zhou, X. E., Ke, J., de Waal, P. W., Gu, X., Tan, M. H., Wang, D., Wu, D., Xu, H. E., & Melcher, K. (2015). Structural basis of AMPK regulation by adenine nucleotides and glycogen. Cell Research, 25, 50–66.
Carling, D., Clarke, P. R., Zammit, V. A., & Hardie, D. G. (1989). Purification and characterization of the AMP-activated protein kinase. Copurification of acetyl-CoA carboxylase kinase and 3-hydroxy-3-methylglutaryl-CoA reductase kinase activities. European Journal of Biochemistry, 186, 129–136.
Zhu, L., Chen, L., Zhou, X. M., Zhang, Y. Y., Zhang, Y. J., Zhao, J., Ji, S. R., Wu, J. W., & Wu, Y. (2011). Structural insights into the architecture and allostery of full-length AMP-activated protein kinase. Structure, 19, 515–522.
Foufelle, F., & Ferre, P. (2005). Role of adenosine monophosphate-activated protein kinase in the control of energy homeostasis. Current Opinion in Clinical Nutrition and Metabolic Care, 8, 355–360.
Gauthier, M. S., O’Brien, E. L., Bigornia, S., Mott, M., Cacicedo, J. M., Xu, X. J., Gokce, N., Apovian, C., & Ruderman, N. (2011). Decreased AMP-activated protein kinase activity is associated with increased inflammation in visceral adipose tissue and with whole-body insulin resistance in morbidly obese humans. Biochemical and Biophysical Research Communications, 404, 382–387.
Peairs, A., Radjavi, A., Davis, S., Li, L., Ahmed, A., Giri, S., & Reilly, C. M. (2009). Activation of AMPK inhibits inflammation in MRL/lpr mouse mesangial cells. Clinical and Experimental Immunology, 156, 542–551.
Gul, T., Balkhi, H. M., & Haq, E. (2013). AMPK: a potent target for treating obesity. International Journal of Pharmaceutics Chemestry Biological Science, 3, 801–813.
Phoenix, K. N., Vumbaca, F., & Claffey, K. P. (2009). Therapeutic metformin/AMPK activation promotes the angiogenic phenotype in the ERalpha negative MDA-MB-435 breast cancer model. Breast Cancer Research and Treatment, 113, 101–111.
Hadad, S. M., Appleyard, V., & Thompson, A. M. (2009). Therapeutic metformin/AMPK activation promotes the angiogenic phenotype in the ERalpha negative MDA-MB-435 breast cancer model. Breast Cancer Research and Treatment, 114, 391.
Shibata, R., Sato, K., Pimentel, D. R., Takemura, Y., Kihara, S., Ohashi, K., Funahashi, T., Ouchi, N., & Walsh, K. (2005). Adiponectin protects against myocardial ischemia-reperfusion injury through AMPK- and COX-2-dependent mechanisms. Nature Medicine, 11, 1096–1103.
Miller, E. J., Li, J., Leng, L., McDonald, C., Atsumi, T., Bucala, R., & Young, L. H. (2008). Macrophage migration inhibitory factor stimulates AMP-activated protein kinase in the ischaemic heart. Nature, 451, 578–582.
Won, J. S., Im, Y. B., Kim, J., Singh, A. K., & Singh, I. (2010). Involvement of AMP-activated-protein-kinase (AMPK) in neuronal amyloido- genesis. Biochemical and Biophysical Research Communications, 399, 487–491.
Morais, A. S., Abarca-Quinones, J., Guigas, B., Viollet, B., Starkel, P., Horsmans, Y., & Leclercq, I. A. (2010). Development of hepatic fibrosis occurs normally in AMPK-deficient mice. Clinical Science, 118, 411–420.
Carling, D., Mayer, F. V., Sanders, M. J., & Gamblin, S. J. (2011). AMP-activated protein kinase: nature’s energy sensor. Nature Chemical Biology, 7, 512–518.
Mor, V., & Unnikrishnan, M. K. (2011). 5′-adenosine monophosphate-activated protein kinase and the metabolic syndrome. Endocrine, Metabolic & Immune Disorders Drug Targets, 11, 206–216.
Baumann, P., Mandl-Weber, S., Emmerich, B., Straka, C., & Schmidmaier, R. (2007). Inhibition of adenosine monophosphate-activated protein kinase induces apoptosis in multiple myeloma cells. Anti-Cancer Drugs, 18, 405–410.
Weisova, P., Davila, D., Tuffy, L. P., Ward, M. W., Concannon, C. G., & Prehn, J. H. (2011). Role of 5′-adenosine monophosphate-activated protein kinase in cell survival and death responses in neurons. Antioxidants and Redox Signaling, 14, 1863–1876.
Filippov, S., Pinkosky, S. L., & Newton, R. S. (2014). LDL-cholesterol reduction in patients with hypercholesterolemia by modulation of adenosine triphosphate-citrate lyase and adenosine monophosphate-activated protein kinase. Current Opinion in Lipidology, 25, 309–315.
Osler, M. E., & Zierath, J. R. (2008). Adenosine 5′-monophosphate-activated protein kinase regulation of fatty acid oxidation in skeletal muscle. Endocrinology, 149, 935–941.
Ropelle, E. R., Pauli, J. R., Zecchin, K. G., Ueno, M., de Souza, C. T., Morari, J., Faria, M. C., Velloso, L. A., Saad, M. J., & Carvalheira, J. B. (2007). A central role for neuronal adenosine 5′-monophosphate-activated protein kinase in cancer-induced anorexia. Endocrinology, 148, 5220–5229.
Huang, H., Chen, C. Y., Chen, H. Y., Tsai, F. J., & Chen, C. Y. C. (2010). Computational screening and QSAR analysis for design of AMP-activated protein kinase agonist. Journal of the Taiwan Institute of Chemical Engineers, 41, 352–359.
Siwiak, M., Edelman, A., & Zielenkiewicz, P. (2012). Structural models of CFTR–AMPK and CFTR–PKA interactions: R-domain flexibility is a key factor in CFTR regulation. Journal of Molecular Modelling, 18, 83–90.
Park, H., Eom, J. W., & Kim, Y. H. (2014). Consensus scoring approach to identify the inhibitors of AMP-activated protein kinase α2 with virtual screening. Journal of Chemical Information and Modeling, 54, 2139–2146.
Zhang, Y., Wang, Y., Bao, C., Xu, Y., Shen, H., Chen, J., Yan, J., & Chen, Y. (2012). Metformin interacts with AMPK through binding to γ subunit. Molecular and Cellular Biochemistry, 368, 69–76.
Raj, C. G. D., Sarojini, B. K., Khan, M. T. H., & Raghavendra, R. (2013). In vivo antidiabetic activity and in silico studies on adenosine monophosphate-activated protein kinase (AMPK) of (2E,5E)-2,5-bis(4-hydroxy-3-methoxybenzylidene) cyclopentanone. Medicinal Chemistry Research, 22, 2430–2436.
Pang, T., Zhang, Z. S., Gu, M., Qiu, B. Y., Yu, L. F., Cao, P. R., Shao, W., Su, M. B., Li, J. Y., Nan, F. J., & Li, J. (2008). Small molecule antagonizes autoinhibition and activates AMP-activated protein kinase in cells. Journal of Biological Chemistry, 283, 16051–16060.
Peng, C., & Head-Gordon, T. (2011). The dynamical mechanism of auto-inhibition of AMP activated protein kinase. PLoS Computational Biology, 7, e1002082.
Chapnik, N., Genzer, Y., Ben-Shimon, A., Niv, M. Y., & Froy, O. (2014). AMPK-derived peptides reduce blood glucose levels but lead to fat retention in the liver of obese mice. Journal of Endocrinology, 221, 89–99.
Chang, Y. H., Ho, T. Y., Wu, C. H., Chen, C. Y., Huang, H. J., Tsai, F. J., Tsai, C. H., & Chen, C. Y. C. (2009). Study of AMP-activated protein kinase agonists by structure-based drug designing. Advances in Materials Research, 79–82, 2187–2190.
Gruzman, A., Shamni, O., Ben Yakir, M., Sandovski, D., Elgart, A., Alpert, E., Cohen, G., Hoffman, A., Katzhendler, Y., Cerasi, E., & Sasson, S. (2008). Novel d-xylose derivatives stimulate muscle glucose uptake by activating AMP-activated protein kinase alpha. Journal of Medicinal Chemistry, 51, 8096–8108.
Wang, Y., Kong, W., Li, Y., Tang, S., Li, Z., Li, Y., Shan, Y., Bi, C., Jiang, J., & Song, D. (2012). Synthesis and structure–activity relationship of berberine analogues in LDLR up-regulation and AMPK activation. Bioorganic and Medicinal Chemistry, 20, 6552–6558.
Doss, C. G. P., & Nagasundaram, N. (2014). Molecular docking and molecular dynamics study on the effect of ERCC1 deleterious polymorphisms in ERCC1-XPF heterodimer. Applied Biochemistry and Biotechnology, 172, 1265–1281.
Roy, D., Kumar, V., Acharya, K. K., & Thirumurugan, K. (2014). Probing the binding of syzygium-derived α-glucosidase inhibitors with N- and C-terminal human maltase glucoamylase by docking and molecular dynamics simulation. Applied Biochemistry and Biotechnology, 172, 102–114.
Sahoo, B. R., Swain, B., Dikhit, M. R., Basu, M., Bej, A., Jayasankar, P., & Samanta, M. (2013). Activation of nucleotide-binding oligomerization domain 1 (NOD1) receptor signaling in labeo rohita by iE-DAP and identification of ligand-binding key motifs in NOD1 by molecular modeling and docking. Applied Biochemistry and Biotechnology, 170, 1282–1309.
Pulaganti, M., Banaganapalli, B., Mulakayala, C., Chitta, S. K., & Anuradha, C. M. (2014). Molecular modeling and docking studies of O-succinylbenzoate synthase of M. tuberculosis—a potential target for antituberculosis drug design. Applied Biochemistry and Biotechnology, 172, 1407–1432.
Pinheiro, A. S., Duarte, J. B. C., Alves, C. N., & Alberto de Molfetta, F. (2015). Virtual screening and molecular dynamics simulations from a bank of molecules of the amazon region against functional NS3-4A protease-helicase enzyme of hepatitis C virus. Applied Biochemistry and Biotechnology, 176, 1709–1721.
Honarparvar, B., Govender, T., Maguire, G. E., Soliman, M. E., & Kruger, H. G. (2014). Integrated approach to structure-based enzymatic drug design: molecular modeling, spectroscopy, and experimental bioactivity. Chemical Reviews, 114, 493–537.
Ramakrishnan, C., & Ramachandran, G. N. (1965). Stereochemical criteria for polypeptide and protein chain conformations. II. Allowed conformations for a pair of peptide units. Biophysical Journal, 5, 909–933.
Lindorff-Larsen, K., Piana, S., Palmo, K., Maragakis, P., Klepeis, J. L., Dror, R. O., & Shaw, D. E. (2010). Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins: Structure, Function, and Bioinformatics, 78, 1950–1958.
SYBYL (2007) Tripos Inc, St Louis.
Molecular Operating Environment (MOE), Chemical Computing Group Inc., 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada. .
Shoichet, B. K., McGovern, S. L., Wei, B., & Irwin, J. J. (2002). Lead discovery using molecular docking. Current Opinion in Chemical Biology, 6, 439–446.
Lipinski, C. A. (2004). Lead- and drug-like compounds: the rule-of-five revolution. Drug Discovery Today: Technologies, 1, 337–341.
Morris, G. M., Huey, R., Lindstrom, W., Sanner, M. F., Belew, R. K., Goodsell, D. S., & Olson, A. J. (2009). AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. Journal of Computational Chemistry, 30, 2785–2791.
Laskowski, R. A., & Swindells, M. B. (2011). LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. Journal of Chemical Information and Modeling, 51, 2778–2786.
Alder, B. J., & Wainwright, T. (1959). Studies in molecular dynamics. I. General method. Journal of Chemical Physics, 31, 459–466.
Christen, M., Hunenberger, P. H., Bakowies, D., Baron, R., Burgi, R., Geerke, D. P., Heinz, T. N., Kastenholz, M. A., Krautler, V., Oostenbrink, C., Peter, C., Trzesniak, D., & van Gunsteren, W. F. (2005). The GROMOS software for biomolecular simulation: GROMOS05. Journal of Computational Chemistry, 26, 1719–1751.
Chen, Q., & Chen, Y. P. (2006). Mining frequent patterns for AMP-activated protein kinase regulation on skeletal muscle. BMC Bioinformatics, 7, 394.
Patrick, G. L. (2005). An introduction to medicinal chemistry. New York: Oxford University Press.
Acknowledgments
The financial support from the School of Health Sciences, University of KwaZulu-Natal, Westville, Durban, South Africa is greatly acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ramesh, M., Vepuri, S.B., Oosthuizen, F. et al. Adenosine Monophosphate-Activated Protein Kinase (AMPK) as a Diverse Therapeutic Target: A Computational Perspective. Appl Biochem Biotechnol 178, 810–830 (2016). https://doi.org/10.1007/s12010-015-1911-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-015-1911-9