
Abstract
Cellular metabolism represents a newly identified checkpoint of effector functions in the immune system. A solid body of work has characterized the metabolic requirements of normal T cells during activation and differentiation into polarized effector subsets. Similar studies have been initiated to characterize the metabolic requirements for B cells and myeloid cells. Only a few studies though have characterized the metabolism of immune cells in the context of autoimmune diseases. Here, we review what is known on the altered metabolic patterns of CD4+ T cells, B cells, and myeloid cells in lupus patients and lupus-prone mice and how they contribute to lupus pathogenesis. We also discuss how defects in immune metabolism in lupus can be targeted therapeutically.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The field of immunometabolism arguably started in 2002 when C. Thompson and colleagues showed that CD28 activation triggered glycolysis in T cells [1]. A few years later, J. Powell showed that mammalian target of rapamycin (mTOR), a kinase that senses the cellular metabolic status, controls the differentiation of effector T cells [2]. Following these seminal findings, a growing number of studies have revealed that cells in the immune system, T cells in particular, are functionally regulated by their metabolic substrate utilization [3–5, 6•]. The simplest model is that resting T cells rely on mitochondrial oxidative respiration (OXPHOS), favoring fatty acid (FA), and glutamine oxidation. Antigen-mediated activation, co-stimulation, and cytokine signaling lead to T cell differentiation into effector subsets and trigger a drastic metabolic reprogramming, with upregulation of glucose utilization, and activation of mitochondria-independent glycolysis, as a source of ATP as well as a major source of building blocks necessary to cope with massive proliferation as well as synthesis of effector molecules. The metabolism of activated T cells has been compared to that of tumor cells, in which the reliance on non-oxidative glycolysis is referred to as the Warburg effect [6•]. In contrast, regulatory Foxp3+ T (Treg) cells and memory T cells, which share an anergic/quiescent phenotype with resting T cells, mostly depend on FA oxidation as a source of energy. Based on these drastic differences in metabolic requirements and the critical role of effector T cells in autoimmune diseases and cancer, T cell metabolism has been proposed as a target for immunotherapy [4]. Here, we review recent studies in this field that are relevant to systemic lupus erythematosus (SLE), first showing cellular metabolic imbalances in the immune cells of lupus patients or mouse models of systemic autoimmunity (Fig. 1), then the attempts that have been made to correct these imbalances for therapeutic purposes. In addition to substrate utilization, the metabolic status of immune cells has also been shown to contribute to SLE. Insufficient energy or metabolite levels, as well as REDOX imbalance, trigger cell death, leading lymphopenia [7], and a large pool of cellular debris providing a rich source of autoantigens and innate immune activators [8]. Finally, the substrates for epigenetic modifications of DNA and histones are derivatives from mitochondrial metabolism, linking cellular metabolism to a well-documented but poorly understood layer of regulation of the immune system, including in SLE [9•].

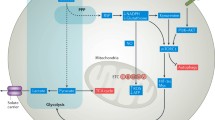
Metabolic pathways that have been involved in SLE. Simplified representation of metabolic pathways affected in lupus, their cellular location, and relationships. Mitochondrial dysfunctions are central to lupus immune cells, including increased oxidative stress, mitochondria membrane potential (Δψm) and electron transport chain (ETC) activity, OXPHOS by the TCA cycle, and regulation by the estrogen receptor-related genes. Glucose metabolism is also increased in T cells and most likely in B cells, with a strong contribution of pyruvate to the TCA cycle, at least in T cells. mTOR activation in T cells has multiple consequences, including the upregulation of glucose transporters and the activation of TCR signaling. Autophagy (which is classically inhibited by mTOR) has been associated with either protecting or enhancing lupus depending on the cell type. Finally, the CD3b receptor CD46 enhances the metabolic output of T cells by upregulating nutrient uptake and activating mTOR. Enhanced pathways in SLE are shown in red, and the protective effect of autophagy is shown in blue
mTORC Activation in Lupus CD4+ T Cells
In normal immune responses, Th1 and Th17 cells require mTORC1 activation while mTORC2 activation favors Th2 differentiation [10•]. Murine mTORC1 −/− T cells fail to activate STAT3 and STAT4 and do not differentiate into Th1, Th2, or Th17 cells under polarizing conditions [2]. There is also data to suggest that mTORC1 directly phosphorylates STAT3 on Ser727 [11, 12], but this result has not been validated for T cells. mTORC1 inhibition by rapamycin promotes Treg expansion; mTORC1 signaling is however required for Treg suppressive function, possibly by inhibiting the mTORC2 pathway [13]. Follicular B helper CD4+ T (Tfh) cells have been recently reported to be relatively independent of mTORC1 activation [14•]. However, ROQIN, which controls the expression of a number of genes involved in Tfh differentiation [15], inhibits AMP-activated protein kinase (AMPK), leading to mTORC1 activation [16•]. In addition, mTORC2 activation has been indirectly linked to Tfh cell differentiation [13]. Finally, the assignment of effector functions during asymmetric cell division in CD8+ T cells is determined by the retention of activated mTORC1 in the synapse-proximal daughter cell, while the synapse-distal mTORC1low daughter cell acquires a memory phenotype [17, 18]. It is likely that a similar asymmetric distribution of mTORC1 exists between effector and memory CD4+ T cells.
mTORC1 activation has been extensively documented in the CD4+ T cells of SLE patients [19] and has been proposed to serve as a biomarker of autoimmune inflammation, as well as cancer, obesity, and aging [10•]. Specifically, mTORC1 activation has been linked to T cell necrosis, the expansion of Th17 and IL-4 producing double negative (DN) T cells, as well as a reduction on the Treg cell pool [20••, 21••]. mTORC1 activation has also been observed in the CD4+ T cells from several strains of lupus-prone mice, including the NZM2410-derived triple congenic (TC) B6.Sle1.Sle2.Sle3 strain [22••, 23•]. Accordingly, treatments with rapamycin or with N-acetylcysteine (a precursor of glutathione that inhibits mTORC1) reduced disease severity in SLE patients [24, 25], as well as in BWF1 mice [26]. mTORC1 activation is not limited to T cells in lupus and may reflect a systemic mitochondrial dysregulation and elevated cellular metabolism. Antiphospholipid antibodies (aPL) have been linked to oxidative stress in the liver, and a recent study found mTORC1 activation in the liver of MRL, MRL/lpr, and B6.lpr lupus-prone mice [27]. Rapamycin treatment reduced oxidative stress in the liver of these mice in correlation with a reduction of aPL levels.
Glycolytic and Oxidative Metabolism in Lupus CD4+ T Cells
The major glucose transporter expressed by T cells is Glut1, which is highly upregulated upon T cell receptor and CD28 signaling [28•]. A direct link between glucose metabolism and systemic autoimmunity was shown by the over-expression of Glut1 in mice that led to the accumulation of activated CD4+ T cells, the production of autoantibodies (autoAbs), and a modest immune complex deposition in the glomeruli of aged mice [29]. Conversely, Glut1 deficiency in CD4+ T cells induced less severe inflammatory diseases such as graft-versus-host disease (GVHD) and IBD [28•], but the effect of Glut1 deficiency was not directly tested in lupus models. In addition, Glut1 expression is decreased in Notch signaling-deficient memory CD4+ T cells due to impaired AKT phosphorylation, which suggests that memory CD4+ T cell survival relies on glucose metabolism [30]. So far, Glut1 expression has not been found to be affected in CD4+ T cells of lupus-prone mice or SLE patients. Other genes involved in the glycolytic pathway were however found to be over-expressed in TC CD4+ T cells [22••, 23•]. Among them, Hif1a, a transcription factor that controls the cellular response to hypoxia, has recently received considerable attention in the context of T cell differentiation and effector functions [31]. The regulation of Hif1a expression is complex, but it is downstream of mTORC1 activation [32]. It is therefore likely that HIF1A is upregulated in T cells from SLE patients. Hif1a activates the expression of a large number of key glycolytic genes such as Glut1 and, accordingly, is required for Th17 cell differentiation [33]. The role of Hif1a in Treg cell differentiation and function is more complex, and both positive and negative regulations have been reported [31]. A hypoxic signature and high levels of Hif1a expression have also been reported in nephritic kidneys [34], but the role of hypoxia and Hif1a in SLE T cells has not yet been specifically addressed. Complement has been shown to regulate Glut1 expression in T cells through the activation of the CD46 (MCP) complement receptor, which also activates amino acid flux, as well as mTORC1 activation in a manner that is necessary for Th1 polarization [35•]. High levels of CD46 activation have been reported in the T cells of SLE patients in association with Treg dysfunctions [36]. It is therefore possible that CD46 signaling also contributes to dysfunctions in cellular metabolism of lupus T cells.
Oxygen consumption is elevated in the splenocytes of BWF1 lupus-prone mice, and a similar high oxygen consumption was found in chronically activated human CD4+ T cells, as opposed to acutely activated T cells that were more glycolytic [37]. This was interpreted as the chronic activation from autoantigen in lupus leading to an oxidative metabolism as opposed to the acute activation induced by foreign antigens (or in vitro supraphysiological activation) leading to a glycolytic metabolism. In support of this hypothesis, oxidative metabolism dominated in CD4+ T cells in a chronic model of GVHD [38], while highly glycolytic T cells drove tissue injury in a hyper-acute model of GVHD [39]. Increased O2 consumption was found in CD4+ T cells from SLE patients [22••, 40] and several lupus-prone mice [22••, 23•]. CD4+ T cells from SLE patients and lupus-prone mice also present an elevated glycolysis [22••, 23•]. Effector memory (EM) CD4+ T cells from healthy donors require high levels of glucose metabolism as well as mitochondrial respiration for survival, proliferation, and IFNγ production [41]. Interestingly, glucose required by EM CD4+ T cells supplies pyruvate for oxidation, increasing mitochondrial membrane potential and ROS production. These results are very similar to what has been reported with SLE CD4+ T cells. Glucose is oxidized in CD4+ T cells from BWF1 [37] and TC mice [23•], and high IFNγ production by these T cells depends on both glucose and mitochondrial metabolism [22••]. These results are consistent with the facts that mitochondrial membrane production and ROS production are elevated in SLE CD4+ T cells [42], and EM CD4+ T cells are expanded in SLE patients [43] and lupus mice [44]. However, a high glycolysis and OXPHOS was also found in naïve CD4+ T cells from lupus mice [22••], suggesting an intrinsic skewing of SLE T cells metabolism that may be a primary effector of autoimmune pathogenesis rather than a mere consequence of immune activation.
Consistent with a dual activation of glucose and oxidative metabolism, IFNγ production by murine lupus CD4+ T cells was normalized (i.e., brought down to levels found in non-autoimmune mice) in vitro by inhibiting glucose metabolism with 2-deoxy-glucose (2DG) or mitochondrial respiration with metformin [22••, 23•]. Metformin also inhibited IFNγ production by CD4+ T cells from SLE patients and healthy controls [22••]. 2DG could not be tested on human T cells because they are very sensitive to glucose inhibition in vitro. A treatment combining metformin and 2DG was very effective in reversing clinical disease in TC, BWF1, as well as B6.lpr mice [22••, 23•]. This was associated with a reduction of CD4+ T cell metabolism as well as a normalization of their activation and effector subset distribution. The significant decrease in the frequency and number of Tfh cells, germinal center (GC) B cells, and plasma cells corresponded to an elimination of anti-dsDNA and anti-chromatin IgG autoAbs. Interestingly, the treatment of these mice with 2-DG and metformin normalized mTORC1 activation concomitant with disease reversal [22••]. Treatment with single drugs, either metformin or 2DG, was not effective at reversing disease in TC mice [22••]. However, treatment with 2DG, and to a lesser extent with metformin, prevented disease development when TC mice were treated before they produce large amounts of autoAbs [23•]. Surprisingly, in regard to the enhanced glycolysis in lupus T cells, treatment with dichloroacetate (DCA), either alone or combined with metformin, did not have a therapeutic effect in TC mice [23•]. DCA is a drug that inhibits pyruvate dehydrogenase kinase (Pdk1) and thereby increases the flux of pyruvate into the mitochondria, promoting glucose oxidation over glycolysis. Interestingly, DCA suppressed Th17 differentiation but increased Th1 polarization in vitro [23•]. Conversely, two inhibitors of the mitochondria pyruvate carrier complex (MPC) [45, 46], UK5099 and troglitazone, that have an opposite effect to DCA, inhibited both Th17 and Th1 polarization, indicating that Th17 and Th1 cells utilize glucose differently. The treatment results indicated that glucose is mostly used through the oxidation of pyruvate by CD4+ T cells of TC lupus-prone mice and suggested that the inhibition of Th17 cells was not critical to their disease reversal. These results should be investigated in other mouse models as well as in the T cells of SLE patients. Intriguingly, troglitazone is a member of the thiazolidinedione (TZD) family, a class of compounds that has been found recently to function as acute MPC inhibitors, effectively shutting down pyruvate oxidation [47, 48]. A treatment with pioglitazone, another TZD, reduced activation of human SLE T cells in vitro, as well as autoAb production and renal pathology in lupus-prone mice [49, 50]. This was interpreted as a protective effect of peroxisome proliferator-activated receptor gamma (PPARγ) activation. It would be interesting, however, to revisit these results and investigate whether the pioglitazone treatment normalized the pyruvate metabolism of lupus T cells. In support of this hypothesis, and in accordance to our results showing a greater dependence of IFNγ production on pyruvate oxidation [23•], pioglitazone normalized IFNγ but not IL-17 production in T cells from lupus patients [49].
Mechanisms of CD4+ T Cell Metabolic Hyper-activity in Lupus
The high glucose and oxygen consumption by lupus CD4+ T cells is not secondary to immune activation since it was detected in naïve cells sorted from young pre-disease mice [22••]. The mechanism responsible for this phenotype is unknown, but it is likely that it is, at least in part, a genetic origin. The many lupus susceptibility genes that have been identified through GWAS have no obvious link to metabolism [51], with the possible exception of two genes involved in autophagy, ATG5 [52] and ATG16L2 [53]. ATG5 controls a non-canonical autophagy known as LC3-associated phagocytosis (LAP), and Atg5-deficient mice present a lupus-like disease associated with an impaired clearance of dead cells by phagocytes [54••]. Little is known about ATG16L2, an isoform of ATG16L that forms a complex with ATG5 [55], and has therefore the potential to regulate LAP. Autophagy is activated by AMPK and inhibited by mTORc1 [56]. It would be of great interest to investigate LAP in the context of metabolic stress in lupus immune cells, as well as the relationship between mTOR activation and autophagy defects in lupus. In addition, a putative association with polymorphisms in CD46/MCP with SLE has been reported [57].
We have identified in the NZM2410 lupus-prone mouse a hypomorphic allele of the estrogen receptor-related gamma gene (Esrrg) that is associated with increased CD4+ T cells activation, IFNγ production, and defective Treg maintenance [58]. In metabolically active tissues, ERRγ transactivates genes that control mitochondrial biogenesis and FA oxidation [59]. Essrg-deficient mice die soon after birth due the critical need for their myocardium to switch from glycolysis to FA oxidation [60]. More recent studies have shown that ERRγ orchestrates the transcriptional program that activates glucose OXPHOS and ATP production that are essential for the function of pancreatic β cells [61], neurons [62], and stem cells [63]. These results strongly suggest that ERRγ regulates T cell function by controlling mitochondrial metabolism. In support of this hypothesis, decreased in mitochondrial functions have been found in the CD4+ T cells expressing the NZM2410 allele of Esrrg [58]. Interestingly, ERRα, another member of the estrogen receptor related gene family, may be playing an opposite role, with Esrra deficiency or inhibition reducing T cell proliferation and effector functions [64]. The interplay between ERR genes in T cells needs to be better understood to evaluate their role in T cell metabolic disturbance in SLE and other immune-related diseases.
Metabolism of Other Immune Cells Involved in Lupus
The characterization of the metabolism of immune cells other than T cells has lagged behind, but a number of recent studies also show metabolic checkpoints that control their activation and effector functions. Activated B cells rely on glucose [65], but their substrate utilization is more balanced than that of activated T cells [66•]. Interestingly, chronic exposure to BAFF increased glycolysis in B cells [66•], and the high level of BAFF associated with SLE predicts that lupus B cells are more glycolytic than non-autoimmune B cells. Germinal center B cells, which are expanded in lupus, are regulated by hypoxia and mTORC1 activity [67•]. Plasma cells as terminally differentiated effector B cells are likely to have specific metabolic requirements [68]. Somewhat as expected, plasma cells require a large amount of glucose, the majority of which is used for immunoglobulin glycosylation. However, a small portion of this glucose is catabolized into pyruvate, which must be imported into the mitochondria for the survival of long-lived plasma cells (LLPC) [69••]. Given that pyruvate oxidation is also required for the survival of memory T cells [41], and pyruvate transport to the mitochondria may represent a biomarker of immune cell longevity, with mitochondrial pyruvate either oxidized for ATP production or used for anaplerotic replenishment of members of the TCA cycle diverted as anabolic intermediates. Given the pathogenic role of LLPC in SLE, targeting MCP, which also modulates lupus T cell functions [23•] may represent a therapeutic venue to eliminate autoAb-producing LLPCs. Autophagy is also required by LLPCs, as shown in mice with a B cell-specific deletion of Atg5 [70]. Interestingly, B cells from lupus-prone BWF1 mice and SLE patients showed increased levels of autophagy [71•], and B cell-specific deletion of Atg5 in B6.lpr mice reduced autoimmune pathology [72]. Finally, mTORC1 constitutive activation promotes plasma cell differentiation [73]. It is unknown whether the high levels of mTORC1 activation in SLE T cells [19] extend to SLE plasma cells.
As T and B cells, dendritic cells (DCs) and macrophages undergo metabolic reprogramming in response to stimulation, and it has been suggested that metabolic checkpoints control the tolerogenic vs. immunogenic effector functions of DCs [74, 75]. Metabolic programming is also critical for the polarization of macrophages, with M1 inflammatory macrophages skewed toward glycolysis and M2 alternatively activated macrophages skewed toward mitochondrial metabolism [75, 76]. Rapamycin treatment of human and mouse plasmacytoid DCs inhibits their production of type I interferon including under TLR7 and TLR9 stimulation [77, 78]. How this impacts the type I interferon signature in lupus remains to be determined. Little is known on the metabolic requirements of neutrophils, which have very few mitochondria. A recent study has shown that asymmetrical ATP production and mTOR signaling are required for neutrophil chemotaxis [79]. Finally, activated NK cells upregulate both glycolysis and respiration [80], and their cytokine production, especially IFNγ, is glucose-dependent [81]. Since each of these cell types has been implicated in SLE, either in patients or in mouse models of the disease, it will be of great interest to characterize the metabolic requirements relative to disease development and activity.
Conclusions
It has been proposed that targeting the unique metabolic features of inflammatory immune cells, and T cells in particular, would offer novel and effective therapeutic venues to treat inflammatory and autoimmune diseases [4]. A number of metabolic abnormalities have now been reported in both SLE patients and lupus-prone mice, as summarized in Table 1. The translation of these findings into therapeutic targets is still a nascent field, but it holds promises as shown in SLE [22••], rheumatoid arthritis (RA) [82], GVHD [38, 39, 83], coronary artery disease [84], and transplantation [85]. There has been intense interest in understanding the molecular basis of tumor metabolism and to apply this knowledge therapeutically [86]. Given the overlap between the metabolic pathways of tumor cells and of rapidly expanding immune cells, there is a lot to learn from our oncology colleagues. The emerging field of immunometabolism is however unraveling an intricate multilayered system of regulation that has common threads, some of which with tumor metabolism, but also cell-specific and stimulus-specific characteristics. One should be cautious, in looking for a universal approach to dampen immune inflammation though metabolic targeting. Indeed two rheumatic autoimmune diseases, SLE and RA, share a dysregulated of CD4+ T cell metabolism, but in opposite directions with hyper-oxidation in SLE and hyper-reduction in RA [87••]. A broader investigation of metabolic dysregulation in the many cell types in the immune system that trigger and sustain lupus pathogenesis should offer a panel of therapeutic targets to be evaluated with the large arsenal of metabolic inhibitors that is available, many of them approved for clinical use (Box 1).
Box 1. The clinician’s corner.
-
Immune cells have high metabolic demands to respond to pathogen challenges with a rapid proliferation and the production of effector molecules. Multiple metabolic defects have been identified in the immune cells of lupus patients and lupus-prone mice, the majority of them in CD4+ helper T cells.
-
The elevated metabolism in lupus CD4+ T cell corresponding to their chronic activation by autoantigens contributes to disease by promoting inflammatory effector functions.
-
The combination of a metabolic inhibitor that limits glucose metabolism and metformin, which inhibits mitochondrial respiration, normalized the metabolism and effector functions of mouse and human lupus T cells in vitro, and reversed disease in mice. This suggests that metabolic inhibitors could be effective in treating patients with lupus.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Frauwirth KA, Riley JL, Harris MH, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity. 2002;16(6):769–77.
Delgoffe GM, Kole TP, Zheng Y, et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. 2009;30(6):832–44.
Buck MD, O’Sullivan D, Pearce EL. T cell metabolism drives immunity. J Exp Med. 2015;212(9):1345–60.
O’Sullivan D, Pearce EL. Targeting T cell metabolism for therapy. Trends Immunol. 2015;36(2):71–80.
Powell JD, Pollizzi KN, Heikamp EB, et al. Regulation of immune responses by mTOR. Ann Rev Immunol. 2012;30(1):39–68.
MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Ann Rev Immunol. 2013;31:259–83. Comprehensive review of the basic mechanisms by which metabolism controls T cell effector functions.
Perl A, Gergely P, Puskas F, et al. Metabolic switches of T-cell activation and apoptosis. Antioxid Redox Signal. 2002;4(3):427–43.
Fernandez D, Perl A. Metabolic control of T cell activation and death in SLE. Autoimmun Rev. 2009;8(3):184–9.
Oaks Z, Perl A. Metabolic control of the epigenome in systemic lupus erythematosus. Autoimmunity. 2014;47(4):256–64. Review of the intersection between epigenetic regulation and cellular metabolism with a focus on T cells in SLE.
Perl A. Activation of mTOR (mechanistic target of rapamycin) in rheumatic diseases. Nat Rev Rheumatol. 2016;12(3):169–82. Comprehensive review of mTOR activition in rheumatic diseases with a focus on T cells, and its potential as therapeutic target.
Yokogami K, Wakisaka S, Avruch J, et al. Serine phosphorylation and maximal activation of STAT3 during CNTF signaling is mediated by the rapamycin target mTOR. Curr Biol. 2000;10(1):47–50.
Kim JH, Yoon MS, Chen J. Signal transducer and activator of transcription 3 (STAT3) mediates amino acid inhibition of insulin signaling through serine 727 phosphorylation. J Biol Chem. 2009;284(51):35425–32.
Zeng H, Yang K, Cloer C, et al. mTORC1 couples immune signals and metabolic programming to establish Treg-cell function. Nature. 2013;499:485–90.
Ray JP, Staron MM, Shyer JA, et al. The interleukin-2-mTORc1 kinase axis defines the signaling, differentiation, and metabolism of T helper 1 and follicular B helper T cells. Immunity. 2015;43(4):690–702. A comparision of Th1 and Tfh cells in virally infected mice showing a dichotomy in metabolism corresponding to the IL-2 and mTORC1 pathways.
Srivastava M, Duan G, Kershaw NJ, et al. Roquin binds microRNA-146a and Argonaute2 to regulate microRNA homeostasis. Nat Commun. 2015;6:6253.
Ramiscal RR, Parish IA, Lee-Young RS, et al. Attenuation of AMPK signaling by ROQUIN promotes T follicular helper cell formation. Elife. 2015;4:e08698. A study that showed that ROQUIN is a gene that regulates Tfh cell differentiation in a complex manner, including through direct inhibition of AMPK.
Pollizzi KN, Sun IH, Patel CH, et al. Asymmetric inheritance of mTORC1 kinase activity during division dictates CD8(+) T cell differentiation. Nat Immunol. 2016;17(6):704–11.
Verbist KC, Guy CS, Milasta S, et al. Metabolic maintenance of cell asymmetry following division in activated T lymphocytes. Nature. 2016;532(7599):389–93.
Fernandez D, Perl A. mTOR signaling: a central pathway to pathogenesis in systemic lupus erythematosus? Discov Med. 2010;9(46):173–8.
Lai ZW, Borsuk R, Shadakshari A, et al. Mechanistic target of rapamycin activation triggers IL-4 production and necrotic death of double-negative T cells in patients with systemic lupus erythematosus. J Immunol. 2013;191(5):2236–46. This study dissects the consequences of activated mTORC1 in human lupus T cells.
Kato H, Perl A. Mechanistic target of rapamycin complex 1 expands Th17 and IL-4+ CD4-CD8- double-negative T cells and contracts regulatory T cells in systemic lupus erythematosus. J Immunol. 2014;192(9):4134–44. This study showed that the activation of mTORC1 has a selective effect on human lupus T cells.
Yin Y, Choi SC, Xu Z, et al. Normalization of CD4+ T cell metabolism reverses lupus. Sci Transl Med. 2015;7(274):274ra18. This study showed that a treatment combining metformin and a glucose inhibitor reversed disease in lupus-prone mice and normalised the metabolism of their CD4+ T cells.
Yin Y, Choi S-C, Xu Z, et al. Glucose oxidation is critical for CD4+ T cell activation in a mouse model of systemic lupus erythematosus. J Immunol. 2016;196(1):80–90. This study showed that murine lupus T cell oxidizes glucose, identifying mitochondrial metabolism as the major mechanism to sustain their activation.
Lai ZW, Hanczko R, Bonilla E, et al. N-acetylcysteine reduces disease activity by blocking mammalian target of rapamycin in T cells from systemic lupus erythematosus patients: a randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2012;64(9):2937–46.
Fernandez D, Bonilla E, Mirza N, et al. Rapamycin reduces disease activity and normalizes T cell activation-induced calcium fluxing in patients with systemic lupus erythematosus. Arthritis Rheum. 2006;54(9):2983–8.
Lui SL, Tsang R, Chan KW, et al. Rapamycin attenuates the severity of established nephritis in lupus-prone NZB/W F1 mice. Nephrol Dial Transplant. 2008;23(9):2768–76.
Oaks Z, Winans T, Caza T, et al. Mitochondrial dysfunction in the liver and antiphospholipid antibody production precede disease onset and respond to rapamycin in lupus-prone mice. Arthritis & Rheumatol. 2016: Jun 22. doi: 10.1002/art.39791. [Epub ahead of print].
Macintyre AN, Gerriets VA, Nichols AG, et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 2014;20(1):61–72. This study established that glucose uptake through Glut1 is essential for CD4+ T cell activation.
Jacobs SR, Herman CE, MacIver NJ, et al. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol. 2008;180(7):4476–86.
Maekawa Y, Ishifune C, Tsukumo S, et al. Notch controls the survival of memory CD4+ T cells by regulating glucose uptake. Nat Med. 2015;21(1):55–61.
Yang ZC, Liu Y. Hypoxia-inducible factor-1alpha and autoimmune lupus, arthritis. Inflammation. 2016;39(3):1268–73.
Chi H. Regulation and function of mTOR signalling in T cell fate decisions. Nat Rev Immunol. 2012;12(5):325–38.
Shi LZ, Wang R, Huang G, et al. HIF1α–dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. 2011;208(7):1367–76.
Davidson A. What is damaging the kidney in lupus nephritis? Nat Rev Rheumatol. 2016;12(3):143–53.
Kolev M, Dimeloe S, Le Friec G, et al. Complement regulates nutrient influx and metabolic reprogramming during Th1 cell response. Immunity. 2015;42(6):1033–47. This study revealed a complex role for complement receptor CD46 in promoting Th1 differentiation through metabolic reprogramming.
Le Buanec H, Gougeon ML, Mathian A, et al. IFN-alpha and CD46 stimulation are associated with active lupus and skew natural T regulatory cell differentiation to type 1 regulatory T (Tr1) cells. Proc Natl Acad Sci U S A. 2011;108(47):18995–9000.
Wahl DR, Petersen B, Warner R, et al. Characterization of the metabolic phenotype of chronically activated lymphocytes. Lupus. 2010;19(13):1492–501.
Glick GD, Rossignol R, Lyssiotis CA, et al. Anaplerotic metabolism of alloreactive T cells provides a metabolic approach to treat graft-versus-host disease. J Pharmacol Exp Ther. 2014;351(2):298–307.
Nguyen HD, Chatterjee S, Haarberg KM, et al. Metabolic reprogramming of alloantigen-activated T cells after hematopoietic cell transplantation. J Clin Invest. 2016;126(4):1337–52.
Doherty E, Oaks Z, Perl A. Increased mitochondrial electron transport chain activity at complex i is regulated by N-Acetylcysteine in lymphocytes of patients with systemic lupus erythematosus. Antioxid Redox Signal. 2014;21(1):56–65.
Dimeloe S, Mehling M, Frick C, et al. The immune-metabolic basis of effector memory CD4+ T cell function under hypoxic conditions. J Immunol. 2016;196(1):106–14.
Perl A, Gergely Jr P, Nagy G, et al. Mitochondrial hyperpolarization: a checkpoint of T-cell life, death and autoimmunity. Trends Immunol. 2004;25(7):360–7.
Sobel ES, Brusko TM, Butfiloski EJ, et al. Defective response of CD4+ T cells to retinoic acid and TGFbeta in systemic lupus erythematosus. Arthritis ResTher. 2011;13(3):R106.
Morel L, Croker BP, Blenman KR, et al. Genetic reconstitution of systemic lupus erythematosus immunopathology with polycongenic murine strains. Proc Natl Acad Sci U S A. 2000;97(12):6670–5.
Bricker DK, Taylor EB, Schell JC, et al. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science. 2012;337(6090):96–100.
Herzig S, Raemy E, Montessuit S, et al. Identification and functional expression of the mitochondrial pyruvate carrier. Science. 2012;337(6090):93–6.
Divakaruni AS, Wiley SE, Rogers GW, et al. Thiazolidinediones are acute, specific inhibitors of the mitochondrial pyruvate carrier. Proc Natl Acad Sci U S A. 2013;110(14):5422–7.
Colca JR, McDonald WG, Cavey GS, et al. Identification of a mitochondrial target of thiazolidinedione insulin sensitizers (mTOT): relationship to newly identified mitochondrial pyruvate carrier proteins. PLoS One. 2013;8(5), e61551.
Zhao W, Berthier CC, Lewis EE, et al. The peroxisome-proliferator activated receptor-γ agonist pioglitazone modulates aberrant T cell responses in systemic lupus erythematosus. Clin Immunol. 2013;149(1):119–32.
Aprahamian T, Bonegio RG, Richez C, et al. The peroxisome proliferator-activated receptor γ agonist rosiglitazone ameliorates murine lupus by induction of adiponectin. J Immunol. 2009;182(1):340–6.
Teruel M, Alarcon-Riquelme ME. Genetics of systemic lupus erythematosus and Sjogren’s syndrome: an update. Curr Opin Rheumatol. 2016;28(5):506–14.
X-j Z, Lu X-l, J-c L, et al. Genetic association of PRDM1-ATG5 intergenic region and autophagy with systemic lupus erythematosus in a Chinese population. Ann Rheum Dis. 2011;70(7):1330–7.
Lessard CJ, Sajuthi S, Zhao J, et al. Identification of a systemic lupus erythematosus risk locus spanning ATG16L2, FCHSD2, and P2RY2 in koreans. Arthritis Rheumatol. 2016;68(5):1197–209.
Martinez J, Cunha LD, Park S, et al. Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature. 2016;533(7601):115–9. A novel role for Atg5 in regulating apototic cell clearance by myeloid cells through non-canonical autophagy.
Ishibashi K, Fujita N, Kanno E, et al. Atg16L2, a novel isoform of mammalian Atg16L that is not essential for canonical autophagy despite forming an Atg12-5-16L2 complex. Autophagy. 2011;7(12):1500–13.
Rockel JS, Kapoor M. Autophagy: controlling cell fate in rheumatic diseases. Nat Rev Rheumatol. 2016;12(9):517–31.
Liszewski MK, Atkinson JP. Complement regulator CD46: genetic variants and disease associations. Human Genomics. 2015;9(1):1–13.
Perry DJ, Yin Y, Telarico T, et al. Murine lupus susceptibility locus Sle1c2 mediates CD4+ T cell activation and maps to estrogen-related receptor gamma. J Immunol. 2012;189(2):793–803.
Eichner LJ, Giguere V. Estrogen related receptors (ERRs): a new dawn in transcriptional control of mitochondrial gene networks. Mitochondrion. 2011;11(4):544–52.
Alaynick WA, Kondo RP, Xie W, et al. ERRgamma directs and maintains the transition to oxidative metabolism in the postnatal heart. Cell Metab. 2007;6(1):13–24.
Yoshihara E, Wei Z, Lin CS, et al. ERRgamma is required for the metabolic maturation of therapeutically functional glucose-responsive beta cells. Cell Metab. 2016;23(4):622–34.
Pei L, Mu Y, Leblanc M, et al. Dependence of hippocampal function on ERRgamma-regulated mitochondrial metabolism. Cell Metab. 2015;21(4):628–36.
Kida YS, Kawamura T, Wei Z, et al. ERRs mediate a metabolic switch required for somatic cell reprogramming to pluripotency. Cell Stem Cell. 2015;16(5):547–55.
Michalek RD, Gerriets VA, Nichols AG, et al. Estrogen-related receptor-alpha is a metabolic regulator of effector T-cell activation and differentiation. Proc Natl Acad Sci U S A. 2011;108(45):18348–53.
Murray PJ, Rathmell J, Pearce E. SnapShot: immunometabolism. Cell Metab. 2015;22(1):190–90.e1.
Caro-Maldonado A, Wang R, Nichols AG, et al. Metabolic reprogramming is required for antibody production that is suppressed in anergic but exaggerated in chronically BAFF-exposed B cells. J Immunol. 2014;192(8):3626–36. This study showed that elevated levels of BAFF push B cells through a highly glycolytic metabolism associated with higher effector functions.
Cho SH, Raybuck AL, Stengel K, et al. Germinal centre hypoxia and regulation of antibody qualities by a hypoxia response system. Nature. 2016;advance online publication. A novel mechanism regulating B cell functions through a graded level of hypoxia in germinal centers.
Aronov M, Tirosh B. Metabolic control of plasma cell differentiation—what we know and what we don’t know. J Clin Immunol. 2016;36 Suppl 1:12–7.
Lam WY, Becker AM, Kennerly KM, et al. Mitochondrial pyruvate import promotes long-term survival of antibody-secreting plasma cells. Immunity. 2016;45(1):60–73. An elegant study showing that pyruvate oxidation is required by plasma cells to be long-lived.
Pengo N, Scolari M, Oliva L, et al. Plasma cells require autophagy for sustainable immunoglobulin production. Nat Immunol. 2013;14(3):298–305.
Clarke AJ, Ellinghaus U, Cortini A, et al. Autophagy is activated in systemic lupus erythematosus and required for plasmablast development. Ann Rheum Dis. 2015;74(5):912–20. This study implicated autophagy as promoting the survival and activation of autoreactive B cells in mice.
Arnold J, Murera D, Arbogast F, et al. Autophagy is dispensable for B-cell development but essential for humoral autoimmune responses. Cell Death Differ. 2016;23(5):853–64.
Benhamron S, Pattanayak SP, Berger M, et al. mTOR activation promotes plasma cell differentiation and bypasses XBP-1 for immunoglobulin secretion. Mol Cell Biol. 2015;35(1):153–66.
Pearce EJ, Everts B. Dendritic cell metabolism. Nat Rev Immunol. 2015;15(1):18–29.
O’Neill LA, Pearce EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med. 2016;213(1):15–23.
Galvan-Pena S, O’Neill LA. Metabolic reprograming in macrophage polarization. Front Immunol. 2014;5:420.
Cao W, Manicassamy S, Tang H, et al. Toll-like receptor-mediated induction of type I interferon in plasmacytoid dendritic cells requires the rapamycin-sensitive PI(3)K-mTOR-p70S6K pathway. Nat Immunol. 2008;9(10):1157–64.
Boor PP, Metselaar HJ, Mancham S, et al. Rapamycin has suppressive and stimulatory effects on human plasmacytoid dendritic cell functions. Clin Exp Immunol. 2013;174(3):389–401.
Bao Y, Ledderose C, Graf AF, et al. mTOR and differential activation of mitochondria orchestrate neutrophil chemotaxis. J Cell Biol. 2015;210(7):1153–64.
Donnelly RP, Loftus RM, Keating SE, et al. mTORC1-dependent metabolic reprogramming is a prerequisite for NK cell effector function. J Immunol. 2014;193(9):4477–84.
Keating SE, Zaiatz-Bittencourt V, Loftus RM, et al. Metabolic reprogramming supports IFN-gamma production by CD56bright NK cells. J Immunol. 2016;196(6):2552–60.
Yang Z, Shen Y, Oishi H, et al. Restoring oxidant signaling suppresses proarthritogenic T cell effector functions in rheumatoid arthritis. Sci Transl Med. 2016;8(331):331–8.
Gatza E, Wahl DR, Opipari AW, et al. Manipulating the bioenergetics of alloreactive T cells causes their selective apoptosis and arrests graft-versus-host disease. Sci Transl Med. 2011;3(67):67ra8–8.
Shirai T, Nazarewicz RR, Wallis BB, et al. The glycolytic enzyme PKM2 bridges metabolic and inflammatory dysfunction in coronary artery disease. J Exp Med. 2016;213(3):337–54.
Lee CF, Lo YC, Cheng CH, et al. Preventing allograft rejection by targeting immune metabolism. Cell Rep. 2015;13(4):760–70.
Kishton RJ, Rathmell JC. Novel therapeutic targets of tumor metabolism. Cancer J. 2015;21(2):62–9.
Yang Z, Shen Y, Oishi H, et al. Restoring oxidant signaling suppresses proarthritogenic T cell effector functions in rheumatoid arthritis. Sci Transl Med. 2016;8(331):331ra38. This study dissected the abormal metabolism of T cells from RA patients, and showed that it can be normalized with oxidizing drugs.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by the authors.
Additional information
This article is part of the Topical Collection on Systemic Lupus Erythematosus
Rights and permissions
About this article

Cite this article
Choi, SC., Titov, A.A., Sivakumar, R. et al. Immune Cell Metabolism in Systemic Lupus Erythematosus. Curr Rheumatol Rep 18, 66 (2016). https://doi.org/10.1007/s11926-016-0615-7
Published:
DOI: https://doi.org/10.1007/s11926-016-0615-7