
Abstract
Purpose of review
To review the diagnosis and current treatments for ocular myasthenia gravis (OMG) with a focus on recent advances.
Recent findings
Novel microRNA biomarkers show promise in predicting which patients with OMG will subsequently develop generalized disease. Newly developed clinical rating scales may enable more effective OMG-specific research. A recent meta-analysis suggested that treatment with prednisone may reduce the risk of developing generalized myasthenia gravis (GMG) in patients with OMG. Multiple new steroid-sparing immunotherapies, including complement inhibitors and neonatal Fc receptor antibodies, have proven effective in GMG and may be an option in refractory OMG patients.
Summary
Ocular myasthenia gravis is a rare disease that causes ptosis and diplopia. Many effective therapies exist, and the prognosis is good. Advances continue to be made in the diagnosis and management of OMG.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Myasthenia gravis (MG) is an autoimmune disease characterized by impaired transmission at the neuromuscular junction [1]. Resultant muscle weakness can affect any skeletal muscle—including ocular, bulbar, cervical, appendicular, and respiratory muscles—and may fluctuate throughout the day. Patients commonly present with double vision, ptosis, limb weakness, dysphagia, or slurred speech [2]. Severe weakness of respiratory or swallowing muscles can be life-threatening but occurs in a minority of patients, and most patients have good outcomes with treatment [3]. MG is a rare disease with an annual incidence between 2 and 22 cases per million and a prevalence around 60–100 per million [4]. It has a bimodal peak, with a female predominant peak between ages 20 and 30 and a male predominant peak between ages 60 and 80 [4].
Ocular symptoms are the most common presenting symptom in patients diagnosed with MG, with over half of patients having double vision or ptosis at presentation [2, 5]. Although many patients with ocular complaints also have evidence of generalized weakness, ~ 1/3 of patients have only ocular symptoms at the time of diagnosis and are classified as ocular myasthenia gravis (OMG) [2, 6]. Approximately 2/3 of patients with OMG will eventually develop systemic weakness and be diagnosed with generalized myasthenia gravis (GMG). Most progression to GMG occurs within 2 years of symptom onset [2, 5, 7], and less than 10% of patients will develop GMG after 2 years [5].
While typically thought of as mild relative to GMG, OMG can be highly distressing and debilitating. Diplopia and ptosis both interfere with vision, including vision-demanding tasks such as driving, and as a result, they negatively impact quality of life and the ability to work [8]. Fortunately, many effective treatments exist for OMG. Here, we review the pathophysiology, diagnosis, and management of OMG with a focus on recent advances.
Pathophysiology
Myasthenia gravis is caused by autoimmune injury to the post-synaptic neuromuscular junction (NMJ). Damage to the post-synaptic membrane decreases the safety factor for neuromuscular transmission, which is the difference between the size of the endplate potential and the amount of depolarization required to generate a muscle action potential [9]. A lower safety factor increases the likelihood that a given nerve signal will fail to generate muscle contraction [9]. With repeated nerve activation, as occurs in exercise, the amount of acetylcholine released into the NMJ decreases, reducing the size of the endplate potential. In the setting of a reduced safety factor, this results in the fatigable weakness characteristic of MG.
The majority (~ 90%) of MG is caused by antibodies to the acetylcholine receptor (AChR) [10]. Anti-AChR antibodies impair NMJ function via a combination of complement-mediated damage to the post-synaptic membrane, crosslinking and internalization of AChRs, and blocking of acetylcholine binding to the AChR [11]. The second most common cause of MG (accounting for 40–70% of AChR-negative GMG patients) is antibodies to muscle-specific tyrosine kinase (MuSK), which prevent clustering of AChR on the post-synaptic membrane [12, 13]. Recently, antibodies to low-density lipoprotein receptor-related protein 4 (LRP4) and agrin, which bind to each other to trigger MuSK activation, have also been shown to be pathogenic in preclinical models [14, 15].
Patients with OMG are less likely than those with GMG to have anti-AChR antibodies (only 50–60% of OMG patients have AChR antibodies) [5, 16] and almost never have anti-MuSK antibodies [13]. Anti-LRP4 or anti-agrin antibodies are only found in 15% of OMG or GMG patients without anti-AChR or anti-MuSK antibodies [17, 18•, 19, 20]. The pathophysiology of “seronegative” patients without an identified pathogenic antibody is not fully understood but is presumed to be autoimmune given patients’ robust response to immunotherapy. Up to 20–50% of seronegative OMG and GMG patients have pathogenic low-titer anti-AChR antibodies that can be detected with more sensitive cell-based assays but not with conventional radioimmunoprecipitation assays [21,22,23,24,25,26]. The remainder of seronegative disease is likely due to antibodies to other components of the post-synaptic membrane. Many additional antibodies have been associated with MG, though most have not been proven to be causal [11].
The reasons why some patients present with purely ocular symptoms remain incompletely understood. Some patients with OMG have evidence of damage to the NMJ in limb muscles in the absence of generalized symptoms [27], so it is possible that the extraocular muscles (EOMs) have a lower threshold for developing clinical weakness as a result of impaired NMJ transmission. The post-synaptic membrane in EOMs has a unique microstructure [28] and lower density of AChRs [29], which may lower the safety factor. Presynaptic nerves at the EOMs fire more rapidly than those at other skeletal muscles and secrete less acetylcholine with each firing [30], making the NMJ more susceptible to fatigability. The NMJ of EOMs may also produce fewer complement regulatory proteins, predisposing the NMJ to increased injury when AChR antibodies activate complement [31].
Diagnosis
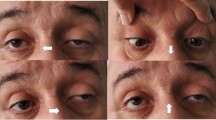
The most common presenting symptoms of OMG are ptosis and diplopia. Ptosis can be unilateral or bilateral but is often asymmetric and variable. Diplopia is binocular (present only with both eyes open) and may be vertical, horizontal, or oblique as a result of weakness in any of the EOMs. Weakness can occur in any or all of the EOMs, and, as a result, OMG can mimic an isolated cranial nerve palsy or intranuclear ophthalmoplegia. Patients may have mild orbicularis oculi weakness, but this usually does not result in incomplete eye closure (lagophthalmos). All symptoms are typically fatigable and therefore worsen as the day progresses or with sustained activity, such as occurs when driving.
Bedside testing
Multiple bedside tests can be helpful in diagnosing OMG, primarily in patients with ptosis. The presence of variable ptosis; fatigable ptosis; or the “curtain sign,” in which raising the ptotic eyelid causes the other eyelid to droop, all suggest OMG but the test characteristics of these findings are not well established. Cogan’s lid twitch, which is elicited by having the patient sustain downgaze for 15 s and observing for a brief upward eyelid twitch on return to primary gaze, has a sensitivity between 50 and 75% and a specificity of > 90% for OMG [32,33,34]. The forced eyelid closure test, in which patients squeeze their eyelids shut for several seconds before opening and assessing for twitch, has been reported to have better sensitivity (> 90%) and specificity, albeit in a small, single-center retrospective study [33]. The ice pack test may be the most studied bedside test and is performed by placing an ice pack over the patient’s closed eyes for 2 min and measuring ptosis before and after via the margin reflex distance (MRD, the distance from the center of the pupil to the upper eyelid margin). A prior systematic review of case–control studies placed the sensitivity and specificity as high as 94% and 97%, respectively [35], though recent prospective studies placed the sensitivity between 77 and 86% and specificity between 79 and 98% [36, 37••]. Performing the ice pack test after sustained upgaze may improve the sensitivity [38]. With the use of prisms to measure ocular alignment, the ice pack test can also be used in patients presenting with diplopia [36]. Edrophonium, a rapidly acting acetylcholinesterase inhibitor, was historically used in the diagnosis of MG by administering it to patients in the office and assessing for clinical improvement but is no longer commercially available in many countries, including the USA.
Serum testing
Serum antibody testing is a cornerstone in the diagnosis of MG. The specificity of anti-AChR antibodies for OMG approaches 100% (indicating that there are few to no false positives), and a positive antibody test is sufficient to make the diagnosis in the right clinical context [35]. Because only around half of OMG patients have anti-AChR or anti-MuSK antibodies [13, 39], the sensitivity of antibody testing for OMG is quite low. Testing for LRP4 or other antibodies can be considered, though the test characteristics are not well established.
Electrodiagnostics
The final widely used method to make a diagnosis of OMG is electrodiagnostic studies. Slow repetitive nerve stimulation (RNS) is highly specific (> 95%) but poorly sensitive (~ 30%) for OMG due to an inability to directly test ocular muscles [35]. Single-fiber EMG (SF-EMG) of the orbicularis oculi has excellent sensitivity (79–97%) and specificity (80–92%) but is technically challenging, uncomfortable, and only available at specialized centers [35, 37••, 40]. A recent prospective, single-center study found that SF-EMG and the ice pack test have similar test characteristics and the combination of the two has improved sensitivity (98%) and specificity (92%) [37••].
Patients with negative testing
In patients for whom clinical, antibody, and electrodiagnostic testing are all negative but a high clinical suspicion for OMG remains, an empiric trial of pyridostigmine can be considered. Subjective improvement with pyridostigmine can be difficult to interpret, so determination of response should rely on objective measures such as MRD or measurement of eye deviation with prisms.
The differential diagnosis for OMG should be revisited in patients in whom all testing is negative or the response to therapy is not as expected. Thyroid ophthalmopathy, chronic progressive external ophthalmoplegia, myotonic dystrophy, and oculopharyngeal muscular should be considered in patients with any combination of ptosis or diplopia. Congenital myasthenic syndromes (CMS), which are caused by genetic abnormalities within the neuromuscular junction, can occasionally present in adulthood and mimic seronegative OMG with abnormal RNS or SF-EMG. Some forms of CMS will even respond to pyridostigmine, but they as a rule do not respond to immunotherapy. Given this differential, genetic testing should be considered in patients who do not respond as expected to therapies.
Therapeutic approach
Supportive care
Supportive strategies can be a useful adjunct in OMG either prior to or in addition to drug therapy. For patients with ptosis, eyelid crutches can be added to glasses to improve vision during the day but are difficult to obtain and adjust. For patients with diplopia, patching of one eye will eliminate the symptom of double vision while worn, though it may reduce depth perception and is not cosmetically pleasing. Prisms can be considered in patients with diplopia, but they are not effective for dynamic and variable ocular misalignment as is typical in OMG.
Acetylcholinesterase inhibitors
First-line therapy for OMG is the acetylcholinesterase inhibitor pyridostigmine. Although there have been no randomized clinical trials of pyridostigmine, decades of use and observation have demonstrated its effectiveness [41]. Pyridostigmine blocks the degradation of acetylcholine, increasing the amount of acetylcholine available in the NMJ and thus improving neuromuscular transmission. It has a rapid (< 30 min) onset of action and lasts for about 3–4 h (see Table 1 for dosing). An extended-release version exists, but absorption is inconsistent and results in variable dosing throughout the day, limiting its usefulness. Pyridostigmine monotherapy resolves symptoms in more than half of patients with ptosis, though it is less effective for diplopia [42]. The major dose-limiting side effects are diarrhea and cramping.
Glucocorticoids
Glucocorticoids are second-line therapy for OMG and should be used in patients who continue to have symptoms while on pyridostigmine. As with pyridostigmine, there is a paucity of randomized data supporting the effectiveness of steroids and the recommendation for glucocorticoids comes primarily from expert opinion and observational studies [42, 43]. In GMG, one randomized study showed no difference in patient outcomes between patients on oral prednisone and placebo at 6 months [44] and another small study showed that a short course of IV methylprednisolone improved muscle function at 2 weeks compared to placebo [45]. In OMG, a single randomized trial of 9 patients found that prednisone increased the chance of minimal disease at 16 weeks versus placebo [46]. Initial and maintenance glucocorticoid dosing is lower for OMG than for GMG (Table 1), and one small, retrospective study suggested that initial dosing as low as 10 mg/day is sufficient [47•]. Initiation at very high doses has been reported to precipitate myasthenic crisis [48], though a recent randomized trial did not show an increased rate of exacerbations with initial high-dose prednisone [49]. Prednisone takes 2–3 weeks to have an effect and the peak effect may take 6 months. Long-term glucocorticoid use has many well-described adverse effects, including weight gain, diabetes, hypertension, peptic ulcer disease, and cataracts [50]. Once clinical remission is achieved, the glucocorticoid dosage should be weaned as low as possible.
Multiple retrospective studies have suggested that glucocorticoids reduce the risk of generalization in OMG. A meta-analysis of 8 studies found a pooled odds ratio for the development of GMG of 0.19 [51]. However, risk factors for generalization [7, 52••] were not completely controlled for in all studies and many patients received other immunosuppressants in addition to steroids. Moreover, steroids may simply mask symptoms of incipient GMG rather than prevent GMG, and a washout period would be required to determine if GMG were truly prevented. Given the low quality of the available evidence, glucocorticoids are not recommended solely for the prevention of generalized disease in patients with OMG.
Nonsteroidal immunosuppressants
Addition of a nonsteroidal immunosuppressant is indicated for patients in whom glucocorticoids do not sufficiently control symptoms, the glucocorticoid dose cannot be sufficiently tapered, or there is a contraindication to glucocorticoids. Nonsteroidal agents typically take at least 6 months to take effect. Because two-thirds of patients with OMG remain symptom-free on prednisone alone [53•], nonsteroidal agents are not typically started at the time of glucocorticoid initiation as is sometimes done in GMG. Azathioprine and mycophenolate are the most used agents, but cyclosporine, methotrexate, and tacrolimus can also be used. Overall, there is little data on nonsteroidal immunosuppressants specifically in OMG, and efficacy is extrapolated from studies in GMG.
Azathioprine is the first-line nonsteroidal agent for MG in many international guidelines [54]. A small, randomized, placebo-controlled trial in GMG showed that the addition of azathioprine to prednisolone reduced the relapse rate after 6–12 months and enabled significant reductions in glucocorticoid doses between 1 and 2 years [55]. There is no other high-quality evidence for azathioprine in GMG and no clinical trial data for azathioprine in OMG. The most common side effects are nausea, vomiting, and bone marrow suppression, and blood counts must be monitored closely at initiation. Idiosyncratic hypersensitivity causing a flu-like syndrome with fever and gastrointestinal symptoms can occur in the first several weeks and warrants discontinuation. Patients with polymorphisms in the TPMT gene are at increased risk of severe bone marrow suppression, though the role of routine screening prior to starting azathioprine is debated. Some studies have suggested a very small increased risk of malignancy, but this was not confirmed in a recent meta-analysis [56]. Xanthine oxidase inhibitors (e.g., allopurinol) used to treat gout lower the clearance of azathioprine and should be avoided.
Mycophenolate is also commonly used as a first-line nonsteroidal agent. There is no randomized data supporting the efficacy of mycophenolate, and, in fact, several double-blind, randomized controlled studies in GMG have shown no benefit in the first 9 months [57, 58]. Evidence supporting the use of mycophenolate comes from retrospective studies, primarily a large single-center retrospective analysis that showed patients treated with mycophenolate had equivalent clinical outcomes to those on prednisone at 6–12 months and that patients on mycophenolate and prednisone were able to reduce their prednisone dose at 1 year [59]. Mycophenolate can cause nausea, diarrhea, and mild leukopenia. There is a single case report of lymphoma after mycophenolate treatment, but high-quality evidence on the risk of malignancy at doses used in MG is lacking [60].
The calcineurin inhibitors cyclosporine and tacrolimus can be used in patients who do not respond to azathioprine or mycophenolate. Cyclosporine has been shown to be effective in improving motor function in GMG at 6 months in two small, randomized trials [61, 62]. A single-center retrospective analysis of cyclosporine use in GMG showed that most patients saw clinical improvement at 6 months and were able to reduce their steroid dose [63]. One small, randomized trial showed that tacrolimus reduced daily glucocorticoid doses and the need for rescue therapy over the course of 1 year [64]. Cyclosporine and tacrolimus are metabolized by the P450 system and therefore have many drug-drug interactions. Both can cause renal toxicity and hypertension and increase the risk of squamous cell cancer and lymphoma.
Methotrexate is the final commonly used oral treatment. There is no high-quality evidence to support its efficacy, and a single randomized trial of methotrexate use in AChR-positive GMG showed no benefit at 1 year [65]. Given the paucity of supporting data, the most recent international consensus recommends considering methotrexate only if patients have not tolerated or responded to any of the previously mentioned treatments [66••].
Intravenous immunoglobulin (IVIg) and plasma exchange (PLEX) are mainstays in the treatment of myasthenic crisis but have a limited role in chronic management of MG. While they are occasionally used as maintenance therapy in refractory GMG patients, they are seldom used in OMG as the side effects, cost, and inconvenience tend to outweigh the benefits.
Thymectomy
The thymus has been implicated in the development of MG because the majority of patients with AChR-positive GMG have a thymoma, thymic hyperplasia, or other thymic abnormalities [67]. For all patients with OMG or GMG in whom a thymoma is found, definitive management of the thymoma is indicated, including thymectomy in patients with resectable disease [68, 69]. Thymomas are less commonly found in OMG than GMG [70] but are found in between 1 and 11% of OMG patients [7, 8, 71, 72].
The data for thymectomy in nonthymomatous MG are less clear. Current guidelines recommend consideration of thymectomy in AChR-positive GMG patients aged 18–50 based on a positive randomized trial of transsternal thymectomy in that population [66••, 73]. In OMG, multiple case–control studies have been performed suggesting up to a 50% rate of complete remission after thymectomy, summarized in a recent meta-analysis [74]. However, the heterogeneity between studies was high and most studies had methodological limitations including use of historical controls (who are likely to be clinically different from those referred to surgery), lack of an appropriate control group, and failure to account for baseline differences in disease severity. A recent single-center study utilizing propensity score methods to account for nonrandomization did not find any difference in prednisone dose or symptom severity after thymectomy [75]. Given the paucity of high-quality data, current guidelines recommend that thymectomy be considered only in patients with OMG who do not respond adequately to pyridostigmine and either are refractory to immunosuppression, have contraindications to immunosuppression, or prefer not to take immunosuppressants [66••]. Efforts to design a clinical trial of thymectomy for early OMG are currently underway.
Surgical symptom management
Surgical interventions are commonly used to treat nonmyasthenic diplopia and ptosis but are more challenging to apply to patients with MG as symptoms tend to fluctuate throughout the day. Despite treatment, some patients with OMG develop a degree of fixed weakness manifesting as unvarying diplopia or ptosis that is likely due to permanent damage to the post-synaptic membrane. In this scenario, several groups have explored the role of surgical interventions. Small case series have reported the resolution of diplopia after strabismus surgery in up to half of patients [76] and a reduction in the degree of ptosis after surgical ptosis repair in a highly selected patient population [77, 78]. In all studies, patients had stable disease and at least 2 years of medical treatment prior to intervention.
New and emerging therapies
In recent years, several promising new therapies have been approved for GMG. Patients with OMG were excluded from all pivotal studies, so it is not clear if the results translate to OMG. Some new therapies cost upwards of $600,000 per year [79] and carry significant risks. As such, they have a limited role in the treatment of OMG but can be considered in severe, refractory cases.
Efgartigimod alfa
Efgartigimod alfa is a human IgG1-derived Fc fragment that binds to and inhibits the neonatal Fc receptor (FcRn). FcRn recycles IgGs in the body and thus prolongs the half-life of IgGs. By blocking FcRn, efgartigimod reduces serum IgG levels [80], thereby reducing disease activity. In a randomized, placebo-controlled trial, efgartigimod significantly improved patient function starting 1 week after therapy [81•]. The main side effects were headache and nasopharyngitis, and there was an increased rate of urinary tract and upper respiratory infections. There is little long-term safety data for efgartigimod.
Complement inhibitors
Ravulizumab and eculizumab are both inhibitors of the complement component C5, whose conversion to C5b initiates the formation of the membrane attack complex that is a major cause of post-synaptic damage in MG. Eculizumab is dosed every 2 weeks while ravulizumab is dosed every 8 weeks. A randomized, placebo-controlled trial of eculizumab failed to demonstrate an improvement in the primary endpoint of MG activities of daily living (MG-ADL) score at 6 months but the rate of exacerbations (a secondary endpoint) was lower in treated patients [82•]. A randomized, placebo-controlled trial of ravulizumab showed improvements in MG-ADL scores at 6 months [83]. Both medications significantly increase the risk of Neisserial infections, particularly meningitis, and require meningococcal vaccination several weeks prior to initiation. In the USA, prescribers must enroll in a Risk Evaluation and Mitigation Strategy (REMS) program to prescribe these medications. A subcutaneously administered C5 inhibitor, zilucoplan, demonstrated improvements in strength and function at 12 weeks in a randomized, double-blind, placebo-controlled trial [84] and is currently under FDA review. It will likely require similar risk monitoring.
Rituximab
Rituximab is a monoclonal antibody to CD20, a protein found on the surface of B cells. It has been widely used in other autoimmune diseases to deplete pathogenic antibody-secreting B cells, though its use in MG remains controversial. Guidelines recommend early consideration of rituximab in MuSK-positive GMG [66••], the data for which will not be discussed here due to the very low rate of MuSK-positive OMG. In AChR-positive GMG patients, the data are more mixed, with a randomized controlled trial in stable patients with mild to moderate disease showing no benefit [85] and a trial in newly diagnosed patients showing improved outcomes and fewer exacerbations at four months [86].
Research advances in ocular myasthenia
Predicting generalization
Predicting which patients with OMG will eventually develop GMG may help identify patients in whom earlier immunosuppression is warranted or guide patient selection for future trials of immunosuppressants. Patients who eventually convert to GMG are more likely to be older, be AChR-positive, have bilateral ptosis, have an abnormal RNS, and have thymic hyperplasia [7, 52••]. One group developed a nomogram for risk of generalization based on retrospective chart review of a large cohort in China that had good test characteristics but has not been validated in other populations where the rate of OMG differs [52••]. Two separate groups have found that levels of the microRNA (miRNA) miR-30e-5p are elevated in serum of patients who present with ocular symptoms and develop GMG compared to those who remain with OMG [72, 87]. This finding is promising but will need to be validated in other cohorts.
Biomarkers of severity and treatment efficacy
miRNAs have also been studied as a biomarker for disease severity in GMG. miR-30e-5p, miR-21-5p, and miR-150-5p have all been shown to be elevated in patients with more severe disease and to decline with immunosuppression [72]. One small study suggested miR-150-5p levels are reduced after thymectomy, though the effect was small and only seen at a single time point [88]. Further validation and study in OMG will be required before miRNAs are used in clinical practice.
OMG-specific disease measurement tools
Current validated measurement tools used in MG trials, such as the MG-ADL or the Quantitative Myasthenia Gravis Score (QMGS), are heavily weighted toward generalized disease [89]. A recently developed tool called OMGRate includes both objective and subjective measures and had excellent test characteristics in a single-center study [90••]. Validation in a larger study would provide helpful tool for measuring relevant outcomes in OMG.
Conclusion
Ocular myasthenia gravis is a rare disease that manifests as diplopia and ptosis and can have a profound impact on quality of life. The diagnosis can be challenging due to the low rate of serum antibody positivity but multiple bedside, serum, and electrodiagnostic tests can be used in concert to make the diagnosis. Treatment consists of stepwise progression from acetylcholinesterase inhibitors to immunotherapies, and the overall prognosis is very good. An abundance of new therapies for generalized myasthenia gravis has recently been approved that may be options in refractory disease.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Gilhus NE, Tzartos S, Evoli A, Palace J, Burns TM, Verschuuren JJGM. Myasthenia gravis. Nat Rev Dis Prim. 2019;5(1):1–19. https://doi.org/10.1038/s41572-019-0079-y.
Grob D, Arsura EL, Brunner NG, Namba T. The course of myasthenia gravis and therapies affecting outcome. Ann NY Acad Sci. 1987;505(1 Myasthenia Gr):472–99. https://doi.org/10.1111/j.1749-6632.1987.tb51317.x
Andersen JB, Gilhus NE, Sanders DB. Factors affecting outcome in myasthenia gravis. Muscle Nerve. 2016;54(6):1041–9. https://doi.org/10.1002/MUS.25205.
Carr AS, Cardwell CR, McCarron PO, McConville J. A systematic review of population based epidemiological studies in myasthenia gravis. BMC Neurol. 2010;10. https://doi.org/10.1186/1471-2377-10-46
Sanders DB, Raja SM, Guptill JT, Hobson-Webb LD, Juel VC, Massey JM. The Duke myasthenia gravis clinic registry: I. Description and demographics Muscle Nerve. 2021;63(2):209–16. https://doi.org/10.1002/mus.27120.
Jaretzki A, Barohn RJ, Ernstoff RM, Kaminski HJ, Keesey JC, Penn AS, et al. Myasthenia gravis. Neurology. 2000;55(1):16–23. https://doi.org/10.1212/WNL.55.1.16.
Feng X, Huan X, Yan C, Song J, Lu J, Zhou L, et al. Adult ocular myasthenia gravis conversion: a single-center retrospective analysis in China. Eur Neurol. 2020;83(2):182–8. https://doi.org/10.1159/000507853.
Suzuki S, Murai H, Imai T, Nagane Y, Masuda M, Tsuda E, et al. Quality of life in purely ocular myasthenia in Japan. BMC Neurol. 2014;14(1):142. https://doi.org/10.1186/1471-2377-14-142.
Ruff RL, Lennon VA. How myasthenia gravis alters the safety factor for neuromuscular transmission. J Neuroimmunol. 2008;201–202(C):13. https://doi.org/10.1016/J.JNEUROIM.2008.04.038
Chan KH, Lachance DH, Harper CM, Lennon VA. Frequency of seronegativity in adult-acquired generalized myasthenia gravis. Muscle Nerve. 2007;36(5):651–8. https://doi.org/10.1002/mus.20854.
Gilhus NE, Skeie GO, Romi F, Lazaridis K, Zisimopoulou P, Tzartos S. Myasthenia gravis - autoantibody characteristics and their implications for therapy. Nat Rev Neurol. 2016;12(5):259–68. https://doi.org/10.1038/nrneurol.2016.44.
Hoch W, Mcconville J, Helms S, Newsom-Davis J, Melms A, Vincent A. Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat Med. 2001;7(3):365–8. https://doi.org/10.1038/85520.
McConville J, Farrugia ME, Beeson D, Kishore U, Metcalfe R, Newsom-Davis J, et al. Detection and characterization of MuSK antibodies in seronegative myasthenia gravis. Ann Neurol. 2004;55(4):580–4. https://doi.org/10.1002/ANA.20061.
Yan M, Liu Z, Fei E, Chen W, Lai X, Luo B, et al. Induction of anti-agrin antibodies causes myasthenia gravis in mice. Neuroscience. 2018;373:113–21. https://doi.org/10.1016/j.neuroscience.2018.01.015.
Shen C, Lu Y, Zhang B, Figueiredo D, Bean J, Jung J, et al. Antibodies against low-density lipoprotein receptor-related protein 4 induce myasthenia gravis. J Clin Invest. 2013;123. https://doi.org/10.1172/JCI66039
Tindall RSA. Humoral immunity in myasthenia gravis: biochemical characterization of acquired antireceptor antibodies and clinical correlations. Ann Neurol. 1981;10(5):437–47. https://doi.org/10.1002/ana.410100506.
Higuchi O, Hamuro J, Motomura M, Yamanashi Y. Autoantibodies to low-density lipoprotein receptor-related protein 4 in myasthenia gravis. Ann Neurol. 2011;69(2):418–22. https://doi.org/10.1002/ana.22312.
• Rivner MH, Quarles BM, Pan JX, Yu Z, Howard JF, Corse A, et al. Clinical features of LRP4/agrin-antibody–positive myasthenia gravis: a multicenter study. Muscle Nerve. 2020;62(3):333. https://doi.org/10.1002/MUS.26985. Largest study on the prevalence and clinical characteristics of LRP4 and agrin antibodies.
Wang S, Yang H, Guo R, Wang L, Zhang Y, Lv J, et al. Antibodies to full-length agrin protein in Chinese patients with myasthenia gravis. Front Immunol. 2021;12. https://doi.org/10.3389/FIMMU.2021.753247
Zhang B, Shen C, Bealmear B, Ragheb S, Xiong WC, Lewis RA, et al. Autoantibodies to agrin in myasthenia gravis patients. PLoS One. 2014;9(3). https://doi.org/10.1371/JOURNAL.PONE.0091816
Mirian A, Nicolle MW, Edmond P, Budhram A. Comparison of fixed cell-based assay to radioimmunoprecipitation assay for acetylcholine receptor antibody detection in myasthenia gravis. J Neurol Sci. 2022;432:120084. https://doi.org/10.1016/J.JNS.2021.120084
Spagni G, Gastaldi M, Businaro P, Chemkhi Z, Carrozza C, Mascagna G, et al. Comparison of fixed and live cell-based assay for the detection of AChR and MuSK antibodies in myasthenia gravis. Neurol Neuroimmunol Neuroinflammation. 2023;10(1). https://doi.org/10.1212/NXI.0000000000200038
Jacob S, Viegas S, Leite MI, Webster R, Cossins J, Kennett R, et al. Presence and pathogenic relevance of antibodies to clustered acetylcholine receptor in ocular and generalized myasthenia gravis. Arch Neurol. 2012;69(8):994–10005. https://doi.org/10.1001/archneurol.2012.437.
Yang L, Maxwell S, Leite MI, Waters P, Clover L, Fan X, et al. Non-radioactive serological diagnosis of myasthenia gravis and clinical features of patients from Tianjin. China J Neurol Sci. 2011;301(1–2):71–6. https://doi.org/10.1016/J.JNS.2010.10.023.
Leite MI, Jacob S, Viegas S, Cossins J, Clover L, Morgan BP, et al. IgG1 antibodies to acetylcholine receptors in ‘seronegative’ myasthenia gravis. Brain. 2008;131(7):1940–52. https://doi.org/10.1093/BRAIN/AWN092.
Cruz PMR, Al-Hajjar M, Huda S, Jacobson L, Woodhall M, Jayawant S, et al. Clinical features and diagnostic usefulness of antibodies to clustered acetylcholine receptors in the diagnosis of seronegative myasthenia gravis. JAMA Neurol. 2015;72(6):642–9. https://doi.org/10.1001/JAMANEUROL.2015.0203.
Tsujihata M, Hazama R, Ishii N, Ide Y, Mori M, Takamori M. Limb muscle endplates in ocular myasthenia gravis: quantitative ultrastructural study. Neurology. 1979;29(5):654–61. https://doi.org/10.1212/WNL.29.5.654.
Spencer RF, Porter JD. Structural organization of the extraocular muscles. Rev Oculomot Res. 1988;2:33–79.
Liu R, Xu H, Wang G, Li J, Gou L, Zhang L, et al. Extraocular muscle characteristics related to myasthenia gravis susceptibility. PLoS One. 2013;8(2). https://doi.org/10.1371/JOURNAL.PONE.0055611
Kaminski HJ, Maas E, Spiegel P, Ruff RL. Why are eye muscles frequently involved in myasthenia gravis? Neurology. 1990;40(11):1663–1663. https://doi.org/10.1212/WNL.40.11.1663.
Kaminski HJ, Li Z, Richmonds C, Lin F, Medof ME. Complement regulators in extraocular muscle and experimental autoimmune myasthenia gravis. Exp Neurol. 2004;189(2):333–42. https://doi.org/10.1016/j.expneurol.2004.06.005.
Singman EL, Matta NS, Silbert DI. Use of the Cogan lid twitch to identify myasthenia gravis. J Neuroophthalmol. 2011;31(3):239–40. https://doi.org/10.1097/WNO.0B013E3182224B92.
Apinyawasisuk S, Zhou X, Tian JJ, Garcia GA, Karanjia R, Sadun AA. Validity of forced eyelid closure test: a novel clinical screening test for ocular myasthenia gravis. J Neuro-Ophthalmology. 2017;37(3):253–7. https://doi.org/10.1097/WNO.0000000000000514.
Van Stavern GP, Bhatt A, Haviland J, Black EH. A prospective study assessing the utility of Cogan’s lid twitch sign in patients with isolated unilateral or bilateral ptosis. J Neurol Sci. 2007;256(1–2):84–5. https://doi.org/10.1016/J.JNS.2007.02.020.
Benatar M. A systematic review of diagnostic studies in myasthenia gravis. Neuromuscul Disord. 2006;16(7):459–67. https://doi.org/10.1016/j.nmd.2006.05.006.
Chatzistefanou KI, Kouris T, Iliakis E, Piaditis G, Tagaris G, Katsikeris N, et al. The ice pack test in the differential diagnosis of myasthenic diplopia. Ophthalmology. 2009;116(11):2236–43. https://doi.org/10.1016/J.OPHTHA.2009.04.039.
•• Giannoccaro MP, Paolucci M, Zenesini C, Di Stasi V, Donadio V, Avoni P, et al. Comparison of ice pack test and single-fiber EMG diagnostic accuracy in patients referred for myasthenic ptosis. Neurology. 2020;95(13):e1800–6. https://doi.org/10.1212/WNL.0000000000010619. Important comparison of test characteristics of the ice pack test and SF-EMG.
Kee HJ, Yang HK, Hwang JM, Park KS. Evaluation and validation of sustained upgaze combined with the ice-pack test for ocular myasthenia gravis in Asians. Neuromuscul Disord. 2019;29(4):296–301. https://doi.org/10.1016/J.NMD.2018.12.011.
Evoli A, Tonali P, Bartoccioni E, Monaco ML. Ocular myasthenia: diagnostic and therapeutic problems. Acta Neurol Scand. 1988;77(1):31–5. https://doi.org/10.1111/J.1600-0404.1988.TB06970.X
Giannoccaro MP, Di Stasi V, Zanesini C, Donadio V, Avoni P, Liguori R. Sensitivity and specificity of single-fibre EMG in the diagnosis of ocular myasthenia varies accordingly to clinical presentation. J Neurol. 2020;267(3):739–45. https://doi.org/10.1007/S00415-019-09631-3.
Mehndiratta MM, Pandey S, Kuntzer T. Acetylcholinesterase inhibitor treatment for myasthenia gravis. Cochrane Database Syst Rev. 2014;2014(10). https://doi.org/10.1002/14651858.CD006986.PUB3
Kupersmith MJ, Ying G. Ocular motor dysfunction and ptosis in ocular myasthenia gravis: effects of treatment. Br J Ophthalmol. 2005;89(10):1330–4. https://doi.org/10.1136/BJO.2004.063404.
Schneider-Gold C, Gajdos P, Toyka K V., Hohlfeld RR. Corticosteroids for myasthenia gravis. Cochrane Database Syst Rev. 2005;2011(6). https://doi.org/10.1002/14651858.CD002828.pub2
Howard FM, Duane DD, Lambert EH, Daube JR. Alternate-day prednisone: preliminary report of a double-blind controlled study. Ann NY Acad Sci. 1976;274(1):596–607. https://doi.org/10.1111/J.1749-6632.1976.TB47718.X.
Lindberg C, Andersen O, Lefvert AK. Treatment of myasthenia gravis with methylprednisolone pulse: a double blind study. Acta Neurol Scand. 1998;97(6):370–3. https://doi.org/10.1111/J.1600-0404.1998.TB05968.X.
Benatar M, Mcdermott MP, Sanders DB, Wolfe GI, Barohn RJ, Nowak RJ, et al. Efficacy of prednisone for the treatment of ocular myasthenia (EPITOME): a randomized, controlled trial. Muscle Nerve. 2016;53(3):363–9. https://doi.org/10.1002/mus.24769.
Shah YS, Henderson AD, Carey AR. Effect of initial prednisone dosing on ocular myasthenia gravis control. J Neuroophthalmol. 2021;41(4):e622–6. https://doi.org/10.1097/WNO.0000000000001058.N. Retrospective study supporting the use of low-dose prednisone in OMG.
Pascuzzi RM, Coslett HB, Johns TR. Long-term corticosteriod treatment of myasthenia gravis: report of 116 patients. Ann Neurol. 1984;15(3):291–8. https://doi.org/10.1002/ANA.410150316.
Sharshar T, Porcher R, Demeret S, Tranchant C, Gueguen A, Eymard B, et al. Comparison of corticosteroid tapering regimens in myasthenia gravis: a randomized clinical trial. JAMA Neurol. 2021;78(4):426–33. https://doi.org/10.1001/JAMANEUROL.2020.5407.
Oray M, Samra KA, Ebrahimiadib N, Meese H, Foster CS. Long-term side effects of glucocorticoids. Expert Opin Drug Saf. 2016;15(4):457–65. https://doi.org/10.1517/14740338.2016.1140743.
Li M, Ge F, Guo R, Ruan Z, Gao Y, Niu C, et al. Do early prednisolone and other immunosuppressant therapies prevent generalization in ocular myasthenia gravis in western populations: a systematic review and meta-analysis. Ther Adv Neurol Disord. 2019;12:175628641987652. https://doi.org/10.1177/1756286419876521.
•• Ruan Z, Sun C, Lang Y, Gao F, Guo R, Xu Q, et al. Development and validation of a nomogram for predicting generalization in patients with ocular myasthenia gravis. Front Immunol. 2022;13. https://doi.org/10.3389/fimmu.2022.895007. Provides a nomogram for predicting generalization of GMG based on commonly collected clinical characteristics.
• Verma R, Wolfe GI, Kupersmith MJ. Ocular myasthenia gravis – how effective is low dose prednisone long term? J Neurol Sci. 2021;420:117274. https://doi.org/10.1016/J.JNS.2020.117274. Provides long-term retrospective data on the efficacy of prednisone in OMG.
Sanders DB, Wolfe GI, Benatar M, Evoli A, Gilhus NE, Illa I, et al. International consensus guidance for management of myasthenia gravis: executive summary. Neurology. 2016;87(4):419–25. https://doi.org/10.1212/WNL.0000000000002790.
Palace J, Newsom-Davis J, Lecky B. A randomized double-blind trial of prednisolone alone or with azathioprine in myasthenia gravis. Neurology. 1998;50(6):1778–83. https://doi.org/10.1212/WNL.50.6.1778.
Zhang Z, Wang M, Xu L, Jiang B, Jin T, Shi T, et al. Cancer occurrence following azathioprine treatment in myasthenia gravis patients: a systematic review and meta-analysis. J Clin Neurosci. 2021;88:70–4. https://doi.org/10.1016/J.JOCN.2021.03.015.
Sanders DB, Hart IK, Mantegazza R, Shukla SS, Siddiqi ZA, De Baets MHV, et al. An international, phase III, randomized trial of mycophenolate mofetil in myasthenia gravis. Neurology. 2008;71(6):400–6. https://doi.org/10.1212/01.WNL.0000312374.95186.CC.
Sanders DB. A trial of mycophenolate mofetil with prednisone as initial immunotherapy in myasthenia gravis. Neurology. 2008;71(6):394–9. https://doi.org/10.1212/01.WNL.0000312373.67493.7F.
Hehir MK, Burns TM, Alpers J, Conaway MR, Sawa M, Sanders DB. Mycophenolate mofetil in AChR-antibody-positive myasthenia gravis: outcomes in 102 patients. Muscle Nerve. 2010;41(5):593–8. https://doi.org/10.1002/mus.21640.
Vernino S, Salomao DR, Habermann TM, O’Neill BP. Primary CNS lymphoma complicating treatment of myasthenia gravis with mycophenolate mofetil. Neurology. 2005;65(4):639–41. https://doi.org/10.1212/01.WNL.0000173031.56429.04.
Tindall RSA, Rollins JA, Phillips JT, Greenlee RG, Wells L, Belendiuk G. Preliminary results of a double-blind, randomized, placebo-controlled trial of cyclosporine in myasthenia gravis. N Engl J Med. 1987;316(12):719–24. https://doi.org/10.1056/NEJM198703193161205.
Tindall RSA, Phillips JT, Rollins JA, Wells L, Hall K. A clinical therapeutic trial of cyclosporine in myasthenia gravis. Ann N Y Acad Sci. 1993;681(1 Myasthenia Gr):539–51. https://doi.org/10.1111/j.1749-6632.1993.tb22937.x
Ciafaloni E, Nikhar NK, Massey JM, Sanders DB. Retrospective analysis of the use of cyclosporine in myasthenia gravis. Neurology. 2000;55(3):448–50. https://doi.org/10.1212/WNL.55.3.448.
Nagane Y, Utsugisawa K, Obara D, Kondoh R, Terayama Y. Efficacy of low-dose FK506 in the treatment of myasthenia gravis–a randomized pilot study. Eur Neurol. 2005;53(3):146–50. https://doi.org/10.1159/000085833.
Pasnoor M, He J, Herbelin L, Burns TM, Nations S, Bril V, et al. A randomized controlled trial of methotrexate for patients with generalized myasthenia gravis. Neurology. 2016;87(1):57–64. https://doi.org/10.1212/WNL.0000000000002795
•• Narayanaswami P, Sanders DB, Wolfe G, Benatar M, Cea G, Evoli A, et al. International consensus guidance for management of myasthenia gravis. Neurology. 2021;96(3):114–22. https://doi.org/10.1212/WNL.0000000000011124. Updated international consensus with specific recommendations regarding thymectomy and OMG.
Marx A, Pfister F, Schalke B, Saruhan-Direskeneli G, Melms A, Ströbel P. The different roles of the thymus in the pathogenesis of the various myasthenia gravis subtypes. Autoimmun Rev. 2013;12(9):875–84. https://doi.org/10.1016/j.autrev.2013.03.007.
Falkson CB, Bezjak A, Darling G, Gregg R, Malthaner R, Maziak DE, et al. The management of thymoma: a systematic review and practice guideline. J Thorac Oncol. 2009;4(7):911–9. https://doi.org/10.1097/JTO.0B013E3181A4B8E0.
Ettinger DS, Akerley W, Bepler G, Blum MG, Chang A, Cheney RT, et al. Thymic malignancies. J Natl Compr Canc Netw. 2010;8(11):1302–15. https://doi.org/10.6004/JNCCN.2010.0096.
Álvarez-Velasco R, Gutiérrez-Gutiérrez G, Trujillo JC, Martínez E, Segovia S, Arribas-Velasco M, et al. Clinical characteristics and outcomes of thymoma-associated myasthenia gravis. Eur J Neurol. 2021;28(6):2083–91. https://doi.org/10.1111/ene.14820.
Kupersmith MJ, Latkany R, Homel P. Development of generalized disease at 2 years in patients with ocular myasthenia gravis. Arch Neurol. 2003;60(2):243–8. https://doi.org/10.1001/ARCHNEUR.60.2.243.
Sabre L, Maddison P, Wong SH, Sadalage G, Ambrose PA, Plant GT, et al. miR-30e-5p as predictor of generalization in ocular myasthenia gravis. Ann Clin Transl Neurol. 2019;6(2):243–51. https://doi.org/10.1002/acn3.692.
Wolfe GI, Kaminski HJ, Aban IB, Minisman G, Kuo HC, Marx A, et al. Randomized trial of thymectomy in myasthenia gravis. N Engl J Med. 2016;375(6):511–22. https://doi.org/10.1056/NEJMOA1602489.
Zhu K, Li J, Huang X, Xu W, Liu W, Chen J, et al. Thymectomy is a beneficial therapy for patients with non-thymomatous ocular myasthenia gravis: a systematic review and meta-analysis. Neurol Sci. 2017;38(10):1753–60. https://doi.org/10.1007/s10072-017-3058-7.
Hamedani AG, Pistilli M, Singhal S, Shindler KS, Avery RA, Tamhankar MA, et al. Outcomes after transcervical thymectomy for ocular myasthenia gravis: a retrospective cohort study with inverse probability weighting. J Neuroophthalmol. 2020;40(1):8–14. https://doi.org/10.1097/WNO.0000000000000814.
Peragallo JH, Velez FG, Demer JL, Pineles SL. Long-term follow-up of strabismus surgery for patients with ocular myasthenia gravis. J Neuro-Ophthalmology. 2013;33(1):40–4. https://doi.org/10.1097/WNO.0b013e318280d630.
Shimizu Y, Suzuki S, Nagasao T, Ogata H, Yazawa M, Suzuki N, et al. Surgical treatment for medically refractory myasthenic blepharoptosis. Clin Ophthalmol. 2014;8:1859. https://doi.org/10.2147/OPTH.S69883.
Brogan K, Farrugia ME, Crofts K. Ptosis surgery in patients with myasthenia gravis: a useful adjunct to medical therapy. Semin Ophthalmol. 2018;33(3):429–34. https://doi.org/10.1080/08820538.2017.1284871.
Lien PW, Agboola F, Joshi M, Nikitin D, Yousif Z, Patel S, et al. Cost-effectiveness of eculizumab and efgartigimod for the treatment of generalized myasthenia gravis (p1–1.virtual). Neurology. 2022;98(18 Supplement).
Ulrichts P, Guglietta A, Dreier T, Van Bragt T, Hanssens V, Hofman E, et al. Neonatal Fc receptor antagonist efgartigimod safely and sustainably reduces IgGs in humans. J Clin Invest. 2018;128(10):4372. https://doi.org/10.1172/JCI97911.
• Howard JF, Bril V, Vu T, Karam C, Peric S, Margania T, et al. Safety, efficacy, and tolerability of efgartigimod in patients with generalised myasthenia gravis (ADAPT): a multicentre, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2021;20(7):526–36. https://doi.org/10.1016/S1474-4422(21)00159-9. Randomized trial supporting the effectiveness of efgartigimod.
• Howard JF, Utsugisawa K, Benatar M, Murai H, Barohn RJ, Illa I, et al. Safety and efficacy of eculizumab in anti-acetylcholine receptor antibody-positive refractory generalised myasthenia gravis (REGAIN): a phase 3, randomised, double-blind, placebo-controlled, multicentre study. Lancet Neurol. 2017;16(12):976–86. https://doi.org/10.1016/S1474-4422(17)30369-1. Randomized trial supporting the effectiveness of eculizumab.
Vu T, Meisel A, Mantegazza R, Annane D, Katsuno M, Aguzzi R, et al. Terminal complement inhibitor ravulizumab in generalized myasthenia gravis. NEJM Evid. 2022;1(5). https://doi.org/10.1056/EVIDoa2100066
Howard JF, Nowak RJ, Wolfe GI, Freimer ML, Vu TH, Hinton JL, et al. Clinical effects of the self-administered subcutaneous complement inhibitor zilucoplan in patients with moderate to severe generalized myasthenia gravis: results of a phase 2 randomized, double-blind, placebo-controlled, multicenter clinical trial. JAMA Neurol. 2020;77(5):582–92. https://doi.org/10.1001/JAMANEUROL.2019.5125.
Nowak RJ, Coffey CS, Goldstein JM, Dimachkie MM, Benatar M, Kissel JT, et al. Phase 2 trial of rituximab in acetylcholine receptor antibody-positive generalized myasthenia gravis: the BeatMG study. Neurology. 2021;98(4):E376–89. https://doi.org/10.1212/WNL.0000000000013121.
Piehl F, Eriksson-Dufva A, Budzianowska A, Feresiadou A, Hansson W, Hietala MA, et al. Efficacy and safety of rituximab for new-onset generalized myasthenia gravis: the RINOMAX randomized clinical trial. JAMA Neurol. 2022;79(11). https://doi.org/10.1001/JAMANEUROL.2022.2887
Beretta F, Huang YF, Punga AR. Towards personalized medicine in myasthenia gravis: role of circulating microRNAs miR-30e-5p, miR-150–5p and miR-21–5p. Cells. 2022;11(4). https://doi.org/10.3390/CELLS11040740
Molin CJ, Sabre L, Weis CA, Punga T, Punga AR. Thymectomy lowers the myasthenia gravis biomarker miR-150–5p. 2018;5:450. https://doi.org/10.1212/NXI.0000000000000450
Barnett C, Herbelin L, Dimachkie MM, Barohn RJ. Measuring clinical treatment response in myasthenia gravis. Neurol Clin. 2018;36(2):339. https://doi.org/10.1016/J.NCL.2018.01.006.
•• Wong SH, Eggenberger E, Cornblath W, Xhepa A, Miranda E, Lee H, et al. Preliminary findings of a dedicated ocular myasthenia gravis rating scale: the OMGRate. Neuro-Ophthalmology. 2020;44(3):148. https://doi.org/10.1080/01658107.2019.1660686. Provides a new, preliminarily validated clinical and research tool to measure outcomes in OMG.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Alexander H. Morrison declares that he has no conflict of interest. Grant T. Liu declares that he has no conflict of interest. Ali G. Hamedani declares that he has no conflict of interest.
Human and Animal Rights and Informed Consent
All reported studies/experiments with human or animal subjects performed by the authors have been previously published and complied with all applicable ethical standards (including the Helsinki declaration and its amendments, institutional/national research committee standards, and international/national/institutional guidelines).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Morrison, A.H., Liu, G.T. & Hamedani, A.G. Ocular Myasthenia Gravis. Curr Treat Options Neurol 25, 151–167 (2023). https://doi.org/10.1007/s11940-023-00753-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11940-023-00753-8