
Abstract
Purpose of Review
Type I interferons (IFNαβ) induce the expression of hundreds of genes; thus, it is unsurprising that the initiation, transmission, and resolution of the IFNαβ-mediated immune response is tightly controlled. Mutations that alter nucleic acid processing and recognition, ablate IFNαβ-specific negative feedback mechanisms, or result in dysfunction of the proteasome system can all induce pathogenic IFNαβ signalling and are the focus of this review.
Recent Findings
Recent advances have delineated the precise cytoplasmic mechanisms that facilitate self-DNA to be recognised by cGAS and self-RNA to be recognised by RIG-I or MDA-5. This helps clarify interferonopathies associated with mutations in genes which code for DNase-II and ADAR1, among others. Similarly, loss of function mutations in Pol α, which lowers the presence of antagonistic ligands in the cytosol, or gain of function mutations in RIG-I and MDA-5, result in increased propensity for receptor activation and therefore IFNαβ induction.
Summary
As the aetiology of monogenic autoinflammatory diseases are uncovered, novel and sometimes unsuspected molecular interactions and signalling pathways are being defined. This review covers developments that have come to light over the past 3 years, with reference to the study of interferonopathies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Monogenic type I interferonopathies comprise a group of heterogeneous autoinflammatory diseases associated with constitutive activation of type I interferon (IFNαβ) signalling. IFNαβ is a multigene cytokine family that comprises multiple IFNα subtypes (13 in human and 14 in mouse), a single IFNβ gene, and other family members (IFN-ω, -ɛ, -δ, and -κ) (reviewed in [1]). Early descriptions of this family characterised IFNαβ as factors released from cells that exhibited potent antiviral activity, and it is now well accepted that IFNαβ induces the expression of gene programs critical for the control and clearance of most viruses (reviewed in [2]).
IFNαβ can be secreted by almost all cell types in the body, primarily in response to activation of pattern recognition receptors (PRRs) that sense foreign or self-derived nucleic acids. Once secreted, IFNαβ acts in both an autocrine and paracrine manner to exclusively engage the ubiquitously expressed IFNαβ Receptor (IFNαβR). Ligand binding to IFNαβR activates a Janus kinase (JAK)/signal transducers and activators of the transcription (STAT) pathway and triggers the transcription of a diverse suite of genes known as IFN-stimulated genes (ISGs) (Fig. 1). In addition to cell intrinsic anti-microbial effectors, IFNαβ induces expression of cytokines and chemokines, pro- and anti-apoptotic proteins, and molecules involved in cellular metabolic processes (reviewed in [3]). Given the extensive range of cellular processes modified by IFNαβ signalling and the universal expression of both receptor and ligands, it is no wonder that IFNαβ signalling has multiple levels of regulation, and that inappropriate activation or propagation of the IFNαβ response can lead to severe autoinflammatory disease.

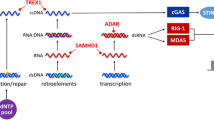
Newly described molecular mechanisms of IFNαβ driven autoinflammation. Mutations leading to loss of function are indicated by black stars and red stars denote gain of function. Mutations which lower the threshold for initiation of the IFNαβ response or result in decreased control of downstream signalling can all drive an IFNαβ-mediated autoinflammatory disease. A. Gain of function mutations in the genes coding for MDA5 and Rig-I result in increased sensitivity of these PRRs to cytosolic nucleic acids, leading to SMS or AGS. Congruently, loss of function mutations in genes that code for sensors of cytoplasmic nucleic acid can lower the threshold for PRR activation or lead to activation by self RNA or DNA molecules. B. DNase II degrades nucleic acids generated during apoptosis and phagocytosis, thus loss of function mutations in the DNase II gene result in a build-up of cytosolic DNA and consequent activation of cGas. C. XLPDR is driven by the loss of the gene product of POLA1. POLA1 deficiency decreases the presence of cytosolic RNA:DNA hybrid molecules, which may act as antagonists to certain PRRs, thereby raising the threshold for signal generation. D. ADAR1 editing of Alu-Alu inverted repeats in dsRNA lowers their binding affinity to MDA5 and PKR, thereby inhibiting inappropriate activation of IFNαβ. Loss of ADAR1 function therefore results in AGS. E. Uncontrolled IFNαβ signalling in Pseudo-TORCH Syndrome 2 is driven by loss of USP18. USP18 negatively regulates IFNαβ signalling by binding to IFNAR2, thereby blocking interaction between this receptor subunit and the signalling molecule: JAK1. F. Mutations which result in proteasome dysfunction drive the build-up of poly ubiquintinated proteins, which activates IFNαβ signalling through an, as yet, undiscovered mechanism. G. Mutations which decrease COPα function result in an immune dysregulatory disease with an ISG signature. H. Gain of function mutations causing the constitutive activation STING drive SAVI
Although the term itself was only coined in 2011, diseases now classed as type I interferonopathies have been reported and studied for almost a century, the prototypic of which being Aicardi-Goutières syndrome (AGS). They are highly heterogeneous, and approximately 20 genes have been implicated in driving IFNαβ expression and pathogenesis in autoinflammatory disorders. Due to the rarity of patients, the variety of genetic causes and the relative infancy of this field, the full spectrum of clinical presentation is yet to be totally described. Interestingly, as clinical presentation generally occurs in patient infancy, parallels have been drawn between monogenic interferonopathies and in utero/early life systemic viral infection, where IFNαβ signalling is pronounced [4]. As per the scope of this review, we will only focus on advances made over the last 3 years; however, we recommend reviews by Rodero et al. 2016 [5] and Kretschmer et al. 2017 [6] for a more comprehensive history and discussion of Interferonopathies.
Interferonopathies Driven by Nucleic Acid Sensors
Mutations causing increased sensitivity of PRRs to nucleic acids can drive constitutive activation of IFNαβ signalling. One example of this is Singleton-Merten Syndrome (SMS), an autosomal-dominant multi-system interferonopathy characterised by progressive calcifications of large blood vessels, dental and skeletal anomalies, osteoporosis, and less commonly: generalised muscle weakness, psoriasis, and early-onset glaucoma [7]. By performing whole-exome sequencing on three unrelated SMS families, Rutsch et al. identified a missense mutation in IFIH1: c.2465G > A (p.R822Q), which encodes the RNA sensor melanoma differentiation-associated protein 5 (MDA5). This arginine to glutamine substitution occurs within one of the core helicase domains of MDA5 (HEL2); MDA5 has two helicase domains which are responsible for binding RNA and RNA-dependent adenosine triphosphate (ATP) hydrolysis. R822Q is proximal to the a highly conserved amino acid motif in HEL2 which mediates MDA5 conformational changes resulting in the creation of a high-affinity nucleic acid binding site [8]. Thus, this mutation may induce conformational changes which enhance MDA5 filament stability and consequent MDA5 induction of IFNαβ [9]. In line with this, whole blood samples from SMS patients exhibited elevated expression of ISGs and in vitro overexpression studies revealed that this mutation conferred MDA5 hyperactivity to self and non-self-dsRNA [9] (Fig. 1A).
Interestingly, six other gain of function point mutations in IFIH1 have been reported in the literature, almost all affecting MDA5 helicase domains. However, patients harbouring these mutations predominantly exhibit neurological symptoms, and as such, are diagnosed with AGS [10, 11]. IFIH1-mediated AGS (AGS7) presents with delayed psychomotor development, spasticity, basal ganglia calcification, cerebral atrophy, and abnormalities of the deep white matter [11], which is distinct from SMS clinical features that manifest predominantly in vessel, cutaneous and osseous tissue [7, 9]. However, phenotypic overlap between the two interferonopathies has been recently described. In 2017, Bursztejn et al. identified three patients from a single family with a pathogenic heterozygous mutation in IFIH1 (c.1465G > A, p.A489T), who presented with an elevated ISG signature in whole blood, and both AGS-like (neurological) and SMS-like (dental) features [12]. Furthermore, Buers et al. reported a single patient who presented with high levels of ISG expression in his peripheral blood mononuclear cells (PBMCs) and a constellation of symptoms highly suggestive of AGS. Yet, upon whole-exome sequencing it was found that this patient harboured the p.R822Q mutation in IFIH1, previously associated with SMS [13•]. Collectively, these studies indicate that there is a phenotypic continuum associated with mutations in IFIH1; however, what other factors influence clinical presentation is an open and exciting question.
Atypical SMS is less severe than classical SMS, presenting with glaucoma yet without dental anomalies [14]. Jang et al. described two mutations in DDX58: c.803G > T (p.C268F) and c.1118A > C (p.E373A) which are located in the ATP-binding motifs I and II of another RNA sensor: retinoic acid-inducible gene I (Rig-I). As ATP binding and hydrolysis on Rig-I prevents induction of IFNαβ upon encounter with host RNA molecules [15], the authors hypothesise that the gain of function effect on Rig-I signalling in these patients is due to an altered interaction between ATP and Rig-I, which lowers the threshold required for induction of IFNαβ (Fig. 1A) [14].
In contrast to gain of function mutations in nucleic acid sensors, a suite of mutations in other genes can increase the abundance of ligands for PRRs, which we will now discuss.
Interferonopathies Caused by Inappropriate Regulation of Nucleic Acids
Constitutive activation of IFNαβ signalling can result from loss of function mutations in genes that code for regulators of nucleic acid presence in the cytosol. Rodero et al. recently identified three patients from two unrelated families demonstrating a spectrum of clinical features including: resolving neonatal anaemia, membranoproliferative glomerulonephritis, liver fibrosis, deforming arthropathy and increased anti-DNA antibodies. This pathology was accompanied by global upregulation of ISGs in whole blood samples and increased serum levels of IFNα. There was also a non-interferon-mediated inflammatory signature in patient serum, characterised by NFκB-driven cytokines including TNFα. Whole-exome sequencing of patient and parental DNA revealed biallelic mutations in DNASE2: c.347G > C (p.G116A) in two siblings and c.362A > T (p.D121V) in the third, unrelated, patient [16]. DNASE2 encodes the lysosomal endonuclease DNase II, which is essential for the digestion of cytosolic DNA generated through apoptosis and the phagocytosis of maturating erythroblast nuclei [17]. Both p.G116A and p.D121V decreased the endonuclease activity of DNase II, lysates from patient fibroblasts exhibited impaired ability to digest plasmid DNA compared to healthy controls and this phenotype could be rescued by the expression of wild-type DNase II [16]. The absence of DNase II in mice leads to the accumulation of undigested DNA in the lysosomes of macrophages, resulting in chronic IFNαβ signalling via the activation of the cytosolic PRR: cyclic GMP-AMP synthase (cGas) (Fig. 1B) and consequently lethal perinatal anaemia [16, 18]. Although not shown directly, Rodero et al. reason that the cGas pathway is similarly activated in mutant DNase II patients leading to IFNαβ driven autoinflammatory disease.
X-linked reticulate pigmentary disorder (XLPDR) is caused by an intronic mutation (c.1375–354A > G) in POLA1, which alters gene splicing. POLA1 encodes the catalytic subunit of DNA polymerase-α (Pol α) and this mutation lowered Pol α expression [19]. Pol α complexes with primase to synthesise the short RNA-DNA primer required for initiating DNA synthesis during DNA replication [20]. XLPDR is characterised by diffuse skin hyperpigmentation with a distinctive reticulate pattern, coupled with recurrent pneumonias, bronchiectasis, chronic diarrhoea, and failure to thrive. Starokadomskyy et al. analysed affected and unaffected individuals from 12 XLPDR families and reported decreased expression of POLA1 mRNA in XLPDR patient-derived cell lines which correlated to constitutive enhancement of IRF- and NFκB-dependent gene expression. Furthermore, analysis of blood samples from XLPDR patients revealed significantly elevated IFNα2 concentrations and elevated ISG expression [19]. This characterisation of XLPDR has revealed a novel role for POLA1 in promoting the synthesis of cytosolic DNA duplexes consisting of one DNA and one RNA strand. In the absence of POLA1 these duplexes were downregulated, the authors suggesting that cytosolic RNA:DNA duplexes could bind to PRRs such as cGas, Rig-I, and MDA5 without triggering their activation (Fig. 1C), competing with their cognate ligands, and thereby raising threshold for signal generation [19].
The long-studied AGS is genetically heterogeneous and can occur due to mutations in various genes including IFIH1, TREX1, or ADAR1 [21]. ADAR1 encodes an adenosine-to-inosine editing enzyme: ADAR1. Excitingly, two recent publications have elucidated the relationship behind MDA5 and ADAR1 in driving AGS. ADAR1 editing of Alu-Alu inverted repeats (IR-Alu) in dsRNA leads to molecule destabilisation, thereby weakening the binding of self dsRNA to MDA5. Ahmad et al. demonstrated that loss of ADAR1’s dsRNA editing ability, through genetic deletion, increases the pool of non-edited IR-Alu in the cytosol and this triggers MDA5-mediated IFNαβ production (Fig. 1D). Similarly, gain of function mutations in MDA5 can render this PRR insensitive to ADAR1 introduced structural irregularities in dsRNA, thereby leading to inappropriate activation of MDA5 [22•]. Chung et al. also observed ADAR1 editing of Alu elements and further demonstrated ADAR1 also inhibits the activation of another dsRNA sensor: protein kinase R (PKR) (Fig. 1D). PKR is an ISG, so activation of this PRR was secondary to MDA5 induced IFNαβ. When activated, PKR mediated shutdown of protein translation and cell death [23•]. Collectively, these two reports indicate that ADAR1 acts as negative regulator for both IFNαβ production and downstream responses to IFNαβ.
Interferonopathies Associated with IFN Signalling
In contrast to enhanced induction of IFNαβ, loss of function mutations in negative regulators of IFNαβ can lead to prolonged or an aberrant response to IFNαβ signalling and therefore pathology. TORCH Syndrome refers to severe inflammatory disease stemming from foetal or early life infection with a variety of pathogens, many of which induce a robust IFNαβ response, the term “TORCH” being an acronym for: (T)oxoplasmosis, (O)ther agents, (R)ubella, (C)ytomegalovirus, and (H)erpes simplex [24]. Pseudo-TORCH Syndrome 2 is a sterile inflammatory disease which manifests similar to TORCH Syndrome, hallmark features include intracranial haemorrhage, calcification, brain malformations, liver dysfunction, and death within days of birth [25•]. This devastating disease stems from loss of function mutations in the gene coding for ubiquitin-specific peptidase 18 (USP18). USP18 negatively regulates IFNαβ signalling by binding to IFNAR2, thereby interrupting a JAK-receptor interaction [26] (Fig. 1E). Two recessive loss-of-function mutations of USP18 in five individuals from two unrelated families have been identified: a c.652C > T and a large deletion at the 3′ UTR region of USP18 [25•]. These mutations result in a complete deletion of USP18 and consequently, prolonged IFNαβ signalling. Patient fibroblasts exhibited enhanced IFNαβR signalling and expression of ISGs upon stimulation with IFNα. Interestingly, no baseline difference was observed between patient and healthy control samples, highlighting that USP18 deficiency does not alter the induction of IFNαβ, rather the cellular response to this family of cytokines. Importantly, transduction with USP18 ameliorated this enhanced induction of ISGs and activation of the IFNαβR signalling pathway in patient cells [25•]. Pseudo-TORCH Syndrome 2 emphasises the importance of negative feedback in the IFNαβ system.
Interferonopathies Caused by Unknown Pathways
Unlike the previously described syndromes, the genetic aetiology of several remaining interferonopathies currently have no known link to IFNαβ induction or signalling. Proteasome-associated autoinflammatory syndrome (PRAAS) is a spectrum of autoinflammatory diseases (previously known as Nakajo-Nishimura syndrome (NNS), Japanese Autoinflammatory Syndrome with Lipodystrophy (JASL), Joint contractures, Muscle atrophy, microcytic anaemia, and panniculitis-induced childhood-onset lipodystrophy (JMP) syndrome, or chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE)) that present in infancy and are characterised by annular erythematous skin lesions with panniculitis-induced lipodystrophy, hepatomegaly, arthralgias, recurrent fever, joint contractions with muscle atrophy, and basal ganglia calcification. From a genetic perspective, PRAAS is particularly interesting; a series of studies identified autosomal recessive homozygous or compound heterozygous loss-of-function mutations in the gene which codes for the inducible proteasome component: β5i (PSMB8) (p.T75M; p.G201V; p.A92T; p.M117V; p.C135X) and PRAAS was therefore thought of as monogenic [27,28,29,30]. However, patients who had no known mutation or were only heterozygous for disease-associated mutations in the PSMB8 gene were later identified, and in an elegant follow-up study, digenic inheritance of errors in other proteasome subunits: PSMB9 (p.G165D), PSMA3 (p.R233del; p.H111Ffs*10) and PSMB4 (5’ UTR: c.-9G > A; p.D212_V2124del; p.P16Sfs*45; p.Y222X), along with another mutation in PSMB8 (p.K105Q) were discovered. Additionally, an autosomal dominant mutation in POMP (p.E115Dfs*20) which encodes the proteasome maturation protein, was also identified [31•]. These loss of function mutations confer a cumulative genetic burden, variably affecting gene transcription, subunit expression or folding and subsequent proteasome assembly, ultimately leading to decreased proteasomal activity.
The proteasome is a degradation system for misfolded or damaged proteins marked for removal by polyubiquitination (reviewed in [32]) (Fig. 1F). Studies on PRAAS patients have demonstrated a high frequency of ubiquitin-rich inclusions in lesional skin biopsies, compared to healthy controls or control inflammatory disorders. Patient-derived cells also exhibit enhanced ISG expression and well as constitutive STAT1 phosphorylation, regardless of genotype [29, 30, 31•]. However, the link between proteotoxic stress and activation of IFNαβ is currently unexplored.
COPA Syndrome is an immune dysregulatory disease which is associated with an ISG signature in peripheral blood cells [33]. This disease presents in early life with cough and tachypnea and is characterised by early onset polyarticular arthritis, progressive lung disease, and often, renal complications [34,35,36]. COPA Syndrome is inherited in an autosomal dominant manner, with variable penetrance, and results from mutations affecting a narrow amino acid stretch in the COPA gene resulting in non-functional gene product. COPA encodes the α subunit of the coatomer complex 1 (COPα), which regulates vesicular retrograde transport between the Golgi and endoplasmic reticulum (ER) [37]. This syndrome is highly complex and whether it should be classed as an autoinflammatory or autoimmune disease is not entirely clear. Aside from the observed ISG signature, COPA syndrome is associated with autoantibody development, increased Th17 cells and proinflammatory cytokine expression [34]. What drives the autoinflammatory component of this disease is unknown; however, patient-associated COPA mutations result in an increase in ER stress which, in turn, can activate proinflammatory transcriptional programs via calcium leakage (Fig. 1G) [38, 39]. In vitro expression of COPA mutations activates the unfolded protein response [34], perhaps indicating a link between PRAAS and COPA syndrome (Fig. 1G).
Treatment
Interferonopathy patients were traditionally treated with broadly immunosuppressive drugs such as high-dose steroids or methotrexate, with limited effectivity. Perhaps unsurprisingly, these patients are also refractory to IL-1 blockade and TNF antagonists. However, antibody-mediated blockade of the proinflammatory cytokine IL-6, which can be induced by IFNαβ, has exhibited some effectiveness in select patients [28,29,30, 40, 41].
In vitro studies of patient cells have demonstrated that the constitutive expression of ISGs and phosphorylation of STAT1 is decreased upon co-culture with JAK inhibitors (Tofacitinib, Ruxolitinib, and Baricitinib) [30, 42]. These studies have led to Baricitinib (a reversible Jak 1 and 2 inhibitor) being approved for compassionate use in patients with presumed interferon-mediated pathology (NCT01724580). Preliminary data has shown that Baricitinib therapy significantly decreases patient symptom scores, daily steroid requirement, and the presence of IFN biomarkers [43, 44].
Jak inhibitors are broadly immunosuppressive because Jak molecules are utilised by a range of cytokines, not just IFNαβ, to induce signalling cascades. This may be particularly advantageous in the case of patients suffering from Stimulator of IFN genes (STING)-associated vasculopathy with onset in infancy (SAVI). SAVI is driven by gain of function mutations leading to the constitutive activation of the cytosolic sensor and adaptor protein: STING (Fig. 1H) [45, 46]. Activation of STING drives inflammation not only through induction IFNαβ, but also by the activation of NFκB signalling [47]. A recent study using the first SAVI mouse model found that limitation of IFNαβ signalling by genetic deletion of IRF3 did not ameliorate the autoinflammatory phenotype [48], indicating that NFκB activation, rather than IFNαβ signalling may be the primary driver of immune dysregulation. Similarly, patients with loss of function mutations in DNASE2, which can lead to activation of STING via cGAS, also exhibit elevated serum levels of NFκB driven cytokines, and deletion of IFNαβR in DNase II-deficient mice does not entirely protect these mice from autoinflammatory complications [49]. Thus, Baricitinib and other JAK inhibitors which can inhibit both IFNαβ and NFκB driven inflammation may provide a more holistic blockade of autoinflammation in these patients. Yet by the same token, patients being treated with such a broadly immunosuppressive therapy are at risk of opportunistic infections. Kim et al. reported 44% of treated interferonopathy patients developed a low level of BK viremia and Montealegre et al. also reported viral, bacterial and fungal infections in some Baricitinib treated patients [43, 44]. These opportunistic infections demonstrate the need for more targeted therapies for long term treatment. Future advances will drive the development of novel compounds specific to targets up or downstream of IFNαβ dysregulation. Indeed, an ongoing study (NCT02363452) is currently assessing the potential therapeutic efficacy of antiretroviral agents in AGS patients with TREX1 deficiency, which aims at inhibiting reverse transcription of retroelements.
Concluding Remarks
Although rare, interferonopathies provide us with a unique glimpse into the regulation of a pleotropic and potent family of cytokines. With the advancement of genomic sequencing, it is becoming easier to identify genes which modulate facets of IFNαβ signalling. In many cases, such as gain of function mutations in PRRs or loss of function mutations in molecules that process cytosolic nucleic acids, the link to IFNαβ induction is logical, with these mutations effectively mimicking viral infection. However, study of PRAAS has revealed a relatively unknown relationship between the build-up of polyubiqutinated proteins and induction of IFNαβ. This is an exciting finding as it indicates there is potentially an as yet undefined, cytosolic sensor for protein aggregation. Indeed, it would be of interest to compare and contrast clinical presentation and cellular dysfunction of PRAAS with other autoinflammatory disorders involving loss of function mutations in deubiquitinases (DUBs), such as OTULIN-related autoinflammatory syndrome (Steiner et al. 2018). It is possible that autoinflammatory diseases associated with the accumulation of ubiquitinated proteins in the cytosol, through either proteasome dysfunction or decreased DUB activity, are driven by the same upstream factors and we are observing a spectrum of clinical manifestations within a single disease. Indeed, although we have separated them for this series of reviews, it is important to remember that IFNαβ and NKκB signalling are generally concomitant. Thus, both of these pathways are likely to contribute, in varying degrees, to a given autoinflammatory disease, as observed in SAVI for example.
Our understanding of monogenic autoinflammatory disease is exponentially growing; however, there remain many open questions. If IFNαβ, IFNαβR, and many of the other proteins discussed in this review are universally expressed, why do certain tissues, e.g., neuronal tissue, suffer a higher degree of inflammatory burden? Why do identical mutations present with distinct phenotypes in different individuals, as observed in IFIH1 driven SMS and AGS? Elucidation of aberrant IFNαβ-mediated inflammation is of great scientific interest as it will likely result in the identification of novel therapeutic targets not only to reduce pathology in interferonopathy patients, but also for individuals diagnosed with other inflammatory or metabolic syndromes, microbial infections or cancer.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines, and their receptors. Immunol Rev. 2004;202:8–32.
McNab F, Mayer-Barber K, Sher A, Wack A, O'Garra A. Type I interferons in infectious disease. Nat Rev Immunol. 2015;15(2):87–103.
Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol. 2014;32:513–45.
Crow YJ, Black DN, Ali M, Bond J, Jackson AP, Lefson M, et al. Cree encephalitis is allelic with Aicardi-Goutieres syndrome: implications for the pathogenesis of disorders of interferon alpha metabolism. J Med Genet. 2003;40(3):183–7.
Rodero MP, Crow YJ. Type I interferon-mediated monogenic autoinflammation: the type I interferonopathies, a conceptual overview. J Exp Med. 2016;213(12):2527–38.
Kretschmer S, Lee-Kirsch MA. Type I interferon-mediated autoinflammation and autoimmunity. Curr Opin Immunol. 2017;49:96–102.
Feigenbaum A, Müller C, Yale C, Kleinheinz J, Jezewski P, Kehl HG, et al. Singleton-Merten syndrome: an autosomal dominant disorder with variable expression. Am J Med Genet A. 2013;161A(2):360–70.
Reikine S, Nguyen JB, Modis Y. Pattern recognition and signaling mechanisms of RIG-I and MDA5. Front Immunol. 2014;5:342.
Rutsch F, MacDougall M, Lu C, Buers I, Mamaeva O, Nitschke Y, et al. A specific IFIH1 gain-of-function mutation causes Singleton-Merten syndrome. Am J Hum Genet. 2015;96(2):275–82.
Oda H, Nakagawa K, Abe J, Awaya T, Funabiki M, Hijikata A, et al. Aicardi-Goutieres syndrome is caused by IFIH1 mutations. Am J Hum Genet. 2014;95(1):121–5.
Rice GI, del Toro Duany Y, Jenkinson EM, Forte GMA, Anderson BH, Ariaudo G, et al. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat Genet. 2014;46(5):503–9.
Bursztejn AC, Briggs TA, del Toro Duany Y, Anderson BH, O'Sullivan J, Williams SG, et al. Unusual cutaneous features associated with a heterozygous gain-of-function mutation in IFIH1: overlap between Aicardi-Goutieres and Singleton-Merten syndromes. Br J Dermatol. 2015;173(6):1505–13.
• Buers I, et al. MDA5-Associated Neuroinflammation and the Singleton-Merten Syndrome: Two Faces of the Same Type I Interferonopathy Spectrum. J Interf Cytokine Res. 2017;37(5):214–9. Buers et al present an interesting case study and discuss the phenotypic continum between AGS and SMS.
Jang MA, Kim EK, Now H, Nguyen NTH, Kim WJ, Yoo JY, et al. Mutations in DDX58, which encodes RIG-I, cause atypical singleton-Merten syndrome. Am J Hum Genet. 2015;96(2):266–74.
Rawling DC, Fitzgerald ME, Pyle AM. Establishing the role of ATP for the function of the RIG-I innate immune sensor. Kowalczykowski SC, ed. eLife. 2015;4:e09391. https://doi.org/10.7554/eLife.09391.
Rodero MP, Tesser A, Bartok E, Rice GI, Della Mina E, Depp M, et al. Type I interferon-mediated autoinflammation due to DNase II deficiency. Nat Commun. 2017;8(1):2176.
Evans CJ, Aguilera RJ. DNase II: genes, enzymes and function. Gene. 2003;322:1–15.
Kawane K, Motani K, Nagata S. DNA degradation and its defects. Cold Spring Harb Perspect Biol. 2014;6(6)
Starokadomskyy P, Gemelli T, Rios JJ, Xing C, Wang RC, Li H, et al. DNA polymerase-alpha regulates the activation of type I interferons through cytosolic RNA:DNA synthesis. Nat Immunol. 2016;17(5):495–504.
Perera RL, et al. Mechanism for priming DNA synthesis by yeast DNA polymerase alpha. elife. 2013;2:e00482.
Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte GM, Gornall HL, et al. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am J Med Genet A. 2015;167A(2):296–312.
• Ahmad S, et al. Breaching self-tolerance to Alu Duplex RNA underlies MDA5-mediated inflammation. Cell. 2018;172(4):797–810 e13. Published back to back with Chung et al (2018) these studies elucidate the role of ADAR1 in controlling IFNαβ induction through editing of self RNA.
• Chung H, et al. Human ADAR1 prevents endogenous RNA from triggering translational shutdown. Cell. 2018;172(4):811–824 e14. Published back to back with Ahmad et al (2018).
Neu N, Duchon J, Zachariah P. TORCH infections. Clin Perinatol. 2015;42(1):77–103. viii
• Meuwissen ME, et al. Human USP18 deficiency underlies type 1 interferonopathy leading to severe pseudo-TORCH syndrome. J Exp Med. 2016;213(7):1163–74. USP18 deficency emphasises the devastating effect of uncontrolled IFNαβ signalling.
Malakhova OA, Kim KII, Luo JK, Zou W, Kumar KGS, Fuchs SY, et al. UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. EMBO J. 2006;25(11):2358–67.
Agarwal AK, Xing C, DeMartino GN, Mizrachi D, Hernandez MD, Sousa AB, et al. PSMB8 encoding the beta5i proteasome subunit is mutated in joint contractures, muscle atrophy, microcytic anemia, and panniculitis-induced lipodystrophy syndrome. Am J Hum Genet. 2010;87(6):866–72.
Arima K, Kinoshita A, Mishima H, Kanazawa N, Kaneko T, Mizushima T, et al. Proteasome assembly defect due to a proteasome subunit beta type 8 (PSMB8) mutation causes the autoinflammatory disorder, Nakajo-Nishimura syndrome. Proc Natl Acad Sci U S A. 2011;108(36):14914–9.
Kitamura A, Maekawa Y, Uehara H, Izumi K, Kawachi I, Nishizawa M, et al. A mutation in the immunoproteasome subunit PSMB8 causes autoinflammation and lipodystrophy in humans. J Clin Invest. 2011;121(10):4150–60.
Liu Y, Ramot Y, Torrelo A, Paller AS, Si N, Babay S, et al. Mutations in proteasome subunit beta type 8 cause chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature with evidence of genetic and phenotypic heterogeneity. Arthritis Rheum. 2012;64(3):895–907.
• Brehm A, et al. Additive loss-of-function proteasome subunit mutations in CANDLE/PRAAS patients promote type I IFN production. J Clin Invest. 2015;125(11):4196–211. Brehm et al unravel the genetic heterogeneity of PRAAS, demonstrating that this disease arises from multiple mutations that result in a cumulative burden on the proteasome system.
Ciechanover A. Intracellular protein degradation: from a vague idea through the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Rambam Maimonides Med J. 2012;3(1):e0001.
Volpi S, Tsui J, Mariani M, Pastorino C, Caorsi R, Sacco O, et al. Type I interferon pathway activation in COPA syndrome. Clin Immunol. 2018;187:33–6.
Watkin LB, et al. COPA mutations impair ER-Golgi transport and cause hereditary autoimmune-mediated lung disease and arthritis. Nat Genet. 2015;47(6):654–60.
Noorelahi R, Perez G, Otero HJ. Imaging findings of Copa syndrome in a 12-year-old boy. Pediatr Radiol. 2018;48(2):279–82.
Jensson BO, Hansdottir S, Arnadottir GA, Sulem G, Kristjansson RP, Oddsson A, et al. COPA syndrome in an Icelandic family caused by a recurrent missense mutation in COPA. BMC Med Genet. 2017;18(1):129.
Letourneur F, Gaynor EC, Hennecke S, Démollière C, Duden R, Emr SD, et al. Coatomer is essential for retrieval of dilysine-tagged proteins to the endoplasmic reticulum. Cell. 1994;79(7):1199–207.
Pahl HL, Baeuerle PA. Activation of NF-kappa B by ER stress requires both Ca2+ and reactive oxygen intermediates as messengers. FEBS Lett. 1996;392(2):129–36.
Deniaud A, Sharaf el dein O, Maillier E, Poncet D, Kroemer G, Lemaire C, et al. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene. 2008;27(3):285–99.
Kuijpers TW. Aicardi-Goutieres syndrome: immunophenotyping in relation to interferon-alpha. Eur J Paediatr Neurol. 2002;6 Suppl A:A59–64. discussion A65–6, A77–86
De Laet C, et al. Phenotypic overlap between infantile systemic lupus erythematosus and Aicardi-Goutieres syndrome. Neuropediatrics. 2005;36(6):399–402.
Liu Y, Holdbrooks AT, de Sarno P, Rowse AL, Yanagisawa LL, McFarland BC, et al. Therapeutic efficacy of suppressing the Jak/STAT pathway in multiple models of experimental autoimmune encephalomyelitis. J Immunol. 2014;192(1):59–72.
Montealegre G, Reinhardt A, Brogan P, et al. Preliminary response to Janus kinase inhibition with baricitinib in chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperatures (CANDLE). Pediatric Rheumatology Online Journal. 2015;13(Suppl 1):O31. https://doi.org/10.1186/1546-0096-13-S1-O31.
Kim H, Brooks KM, Tang CC, Wakim P, Blake M, Brooks SR, et al. Pharmacokinetics, pharmacodynamics, and proposed dosing of the oral JAK1 and JAK2 inhibitor baricitinib in pediatric and young adult CANDLE and SAVI patients. Clin Pharmacol Ther. 2017;
Jeremiah N, Neven B, Gentili M, Callebaut I, Maschalidi S, Stolzenberg MC, et al. Inherited STING-activating mutation underlies a familial inflammatory syndrome with lupus-like manifestations. J Clin Invest. 2014;124(12):5516–20.
Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Montealegre Sanchez GA, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. 2014;371(6):507–18.
Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461(7265):788–92.
Warner JD, Irizarry-Caro RA, Bennion BG, Ai TL, Smith AM, Miner CA, et al. STING-associated vasculopathy develops independently of IRF3 in mice. J Exp Med. 2017;214(11):3279–92.
Kawane K, Ohtani M, Miwa K, Kizawa T, Kanbara Y, Yoshioka Y, et al. Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature. 2006;443(7114):998–1002.
Acknowledgements
The authors would like to thank members of the Masters Lab, particularly Dr. Paul Baker and Dr. Fiona Moghaddas, for discussion and advice on this review.
Funding
S.L.M acknowledges funding from NHMRC grants (1144282, 1142354, and 1099262), The Sylvia and Charles Viertel Foundation, HHMI-Wellcome International Research Scholarship, and Glaxosmithkline. S.D. acknowledges funding from NHMRC ECF: GNT1143412.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
Dr. Masters reports grants from NHMRC, grants from Viertel Foundation, grants from HHMI-Wellcome Trust, grants from Glaxosmithkline, outside the submitted work.
Dr. Davidson reports grants from NHMRC, outside the submitted work.
Drs. Steiner and Harapas declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Pediatric Rheumatology
Rights and permissions
About this article

Cite this article
Davidson, S., Steiner, A., Harapas, C.R. et al. An Update on Autoinflammatory Diseases: Interferonopathies. Curr Rheumatol Rep 20, 38 (2018). https://doi.org/10.1007/s11926-018-0748-y
Published:
DOI: https://doi.org/10.1007/s11926-018-0748-y