
Abstract
For xylooligosaccharide (XO) production, endo-xylanase from Thermobifida fusca was modified by error-prone PCR and DNA shuffling. The G4SM1 mutant (S62T, S144C, N198D, and A217V) showed the most improved hydrolytic activity and was two copies expressed in Pichia pastoris under the control of GAP promoter. The maximum xylanase activity in culture supernatants was 165 ± 5.5 U/ml, and the secreted protein concentration reached 493 mg/l in a 2-l baffled shake flask. After 6× His-tagged protein purification, the specific activity of G4SM1 was 2036 ± 45.8 U/mg, 2.12 times greater than that of wild-type enzyme. Additionally, G4SM1 was stable over a wide pH range from 5.0 to 9.0. Meanwhile, half-life of G4SM1 thermal inactivation at 70 °C increased 8.5-fold. Three-dimensional structures suggest that two amino acid substitutions, S62T and S144C, located at catalytic domain may be responsible for the enhanced activity and thermostability of xylanase. Xylobiose was the dominant end product of xylan hydrolysis by G4SM1. Due to its attractive biochemical properties, G4SM1 has potential value in commercial XO production.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
To improve host health, prebiotics had been used to modulate gut microbiota and their fermentation processes [1]. As sources of novel prebiotic oligosaccharides, the demand of xylooligosaccharides (XOs) increased dramatically since 1994 for their unique properties and the effects on health [2]. The XOs are sugar oligomers made up of xylose units, which were nondigestible oligosaccharides [3]. Xylooligomers are effective to stimulate the growth of intestinal Bifidobacteria [4, 5] thus decreasing systemic inflammation [6], enhance cecal epithelial cell proliferation [7], improve calcium absorption [8], increase growth performance, enhance endocrine metabolism, and improve immune function [9].
Enzyme treatments of lignocellulosic materials or isolated xylan are the main approaches for XO production [10–12]. For XO production, the desired enzyme is low exo-xylanase and/or β-xylosidase activity to avoid the production of xylose [13]. Furthermore, xylanases exhibiting high catalytic activity and thermo-alkalistability have particular value in industrial application [14–16]. In contrast to other glycoside hydrolase (GH) families 5, 8, and 10, GH11 consists solely of endo-1,4-β-xylanases and usually gives larger end products [17]. Thermobifida fusca xylanase (Tfx, GenBank No. U01242) was reported to be one of the most thermostable GH11 xylanases. Previous study also suggested that Tfx was able to retain almost all activity after incubation at 75 °C for 18 h [18]. However, the specific catalytic activities (117–686 U/mg) were still unable to satisfy the demands for practical applications [14, 18, 19].
In the current study, we employed directed evolution strategy, including error-prone PCR and DNA shuffling, to generate and assemble mutants. The mutated gene showing increased catalytic activity was subjected to construction of recombinant yeast for producing endo-xylanase. Good thermo-alkaliphilic stability and high specific activity and the xylobiose dominant production made the recombinant xylanase more useful in industrial XO production.
Methods
Materials
Plasmid pET-22b (+) (Novagen, Madison, WI, USA) and pGAPZαA (Invitrogen, Shanghai, China) were used as the expression vectors for recombinant xylanase in Escherichia coli BL21 (DE3) and Pichia pastoris GS115 (his4) (Invitrogen, Shanghai, China), respectively. The plasmid pUCm-T/tfx was conserved in our laboratory. Oligonucleotides were synthesized by Sangon (Shanghai, China). Birchwood xylan was purchased from Sigma (St. Louis, MO, USA), and restriction endonucleases were purchased from TaKaRa (Dalian, China). Standard XOs were purchased from Megazyme (Wicklow, Ireland).
Mutagenesis, Recombination, and Library Construction
Error-prone PCR (EP-PCR) was employed to generate random mutants by varying the concentration of Taq polymerase (TaKaRa, Dalian, China), dNTPs, Mg2+, and Mn2+. The tfx gene (approximately 20 ng of 3.7 kb plasmid pUCm-T/tfx) was amplified using primers 22b-TF and 22b-TR (20 pmol each, Table 1). The PCR program was performed on a Mastercycler (Eppendorf, Germany) for 30 cycles consisting of 94 °C for 50 s, 55 °C for 30 s (−0.1 °C per cycle), and 72 °C for 1 min. The PCR products were purified and ligated into pET-22b (+) vector between the EcoRI and XhoI sites. Cell transformation, induction, and cell lysis by PopCultureTM (Novagen, Madison, USA) were carried out as described previously [20].
Six mutants with improved xylanase activity were chosen and mixed as the template for DNA shuffling. The DNA shuffling procedure was performed as described by Stemmer [21] and Zhang et al. [15]. Reassembly of the full-length gene was achieved using primers 22b-TF and 22b-TR (20 pmol each, Table 1). The reassembled gene was ligated into pET-22b (+) between the EcoRI and XhoI sites, followed by transformation and manipulation of the cell suspension as described by Wang et al. [20].
Library Screening
Xylanase activity was determined using 1 % (w/v) birchwood xylan (Sigma) substrate and the DNS method [22]. Cell-free supernatants were used for the enzymatic assays in 96-well plates [20]. One unit (U) of xylanase activity was defined as the amount of enzyme that produced reducing sugar equivalent to 1 μmol of xylose per minute. d-xylose was used as the standard. Those transformants exhibiting higher catalytic activity than their parents were sequenced by Invitrogen (Shanghai, China). Protein concentration was determined using the Bradford method [23], and bovine serum albumin was used as the standard.
Heterologous Expression in P. Pastoris
Both wild-type tfx (WT) and G4SM1 genes were amplified using Gap-TF and Gap-TR (Table 1). After restriction digestion, the fragments were inserted into a shuttle vector pGAPZαA. The resultant plasmids were transformed into E. coli TOP10F′ by heat shock and the resulting positive transformants, designated pGAPZαA/tfx and pGAPZαA/G4SM1, and were selected on low-salt LB plates (0.5 % yeast extract, 1 % peptone, 0.5 % NaCl, and 2 % agar) containing 25 μg/ml zeocin. For P. pastoris integration, approximately 10 μg of recombinant plasmids was AvrII-linearized and transformed into GS115 by electroporation (1500 V, 4.6 ms) (Eppendorf 2510, USA). The cuvette contents were spread onto YPDS plates (1 % yeast extract, 2 % peptone, 2 % glucose, 1 M sorbitol, and 2 % agar) containing 100 μg/ml zeocin and incubated at 30 °C for 2–3 days. Positive transformants were then spread onto YPDS plates supplemented with 500, 1000, 1500, and 2000 μg/ml zeocin to obtain multi-copy strains. Scale-up expression was achieved using 2-l baffled shake flasks containing 100-ml YPD medium that were incubated by shaking at 30 °C for 120 h.
Southern Blot and Quantitative Real-Time PCR Analysis
Yeast genomic DNA was prepared from 5-ml YPD cultures with modifications to the benzyl chloride method [24]. Briefly, the cells were suspended in wash buffer (100 mM Tris-HCl and 40 mM EDTA) and incubated with benzyl chloride and sodium dodecyl sulfate (SDS) to generate spheroplasts and deproteinize the DNA. The genomic DNA was recovered by ethanol precipitation. Equal amounts (approximately 100 μg) of genomic DNA samples were digested with DraI. Smears were observed on 1 % agarose gels and transferred to nylon membranes (GE Healthcare, Piscataway, NJ, USA) overnight in 20× SSC (3 M NaCl, 0.3 M sodium citrate, pH 7.0). The Gap-TF/Gap-TR primer pair was used to amplify the full-length xylanase gene, and the PCR product was radiolabeled with [α32P]-dCTP as the probe. After UV cross-linking, the membrane was hybridized with the probe, and hybridization signals were detected by phosphorimaging using a Typhoon 9200 imager (GE Healthcare).
Quantitative real-time PCR (qRT-PCR) was carried out using the SYBR Green PCR master mix kit (TaKaRa, Dalian, China) in a Roche LightCycler® 480 System II (Roche, Rotkreuz, Switzerland). Genomic DNA (approximately 100 ng each) served as the template and was amplified using either the his4-F/his4-R primer pair (for amplification of the his4 gene) or the Gap-TF/tfx-R primer pair (for amplification of the tfx gene). The relative gene expression of tfx was estimated as 2−ΔΔCT as described by Livak and Schmittgen [25].
Protein Purification, Deglycosylation, and SDS-PAGE
The culture supernatant obtained at 120 h was used for 6× His-tagged affinity purification with nickel-nitrilotriacetic acid (Ni-NTA) agarose (Qiagen, GmbH, Hilden, Germany). Both crude and purified enzymes were added to 0.05 U endoglycosidase H (Roche, Indianapolis, IN, USA) and incubated at 37 °C for 24 h. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE, 12 % running gel and 5 % stacking gel) was performed according to the method of Laemmli [26]. Protein bands were visualized by Coomassie Brilliant Blue R-250 staining.
Properties of Tfx and Its Mutants
In this section, equal amounts (approximately 5 μg) of purified xylanases were used to estimate enzymatic properties, unless otherwise stated. To determine the optimal pH, the enzymatic assays were carried out over a range of pH 3.0–10.0 (pH 3.0–8.0, citrate/phosphate buffer; pH 9.0–10.0, 0.2 M glycine, and 0.2 M NaOH buffer) at 60 °C for 5 min. To determine pH stability, the enzymes were preincubated in various pH buffers at 37 °C for 2 h, and the residual xylanase activities were measured under standard conditions (60 °C, pH 6.0).
The effect of temperature on xylanase activity was evaluated by incubating the enzyme at a range of temperatures (40–90 °C) in citrate/phosphate buffer (pH 6.0) for 5 min. To estimate thermostability, enzymes were preincubated in citrate/phosphate buffer (pH 6.0) at different temperatures (60–90 °C) for 1 h, and samples were obtained at different time intervals (2, 10, 30, and 60 min), followed by immediate chilling on ice for 5 min before assaying their residual activities. The half-lives of thermal inactivation were determined as described by Zhang et al. [15]. The residual activities were measured under standard conditions (60 °C, pH 6.0). The kinetic constants were evaluated under standard conditions (60 °C, pH 6.0) for 10 min, using different concentrations of birchwood xylan (from 0.2 to 15 mg/ml). Data were fitted with nonlinear regression for the Michaelis-Menten equation using GraphPad Prism v5.0 (GraphPad Software, San Diego, CA, USA).
Structural Modeling
Homology-based modeling of xylanase proteins was performed by Swiss-Model (http://swissmodel.expasy.org/) according to the template (PDB code 1M4W, chain A) with 84 % amino acid sequence identity and an E value of 2.70 × 10−67 [27, 28]. The three-dimensional structure of the catalytic domain of xylanases was displayed using Chimera1.4.1 [29]. The potential N-glycosylation site and disulfide bond were predicted using NetNGlyc 1.0 (http://www.cbs.dtu.dk/services/NetNGlyc/) and DiANNA Server (http://clavius.bc.edu/~clotelab/DiANNA/main.html), respectively.
Enzymatic Hydrolysis
The 1 % birchwood xylan solution in distilled water was incubated with purified G4SM1 (200 U) at 40 °C. The portions were withdrawn at different time intervals (5 min, 30 min, 1 h, 2 h, 6 h, 12 h, and 24 h) and boiled for 10 min. Samples and standard XOs (xylose, xylobiose, xylotriose, xylotetroase, and xylopentaose) were subjected to a Waters Alliance HPLC system (separations module e2695, Waters, Milford, MA, USA) equipped with a Sugar-Pak TM 1 column (300 mm × 6.5 mm) and refractive index detector (Waters 2414). Distilled water was used as the mobile phase with a flow rate of 0.3 ml/min. Released sugars from xylan were quantified according to the standard curves of XO concentrations compared to the area of each peak.
Results and Discussion
Screening Mutants with Improved Xylanase Activity
Two rounds of EP-PCR were performed to generate random mutants. In the initial round, approximately 3000 transformants were screened for high-activity mutants. Five transformants were obtained and subjected to a second round of EP-PCR, yielding 6 out of 4000 transformants that exhibited improved activity. Next, DNA shuffling was employed to recombine the beneficial mutagenesis. Of the 3000 transformants, the G4SM1 mutant (S62T, S144C, N198D, and A217V) showed the highest xylanase activity and was used for further study.
Construction of Recombinant Yeast Containing Two Copies of the Integrated Xylanase Gene
Following growth on YPDS plates containing 100 μg/ml zeocin, positive cells that integrated the G4SM1 gene were then spread onto plates with higher concentrations of zeocin (500–2000 μg/ml) to obtain multi-copy strains. It was reported that multiple gene insertion events occurred at a frequency of 1–10 % of total zeocin-resistant transformants [30]. In this study, seven transformants (2 from the 2000 μg/ml zeocin plates and 5 from the 1500 μg/ml zeocin plates) were picked and further analyzed to determine gene copy number by Southern blot and qRT-PCR. Southern blot analysis revealed that strain No. 28, which was resistant to 1500 μg/ml zeocin, showed two hybridizing bands, while only a single band was observed in other transformants (Fig. 1a), suggesting that strain No. 28 likely is a two-copy strain. The genomic DNA was analyzed by qRT-PCR to further quantify the expression level of the xylanase gene, and wild-type P. pastoris GS115 was used as a control. As shown in Fig. 1b, the expression patterns of the xylanase gene in the other six transformants were similar (approximately half of his4). However, the xylanase gene was expressed at greater than 0.85 of his4 levels in strain No. 28, nearly 2-fold compared to the other transformants, which was in agreement with our results from the Southern blot analysis. We concluded that strain No. 28 was a two-copy strain and used it for xylanase expression.

Determination of the tfx gene copy number in the genome of recombinant Pichia pastoris GS115 by Southern blot analysis (a) and quantitative real-time PCR (b). a Genomic DNAs were digested with DraI, and a fragment of tfx gene was radiolabeled with [α32P]-dCTP as a probe. b The expression of the tfx gene in recombinant yeast was represented as relative to his4. The genome of P. pastoris GS115 was used as the control (Con)
Heterologous Expression in P. Pastoris
The yeast strain No. 28 was able to produce a maximum activity of 165 ± 5.5 U/ml in the culture supernatant of YPD medium after shaking at 30 °C for 120 h. After 6× His-tagged protein purification with Ni-NTA agarose, the specific activity of G4SM1 reached 2036 ± 45.8 U/mg, 2.12 times that of WT (Table 2). Using SDS-PAGE, a distinguishable band was visualized around 40 kDa (Fig. 2), which is heavier than the molecular mass of the mature peptide (32.0 kDa). This disparity may be attributed to the N-glycosylation modification, which contributes 1–3 kDa per N-glycosylation site in eukaryotic cells [31] and is important for protein folding and maintaining enzymatic activity and stability. There were seven potential N-glycosylation sites (N5, N34, N66, N183, N230, N236, and N285) within the G4SM1 sequence. Among them, three sites (N5, N183, and N230) had a greater than 66 % likelihood of being glycosylated. To examine the N-glycosylation of G4SM1, the purified enzyme was treated with endoglycosidase H. As expected, the protein mass decreased by approximately 10 kDa (Fig. 2).

Purification and deglycosylation of G4SM1 expressed in Pichia pastoris GS115. M, marker from bottom to top is 15, 20 (red), 30, 40, 50, 60, 75 (red), 100, 135, and 175 kDa. Lane 1 culture supernatant obtained at 120 h of protein expression. Lane 2 crude enzyme treated with endoglycosidase H for 24 h. Lane 3 purification of G4SM1 by Ni-NTA agarose affinity chromatography. Lane 4 purified enzyme treated with endoglycosidase H for 24 h
In our previous study, the tfx gene was expressed under the control of AOX1 promoter [14]. However, the AOX1 promoter is induced by methanol to drive expression, which is toxic and a potential fire hazard [32]. Taking safety into consideration, both the tfx and G4SM1 genes were electroporated into P. pastoris GS115 to target the GAP promoter locus, which can constitutively express recombinant proteins at high levels without methanol [33]. The expressed xylanase protein accumulated up to 493 mg/l after incubation for 120 h and could be further augmented under high cell density fermentation conditions.
Characterization of the Enzymatic Properties of Tfx and G4SM1
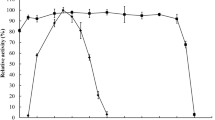
As shown in Fig. 3a, the optimal pH for the activities of Tfx and G4SM1 was pH 6.0. The specific activity of G4SM1 was 2036 ± 45.8 U/mg, or 2.12-fold of WT, while the Km value was 27.8 % lower (P < 0.05). Furthermore, the k cat/K m value of G4SM1 improved by 75.9 %, indicating that the catalytic efficiency or the affinity for the birchwood xylan substrate may be enhanced (Table 2). The xylanases secreted by P. pastoris were observed to be quite stable (>80 % initial activity was retained) over the pH range from 5.0 to 9.0 (Fig. 3b), suggesting that the pH profile of the mutant was not greatly altered compared with our previous study [14]. The effect of temperature on WT or G4SM1 xylanase activity was addressed in Fig. 3c–e. Both WT and G4SM1 showed their highest activities at 60 °C (Fig. 3e), but the relative activity of G4SM1 (90.3 % of maximum activity) was significantly higher (P < 0.01) than that of WT (59.5 % of maximum activity).

Characterization of the effects of pH and temperature on wild-type Tfx and the G4SM1 mutant. a The optimal pH of Tfx and G4SM1. b The pH stability of Tfx and G4SM1. c Thermostability of Tfx from 60 to 90 °C. d Thermostability of G4SM1 from 60 to 90 °C. e The optimal temperature of Tfx and G4SM1. The assays were performed as described in the “Methods” using 1 % birchwood xylan as the substrate. At the optimal pH and temperature, the highest xylanase activity was set as 100 %. The xylanase activity under optimal conditions (60 °C, pH 6.0) was set as 100 % in the assay to determine pH stability. For thermostability, the xylanase activity of enzyme without a heat challenge was set as 100 %. All of the assays were carried out in triplicate
Heat challenges were carried out in 96-well plates at 50–80 °C for either WT (Fig. 3c) or G4SM1 (Fig. 3d). On the whole, the thermostability of Tfx was improved by amino acid substitutions. Both WT and G4SM1 were stable at 60 °C for 1 h. Nevertheless, WT retained only 45.9 % of initial activity after incubation at 70 °C for 10 min and was almost inactive after incubation at 80 °C for 10 min. The G4SM1 mutant maintained most of its activity (89.9 % of initial activity) after treatment at 70 °C for 10 min and retained 45.3 % of initial activity after treatment at 80 °C for 10 min. Half-life of thermal inactivation of G4SM1 displayed an 8.5-fold increase (Table 2), suggesting that the mutant is an attractive alternative for application in feed pelleting process.
Structural Analysis of Tfx and G4SM1
The G4SM1 mutant with enhanced xylanase activity and thermostability contained four amino acid substitutions (S62T, S144C, N198D, and A217V). Irwin et al. [18] reported that the mature peptide of T. fusca xylanase (GenBank No. U01242) was composed of a catalytic domain (1–187 AA), a linker peptide (188–210 AA), and a carbohydrate-binding module (CBM, 211–296 AA, also called the xylan binding domain) from the N- to C-terminus. Both the results of our research (data not published) and those observed by Irwin et al. [18] showed that the CBM sequence was not essential to catalyze the hydrolysis of soluble xylan, although it exhibited the ability to bind to the xylan substrate.
To investigate the relationship between amino acid substitutions and enzymatic activity and stability, we established putative three-dimensional structures of the catalytic domains of Tfx and G4SM1 by homology modeling (Fig. 4). The catalytic structure of Tfx is composed of one α-helix and several β-sheets, which is typical for a family 11 xylanase. The two conserved catalytic residues (E85 and E174) were located inside the pocket cavity [34]. It was inferred that S62T and S144C mutations, located at the edge of the cleavage, may contribute to the improved catalytic activity and thermostability. It is also believed that increasing the number of charged residuals and enhancing polar interactions, hydrophobic surfaces and structural configurations were responsible for protein stability [19, 35]. Interestingly, all of the amino acid changes (serine, threonine, and cysteine) in this study belong to the polar but uncharged R-group. As a result, the number of charged residuals was not altered. A closer view of the mutation sites indicated that S62 shared two hydrogen bonds with T185 (3.450 and 2.929 Å) and one with Y61 (3.163 Å) (Fig. 4a). After the S62T and S144C substitutions, T62 shared one hydrogen bond with T185 (3.544 Å), Y61 (2.809 Å), and C144 (3.187 Å) (Fig. 4b). Although one hydrogen bond between S62 and T185 was sacrificed, a new hydrogen bond between T62 and C144 was generated, as well as a new disulfide bond between C64 and C144. Furthermore, cysteine interactions are more hydrophobic than serine interactions. Taken together, the disulfide bond and hydrophobic interactions at the surface stabilized the protein structure [36, 37], which was in agreement with our thermostability findings (Table 2). Noticeably, E90 located at the edge of the pocket was set free after the amino acid substitution, while a hydrogen bond of 2.617 Å was observed in WT (Fig. 4). We speculate that the improved catalytic activity can be attributed to the release of E90, which may promote the transfer of the substrate to the catalytic centers inside the cavity (E85 and E174).

Homology modeling and structural comparison of Tfx (a) and the G4SM1 mutant (b). The catalytic residues (E85 and E174) and amino acid residuals near the mutation sites (S62T and S144C) were depicted in stick form. The relevant hydrogen bonds were displayed as dashed lines. The figures were generated using Chimera1.4.1 (http://www.cgl.ucsf.edu/chimera/)
Xylooligosaccharides Released from Birchwood Xylan
The hydrolysis products of birchwood xylan by endo-xylanase were analyzed by HPLC (Fig. 5a, b). When G4SM1 was added to the substrate, all XOs accumulated rapidly, except for xylose (Fig. 5b). As the hydrolysis reaction proceeded, those XOs with low polymerization degrees (xylobiose and xylotriose) increased, while xylotriose, xylotetroase, and xylopentaose decreased 2 h later. After 24 h of hydrolysis by endo-xylanase, the major product was xylobiose with a yield of 0.954 ± 0.093 mg/ml (9.54 % of initial xylan substrate). Negligible amounts of xylose were produced (0.037 ± 0.001 mg/ml), which was consistent with previous studies [4, 14] and satisfactory for food-related application. The preferred polymerization degree of XOs used in “functional food” is 2 to 4 [38].

HPLC analysis of xylooligosaccharides released from birchwood xylan. a Hydrolysis products after incubation for 24 h. b Profiles of samples withdrawn at different time intervals (5 min, 30 min, 1 h, 2 h, 6 h, 12 h, and 24 h). Xylan substrates were incubated with G4SM1 at 40 °C. Aliquots collected were boiled for 10 min and subjected to HPLC analysis. ×–×5 were xylose, xylobiose, xylotriose, xylotetroase, and xylopentaose, respectively
Conclusion
In this study, we demonstrated a strategy for xylanase modification using directed evolution. The G4SM1 mutant exhibited the most improved xylanase activity and thermostability and was heterologously expressed in a P. pastoris strain as an integrated two-copy gene. The characterization of the thermo-alkaliphilic stability and high specific activity and the xylobiose dominant production made the recombinant xylanase potentially useful in commercial XO production.
References
Vieira, A. T., Teixeira, M. M., & Martins, F. S. (2013). The role of probiotics and prebiotics in inducing gut immunity. Frontiers in Immunology, 4, 445. doi:10.3389/fimmu.2013.00445.
Vázquez, M. J., Alonso, J. L., Domínguez, H., & Parajó, J. C. (2000). Xylooligosaccharides: manufacture and applications. Trends in Food Science & Technology, 11, 387–393.
Joo, G. J., Rhee, I. K., Kim, S. O., & Rhee, S. J. (1998). Effect of dietary xylooligosaccharide on indigestion and retarding effect of bile acid movement across a dialysis membrane. Journal-Korean Society of Food Science and Nutrition, 27, 705–711.
Chapla, D., Pandit, P., & Shah, A. (2012). Production of xylooligosaccharides from corncob xylan by fungal xylanase and their utilization by probiotics. Bioresource Technology, 115, 215–221.
Finegold, S. M., Li, Z., Summanen, P. H., Downes, J., Thames, G., Corbett, K., Dowd, S., Krak, M., & Heber, D. (2014). Xylooligosaccharide increases bifidobacteria but not lactobacilli in human gut microbiota. Food & Function. doi:10.1039/C3FO60348B.
Hansen, C. H., Frøkiær, H., Christensen, A. G., Bergström, A., Licht, T. R., Hansen, A. K., & Metzdorff, S. B. (2013). Dietary xylooligosaccharide downregulates IFN-γ and the low-grade inflammatory cytokine IL-1β systemically in mice. Journal of Nutrition, 143, 533–540.
Howard, M. D., Gordon, D. T., Garleb, K. A., & Kerley, M. S. (1995). Dietary fructooligosaccharide, xylooligosaccharide and gum arabic have variable effects on cecal and colonic microbiota and epithelial cell proliferation in mice and rats. Journal of Nutrition, 125, 2604–2609.
Mussatto, S. I., & Mancilha, I. M. (2007). Non-digestible oligosacharides: a review. Carbohydrate Polymers, 68, 587–597.
Sun, Z. P., Lv, W. T., Yu, R. K., Li, J., Liu, H. H., Sun, W., Wang, Z. M., Li, J. P., Shan, Z., & Qin, Y. L. (2013). Effect of a straw-derived xylooligosaccharide on broiler growth performance, endocrine metabolism, and immune response. Canadian Journal of Veterinary Research, 77, 105–109.
Haddar, A., Driss, D., Frikha, F., Ellouz-Chaabouni, S., & Nasri, M. (2012). Alkaline xylanases from Bacillus mojavensis A21: production and generation of xylooligosaccharides. International Journal of Biological Macromolecules, 51, 647–656.
Samanta, A. K., Jayapal, N., Kolte, A. P., Senani, S., Sridhar, M., Suresh, K. P., & Sampath, K. T. (2012). Enzymatic production of xylooligosaccharides from alkali solubilized xylan of natural grass (Sehima nervosum). Bioresource Technology, 112, 199–205.
Bian, J., Peng, F., Peng, X. P., Peng, P., Xu, F., & Sun, R. C. (2013). Structural features and antioxidant activity of xylooligosaccharides enzymatically produced from sugarcane bagasse. Bioresource Technology, 127, 236–241.
Boler, B.M.V, & Fahey, G.C. Jr. (2012) Prebiotics of Plant and Microbial Origin. Direct-Fed Microbials and Prebiotics for Animals (Callaway, T.R. and Ricke, S.C., ed.), Springer, New York, NY, pp13-26.
Sun, J. Y., Liu, M. Q., Weng, X. Y., Qian, L. C., & Gu, S. H. (2007). Expression of recombinant Thermomonospora fusca xylanase A in Pichia pastoris and xylooligosaccharides released from xylans by it. Food Chemistry, 104, 1055–1064.
Zhang, Z. G., Yi, Z. L., Pei, X. Q., & Wu, Z. L. (2010). Improving the thermostability of Geobacillus stearothermophilus xylanase XT6 by directed evolution and site-directed mutagenesis. Bioresource Technology, 101, 9272–9278.
Verma, D., Kawarabayasi, Y., Miyazaki, K., & Satyanarayana, T. (2013). Cloning, expression and characteristics of a novel alkalistable and thermostable xylanase encoding gene (Mxyl) retrieved from compost-soil metagenome. PLoS one, 8, e52459.
Collins, T., Gerday, C., & Feller, G. (2005). Xylanases, xylanase families and extremophilic xylanases. FEMS Microbiology Reviews, 29, 3–23.
Irwin, D., Jung, E. D., & Wilson, D. B. (1994). Characterization and sequence of a Thermomonospora fusca xylanase. Applied Environmental Microbiology, 60, 63–770.
Wang, Q., & Xia, T. (2008). Enhancement of the activity and alkaline pH stability of Thermobifida fusca xylanase A by directed evolution. Biotechnology Letters, 30, 937–944.
Wang, Q., Zhao, L. L., Sun, J. Y., Liu, J. X., & Weng, X. Y. (2012). Enhancing catalytic activity of a hybrid xylanase through single substitution of Leu to Pro near the active site. World Journal of Microbiology and Biotechnology, 28, 929–935.
Stemmer, W. P. C. (1994). Rapid evolution of a protein in-vitro by DNA shuffling. Nature, 370, 389–391.
Bailey, M. J., Biely, P., & Poutanen, K. (1992). Interlaboratory testing of methods for assay of xylanase activity. Journal of Biotechnology, 23, 257–270.
Bradford, M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anaytical Biochemistry, 72, 248–254.
Zhu, H., Qu, F., & Zhu, L. H. (1993). Isolation of genomic DNAs from plants, fungi and bacteria using benzyl chloride. Nucleic Acid Research, 21, 5279–5280.
Livak, K. J., & Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods, 25, 402–408.
Laemmli, U. K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227, 680–685.
Arnold, K., Bordoli, L., Kopp, J., & Schwede, T. (2006). The SWISS-MODEL Workspace: a web-based environment for protein structure homology modelling. Bioinformatics, 22, 195–201.
Kiefer, F., Arnold, K., Künzli, M., Bordoli, L., & Schwede, T. (2009). The SWISS-MODEL repository and associated resources. Nucleic Acids Research, 37, D387–D392.
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., & Ferrin, T. E. (2004). UCSF Chimera-a visualization system for exploratory research and analysis. Journal of Computational Chemistry, 25, 1605–1612.
Vassileva, A., Chugh, D. A., Swaminathan, S., & Khanna, N. (2001). Expression of hepatitis B surface antigen in the methylotrophic yeast Pichia pastoris using the GAP promoter. Journal of Biotechnology, 88, 21–35.
Han, Y., & Lei, X. G. (1999). Role of glycosylation in the functional expression of an Aspergillus niger phytase (phyA) in Pichia pastoris. Archives of Biochemistry and Biophysics, 364, 83–90.
Cereghino, J. L., & Cregg, J. M. (2000). Heterologous protein expression in the methylotrophic yeast Pichia pastoris. FEMS Microbiology Reviews, 24, 45–66.
Waterham, H. R., Digan, M. E., Koutz, P. J., Lair, S. V., & Cregg, J. M. (1997). Isolation of the Pichia pastoris glyceraldehyde-3-phosphate dehydrogenase gene and regulation and use of its promoter. Gene, 186, 37–44.
Sapag, A., Wouters, J., Lambert, C., De Ioannes, P., Eyzaguirre, J., & Depiereux, E. (2002). The endoxylanases from family 11: computer analysis of protein sequences reveals important structural and phylogenetic relationships. Journal of Biotechnology, 95, 109–131.
Miyazaki, K., Takenouchi, M., Kondo, H., Noro, N., Suzuki, M., & Tsuda, S. (2006). Thermal stabilization of Bacillus subtilis family-11 xylanase by directed evolution. Journal of Biological Chemistry, 281, 10236–10242.
Van den Burg, B., Dijkstra, B. W., Vriend, G., Van der Vinne, B., Venema, G., & Eijsink, V. G. (1994). Protein stabilization by hydrophobic interactions at the surface. European Journal of Biochemistry, 220, 981–985.
Funahashi, J., Takano, K., Yamagata, Y., & Yutani, K. (2000). Role of surface hydrophobic residues in the conformational stability of human lysozyme at three different positions. Biochemistry, 39, 14448–14456.
Loo, J. V., Cummings, J., Deizenne, N., Englyst, H., Franck, A., Hopkins, M., Kok, N., Macfariane, G., Newton, D., Quigley, M., Roberfroid, M., van Vliet, T., & van den Heuvel, E. (1999). Functional food properties of non-digestible oligosaccharides: a consensus report from the ENDO Project (DGXII-AIRII-CT94-1095). British Journal of Nutrition, 81, 121–132.
Acknowledgments
This work was supported in part by IAEA Coordinated Research Projects (No. 16327/R0), the National Natural Science Foundation of China (30971702), the Natural Science Foundation of Zhejiang Province (3090247), and research grants from the Science and Technology Department of Zhejiang Province, China (2010C32008).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Wang, Q., Du, W., Weng, XY. et al. Recombination of Thermo-Alkalistable, High Xylooligosaccharides Producing Endo-Xylanase from Thermobifida fusca and Expression in Pichia pastoris . Appl Biochem Biotechnol 175, 1318–1329 (2015). https://doi.org/10.1007/s12010-014-1355-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-014-1355-7