
Abstract
It is becoming increasingly clear that cellular metabolism plays a critical role in the propagation of appropriate, effective, and pathologic immune responses. In this chapter, we detail the metabolic pathways involved in T cell activation and differentiation, highlighting specific factors responsible for directing the processes that lead to metabolic programming at important stages in the dynamic life cycle of this immune cell lineage. Additionally, this chapter will discuss how key metabolites are acquired, touching on the factors and conditions regulating the expression of crucial transporter molecules in response to activation and pathological states.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
2.1 Introduction to Cellular Metabolism
The diverse cell types of the immune system perform a range of specialized functions that are critical for host defense. They also actively participate in the processes of wound healing, tissue remodeling, regulation of human metabolism, and restraint of potentially harmful (overzealous or misdirected) immune responses. These many and distinct roles are largely orchestrated through signaling cascades that are triggered by the ligation of extracellular and intracellular receptors. Such stimuli may dictate the migration of immune cells to drastically different microenvironments or the initiation of cellular proliferation and differentiation. As such, proper leukocyte function requires adaptation to varying nutrient levels and a flexible metabolism capable of accommodating changes in cellular demands for energy and biosynthesis. Therefore, the metabolic profile of leukocytes is central to multiple aspects of their function. Recently, it has become clear that elements of cellular metabolism are not only integral to immune cell development. Indeed, they may act as distinct switches capable of reshaping the nature of an immune response. As will be discussed in this chapter, pathways that program cellular metabolism also regulate significant signaling cascades that control the effector functions of immune cells.
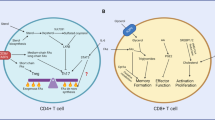
The T lymphocyte cell type is a heterogeneous population encompassing an expansive number of subsets capable of highly specialized functions in response to diverse stimuli. Yet some characteristics are widely shared among these and other leukocyte populations. Among these commonalities is the ability to detect and respond to extracellular and extraorganismal threats. When a mature, yet antigen-inexperienced, T cell emerges from the thymus to enter the circulation, they are relatively inert or quiescent. Prior to receiving activating signals, the modest energetic and biosynthetic needs of naïve T cells are met chiefly through the acquisition and metabolism of pyruvate derived from glucose (via mitochondrial oxidative phosphorylation) or fatty acids (via fatty acid oxidation) to generate adenosine triphosphate (ATP) through the tricarboxylic acid (TCA) cycle. When a naïve T cell encounters activating signals, a dramatic metabolic reprogramming occurs, allowing for the increased production of energy and biological raw materials – e.g., amino acids, fatty acids, and nucleotides [1] (Fig. 2.1). The significance of this metabolic reprogramming cannot be understated, as the robust production of effector molecules (e.g., cytokines, tissue adhesion molecules, cytotoxic factors) and cell division is as costly in terms of cellular resources as it is necessary for the establishment of an effective immune response. Moreover, specific alterations to a naïve T cell’s metabolic profile – or inadequacies therein – during activation may affect the ability of a cell to differentiate into the appropriate effector T cell lineage capable of responding to a specific biological or chemical insults.

Metabolic reprogramming in T cell activation/differentiation. The characteristically quiescent naïve T cell consumes meager amounts of glucose and fatty acids, processing their catabolic byproducts through OXPHOS. Upon TCR ligation and costimulation, T cells engage a rapidly proliferative profile, increasing the surface expression of glucose and amino acid transporters to accommodate this increased demand for products for cellular growth and effector function. Aerobic glycolysis is prioritized at the expense of OXPHOS. During the contraction phase of an immune response, memory lineages persist, relying on a metabolic profile dominated by the metabolism of fatty acids through OXPHOS. Tregs possess a metabolic profile similar to Tmem. Acronyms: FAO fatty acid oxidation, Gly glycolysis, OXPHOS oxidative phosphorylation, AA amino acids, Nuc nucleotides, iTreg peripherally induced Treg, nTreg thymically derived natural Treg
2.1.1 T Cell Differentiation
Engagement of the T cell receptor (TCR) by antigen/MHC complexes and the interaction of costimulatory receptor and ligands (e.g., CD28 and B7 family members) bring about T cell activation. Stimulation of naïve CD4+ T cells not only leads to expansion of these cells but also to their acquisition of highly specialized effector functions accompanying their commitment to defined T helper (Th) lineages. Th differentiation is driven by lineage-specific cytokines present during naïve CD4+ T cell activation. Signaling events downstream of cytokine-cytokine receptor interactions promote the expression of “master regulator” transcription factors responsible for establishing and enforcing Th-specific programs of gene expression. These, in turn, underlie the unique effector molecules and functions of Th cells [2].
For example, Th1 cells are well known for their production of interferon-γ (IFNγ) which, along with other characteristic functions, is driven by the transcription factor T-bet. These cells are important for the cell-mediated immunity necessary to resist intracellular viral, bacterial, and parasitic infections. A robust Th1 response is also needed for effective antitumor immune responses. Cells of the Th2 lineage, on the other hand, produce interleukin-4 (IL-4) and direct immunity to extracellular parasites (e.g., helminthes). GATA3 is a critical regulator of Th2-accociated gene expression [2]. Meanwhile Th17 cell differentiation is driven by STAT3-activating cytokines (such as IL-6) and the transcriptional regulator RORγt. Th17 cells are responsible for fighting extracellular bacterial and fungal infections through secretion of the characteristic cytokines IL-17A, IL-17F, and IL-22. These cells have considerable inflammatory potential and their involvement in autoimmune and inflammatory diseases is well established [3].
Another important subset of CD4+ T cells is known for their ability to suppress the activity of other leukocytes. While several types of T cells can exert regulatory functions, the most recognized and arguably the most important of these is CD4+ T cell subset marked by constitutively high expression of the transcription factor Foxp3 [4]. Foxp3+ regulatory T cells (Tregs) utilize a variety of mechanisms to carry out this suppressive function. These include the production of anti-inflammatory cytokines (e.g., IL-10, TGFβ, and IL-35), the expression of coinhibitory molecules (e.g., CTLA-4 and LAG3), subversion of antigen-presenting cell activity to perpetuate tolerance, and the sequestration of growth factors and resources needed for effector cell expansion and function [5]. The action of these cells prevents overzealous immune activation that can lead to the collateral damage of healthy tissues. Tregs also suppress autoreactive T cells that would otherwise trigger autoimmune disease. While necessary mediators of immune homeostasis, these cells can also oppose the mounting of effective anti-tumor immune responses [6]. Foxp3+ Tregs can arise in the thymus or can be induced from Foxp3− naïve CD4+ precursors in peripheral tissues or ex vivo upon activation in the presence of the cytokines IL-2 and TGF-β [7] (Fig. 2.1). Just as with the aforementioned Th subsets, cellular metabolism plays a role both in the differentiation of these suppressor cells. While cytokines and transcription factors hold incredible sway over the Th decision-making process, as will be discussed later, a number of metabolic factors also influence the differentiation of naïve CD4+ T cells.
CD8+ T cells also differentiate into distinct subsets. These include effector cells capable of killing infected or transformed cells (the so-called cytotoxic T lymphocytes or CTLs) as well as long-lived memory CD8+ T cells that respond with the kinetics of innate immune cells to repeat encounters with antigens. Metabolic factors play a role in the distinct biology of these subsets as well.
2.2 Key Metabolic Pathways of T Cells
2.2.1 Glycolysis and the Tricarboxylic Acid Cycle
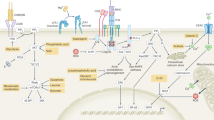
Within T cells, production of ATP largely results from the catabolism (breakdown) of glucose or fatty acids. There are a multitude of potential fates of glucose once it is transported into the cell, although many of the initial processing steps of the pathway termed glycolysis are shared by these paths. Upon entering the cell, glucose is rapidly phosphorylated at the sixth carbon position by the enzyme, hexokinase, which expends a molecule ATP and produces glucose-6-phosphate. In glycolysis, subsequent steps of rearrangement, isomerization, and bond-breaking follow resulting in the generation of the high-energy molecules, nicotinamide adenine dinucleotide (NADH), and ATP, as well as two identical three-carbon pyruvate molecules. Additionally, intermediate products of this process may be shuttled off to the pentose phosphate or serine biosynthesis pathways, the β-oxidation pathway, or the glycogenesis pathway, allowing for the production of nucleotides, fatty acids, or the energy storage molecule, glycogen, respectively (Fig. 2.2).

Pathways in T cell metabolism. This flowchart details the acquisition of extracellular nutrients and the de novo synthesis of anabolic products. Many of these processes are readily reversible, despite their being represented with an unidirectional arrow. Key intermediates of these pathways and enzymes targeted for regulation are highlighted; some intermediates are excluded for simplicity’s sake. 1 Glycolysis; 2 Pentose Phosphate Pathway; 3 Serine Biosynthesis Pathway; 4 β-Oxidation; 5 Glutaminolysis. Acronyms: 3PG 3-phosphoglyceric acid, R5P ribose-5-phosphate, E4P erythrose-4-phosphate, FATPs:fCPS carnitine palmitoyltransferase system
Once pyruvate is generated, it can be shuttled into the mitochondria where further breakdown occurs via the tricarboxylic acid (TCA) cycle – also known as the citric acid or Krebs cycle [8]. Inside the mitochondrial matrix, the three-carbon molecule is either carboxylated or decarboxylated and bound to coenzyme a (CoA), yielding the four-carbon molecule, oxaloacetate, or the two-carbon molecule, acetyl-CoA, respectively. Molecules of oxaloacetate and acetyl-CoA, generated separately from molecules of pyruvate, are bound through the action of citrate synthase, producing the six-carbon molecule, citrate. Seven successive steps of the TCA cycle follow, expelling two-carbon molecules such as carbon dioxide (CO2) and regenerating oxaloacetate to be rebound to a new molecule of acetyl-CoA. Two reducing agents are generated through this cycle, NADH and flavin adenine dinucleotide (FADH2), which donate electrons to cytochromes of the electron transport chain, resulting in the robust production of ATP through oxidative phosphorylation (OXPHOS). Three molecules of NADH (roughly equal to ~2.5 molecules of ATP) and one of FADH2 (~1.5 molecules of ATP) are produced from the consumption of one molecule of acetyl-CoA yielding ~9 molecules of ATP (~30 to 32 molecules of ATP are produced per molecule of glucose through this entire pathway). This process relies on the availability of both NAD+ and FADH+ to oxidize the byproducts of glucose and, importantly, oxygen to serve as the final acceptor of electrons. Additionally, a molecule of GTP is proceeded during one step of the TCA cycle, which, like ATP, contains a high-energy phosphate bond that is broken to supply energy or to activate/deactivate enzymatic processes.
Alternatively, in a pathway that avoids mitochondrial involvement, pyruvate can be processed into lactate through the action of lactate dehydrogenase. The reaction consumes a molecule of NADH and bypasses the requirement for oxygen as the terminal electron acceptor, thereby allowing for a rapid, albeit less efficient, production of ATP from glucose in environments where little oxygen may be present. When oxygen is scarce, glycolysis alone can be used to sustain cellular requirements for ATP, with lactate expelled and NAD+ recycled to restart the catabolic process.
2.2.2 Aerobic Glycolysis
Certain immune cell subsets including T cells can preferentially utilize glycolysis to generate ATP, even when oxygen is abundant. This particular metabolic profile is characteristic of tumor cells, and in 1956, Otto Warburg postulated that this shift in cancer cell metabolism was the fundamental cause of tumorigenesis [9]. Although we now understand a more elaborate basis for cellular transformation and tumor formation, this metabolic shift still bears his name, termed Warburg metabolism (also, aerobic glycolysis; the remainder of the chapter will use the latter terminology). As will be discussed, aerobic glycolysis is utilized by subsets of T cells post-activation to support their effector functions and characteristically rapid proliferation, a phenotype similar to the rampant cellular replication of tumor cells [10, 11].
Although aerobic glycolysis is less efficient for generating ATP from a single molecule of glucose, the use of these pathways confers a significant survival advantage to the cells that employ in it – namely, through the generation of molecular “raw materials” for biosynthetic processes. Two intermediates of glycolysis, namely, glucose-6-phosphate and 3-phosphoglyceric acid (3PG), can be fed into the pentose phosphate or serine biosynthesis pathways, respectively [1, 12, 13]. The resulting products from these pathways provide the cell with essential precursors of anabolic (synthetic) processes. The pentose phosphate pathway can be divided into two phases, an oxidative phase and a subsequent non-oxidative phase, each producing distinct precursors for biosynthetic and effector processes. The first generates the reducing agent, nicotinamide adenine dinucleotide phosphate (NADPH; distinct from NADH), which catalyzes numerous anabolic processes in lipid and cholesterol biosynthesis pathways and facilitates the production of superoxide anions (later, free radicals) within macrophages and neutrophils (see the section on reactive oxygen signaling below) [14]. The non-oxidative phase results in the production of the sugars, ribose-5-phosphate and erythrose-4-phosphate. This allows for the synthesis of nucleotides and nonessential aromatic amino acids, respectively. The serine biosynthesis pathway is essential for the de novo production of serine and glycine for protein and nucleotide biosynthesis. 3PG is converted in three steps to serine, which in turn allosterically inhibits the first enzyme of the series, aptly named, 3PG dehydrogenase. 3PG dehydrogenase requires the cofactor NAD+, which accepts a hydride from 3PG to become NADH. One final step in the process synthesizes glycine from serine, in a readily reversible fashion.
Many activated immune cells – e.g., effector T cells (Teff; namely, Th1, Th2, and Th17 CD4+ subsets and cytotoxic CD8+ cells), neutrophils, M1 macrophages, and some DC subsets – will predominantly utilize aerobic glycolysis to provide energy to the cell. Here the majority of pyruvate molecules produced are rapidly converted into lactate and expelled (see below for distinct regulatory and memory T cell metabolism) [12]. During this process, glycolytic intermediates are diverted through the pentose phosphate and serine biosynthesis pathways, promoting the generation of amino acids, nucleotides, and NADPH. Within CD4+ Teff cells, there is evidence suggesting that a portion of the pyruvate generated during activation-induced glycolysis is still metabolized through the TCA cycle and OXPHOS. CD8+ cells, on the other hand, do not appear to enhance OXPHOS following activation [15,16,17].
Reflecting the importance of glycolytic metabolism in activated Teff cells, many studies have shown that small-molecule inhibitors interfering with this process can be immunomodulatory. The glucose analogue, 2-deoxyglucose (2-DG) actively competes for the active site of hexokinase II (one of the three known isoforms of hexokinase), the enzyme that catalyzes the initial phosphorylation of glucose to G6P. 2-DG treatment selectively inhibits the glycolysis-dependent development of Th17 cells while reciprocally promoting the generation of Foxp3+ regulatory T cells (Tregs) in vitro. Furthermore, adoptive transfer of 2-DG-conditioned Th17 cells results in a less efficient induction of neuropathology in the murine autoimmune model of experimental autoimmune encephalomyelitis (EAE) as compared to untreated Th17 cells [18, 19]. EAE mimics multiple sclerosis, with mice developing ascending paralysis as self-reactive Th1 and Th17 cells assault the myelin sheaths of neurons within the central nervous system (CNS). These results demonstrate the importance of glycolysis for (in this case pathologic) Teff responses. Memory T cells, on the other hand, do not rely on glycolytic metabolism. Reflecting this, 2-DG treatment augments the generation of memory T cells in mice and promotes the execution of memory T cell-mediated antitumor immunity [20].
2.2.3 Fatty Acid Oxidation (FAO)
By mass, the oxidation of fatty acids is the most energetically efficient metabolic process described in this chapter. Due to the negative charge on the polar head group, fatty acids must first be transported across the cellular membrane before they can be metabolized [21]. Once in the cytosol, ligases facilitate the association of a fatty acid to a molecule of CoA at the proximal carbon (C-1; α carbon) of the fatty acid chain. In the case of long chain fatty acids, the newly formed acyl-CoA undergoes a temporary subunit switch, in which CoA is replaced with a carnitine molecule to facilitate its transport across the mitochondrial matrix. Within the matrix, the reverse reaction occurs and further catabolism of the fatty acid proceeds. This reaction is catalyzed by the carnitine palmitoyltransferase system and is essential for the efficient metabolism of fatty acids. Once within the mitochondrial matrix, acyl-CoA dehydrogenase catalyzes the initial catabolic step of the process termed β-oxidation, where the bond between C-2 (β carbon) and C-3 is manipulated by successive oxidations and is eventually cleaved. The process results in the isolation of the two-carbon molecule, acetyl-CoA, from the fatty acid chain and the production of two high-energy reducing molecules, NADH and flavin adenine dinucleotide (FADH2). If a single acetyl-CoA molecule were to continue through the TCA cycle and OXPHOS, it would produce ~10 molecules of ATP. Should the consumption of the NADH and FADH2 molecules produced by β-oxidation be included in this calculation, ~14 molecules of ATP would be produced from a single β-oxidation step. The most common naturally occurring fatty acids are saturated and even numbered, between 4 and 28 carbons in length. Thus, a single fatty acid molecule from this category can result in the production of 28–196 molecules of ATP. Two by two, a fatty acid chain can be shrunk down, each cycle turning over molecules that can be shuttled through OXPHOS to produce ATP, until its last acetyl-CoA molecule remains. Mechanisms exist to account for odd-numbered and unsaturated fatty acids in this process, but these topics are beyond the scope of this chapter. Besides serving as high-energy metabolites, fatty acids also provide the key components of the cellular membranes, namely, phospholipids and steroids. In T cells and a great many other cell types, fatty acid synthesis is used to promote both cellular growth and organelle biogenesis [22].
Tregs and memory T cells (Tmem), as well as M2 macrophages, adopt a vastly different immune profile compared to their naïve and effector counterparts, primarily metabolizing lipids through fatty acid oxidation (FAO; β-oxidation) to support both their function and survival [17, 23] (Fig. 2.1). Tregs are critical for the maintenance of immune homeostasis as they counteract the milieu of proinflammatory immune cells through suppression of leukocyte activation and function. As previously mentioned with the glycolytic small-molecule inhibitor, 2-DG, impairment of glucose metabolism can promote a Treg fate and hinder proinflammatory Teff differentiation. Excess exogenous fatty acids in vitro have a decidedly negative effect on acquisition of the Th17 phenotype, illustrating a metabolic mechanism for the reciprocal regulation of Treg and Th17 cell fates. Indeed, forced reliance on FAO during in vitro T cell differentiation favors the generation of Tregs (as evidenced by the heightened induction of Foxp3 expression and the suppression of responder cell proliferation) at the marked expense of Teff differentiation [23]. The genetic or chemical inhibition of the metabolic regulator, mechanistic target of rapamycin (mTOR; also, FRAP1, which will be discussed in detail below), during CD4+ T cell activation decreases aerobic glycolysis and augments FAO favoring the generation of Foxp3+ Tregs [24] (see the mTOR section below for more details). However, when FAO is simultaneously interrupted through the etomoxir-mediated inhibition of the carnitine palmitoyltransferase system (and thus inhibition of fatty acid transport), T cells are no longer preferentially shunted toward a Treg fate by suppressed mTOR activity.
While Teff cells expand clonally upon activation during the early stages of an immune response, relatively small numbers of long-lived Tmem cells persist during and after the contraction phase [25]. A defining aspect of Tmem is their capacity to respond, with accelerated kinetics and cytokine release, to a repeat antigen encounter (i.e., a secondary infection). These cells transition abruptly from a state of quiescence requiring minimal energetic demands, to one typified by full-bore effector function and proliferation. The rapid reactivation of Tmem cells must be accounted for metabolically. Within the CD8+ memory lineage, it has been well established that both Tmem generation and persistence, as well as repeat-antigen-induced reactivation, are dependent on the modulation of FAO [17, 26, 27]. Compared to their effector counterparts, Tmem cells possess a greater mitochondrial mass (enhanced mitochondrial biogenesis) and correspondingly express FAO-associated enzymes and carnitine palmitoyltransferase system proteins at a substantially higher level [22, 26, 28] imparting them with a greater spare respiratory capacity (SRC). SRC is defined as the enhanced ability of a cell to generate energy in response to cellular activation or stress [26, 27]. Thus, the development of SRC within Tmem populations effectively preps the cells for reactivation. Additionally, the advantage of augmented SRC is thought to facilitate the survival of Tmem cells under conditions of energetic stress [29].
2.3 Metabolite Acquisition Associated with T Cell Activation
Following TCR ligation and the triggering of costimulation pathways, T cells undergo clonal expansion and transcriptome reprograming to express effector molecules including cytotoxins, cytokines, and cell-surface molecules. This robust increase in cellular growth (accumulation of biomass), proliferation, and protein anabolism demands a matched increase in metabolite uptake to provide components for the generation of cellular machinery and free energy required to catalyze chemical reactions [1, 30, 31]. T cells are incredibly reliant on nutrient absorption for replication and function, and as such, starvation from glucose or amino acids represents one mode of regulation controlling T cell population size and activity [32]. Interestingly, in sites resistant to immune activation (a.k.a. immune privileged sites) – e.g., the brain, eye, testicles, and placenta – FAO is the primary source of energy production. Increased FAO supports suppressive immune cell lineages, thereby protecting these vital organs from autoimmunity [33]. T cells control their intake of nutrients through the expression of transporter proteins on the cellular membrane. As will be described, the enhanced expression of the following transporters is essential for the proliferation, differentiation, and effector functions of T cells.
2.3.1 Glucose Uptake
As stated previously, the transition of T cells out of quiescence and into an activated state requires an increased uptake of nutrients to fuel augmented cellular growth and proliferation and meet the anabolic needs of effector molecule production. For Teff subsets, the increased import of glucose is essential for activation-associated functions [34]. A family of glucose transporters (Glut1–14) facilitate the uptake of glucose and related sugars in mammalian cells [35,36,37]. T cells have been reported to express a select few Glut family transporters, namely Glut1, 2, 3, 6, and 8. Chief among these transporters is the facilitative transporter, Glut1, which is responsible for the basal level of glucose uptake in all resting immune cells [34].
Costimulatory signals received via CD28 ligation and propagated through PI3K/Akt signaling results in the upregulation of Glut1 (and a corresponding downregulation of the carnitine palmitoyltransferase system critical for FAO) [38]. It is the increase in cell-surface expression of Glut1 alone that facilitates the dramatic increase of glucose influx required to manage the cells’ heightened glycolytic demands [39, 40]. Interruption of this process has profound effects on T cell function, including their ability to differentiate into proinflammatory lineages. Irrespective of the expression of other glucose transporters, the deletion of Glut1 alone confers a significant deficit to the growth, clonal expansion, and persistence of murine Teff subsets when activated in vivo [34]. Conversely, an overabundance of Glut1 promotes the accumulation of biomass (i.e., cellular growth) in naïve T cells and the acquisition of an activated phenotype [41]. Illustrating the importance of glycolysis in effector molecule anabolism, glucose deficiency diminishes the capacity of cytotoxic CD8+ cells (CTLs) to produce effector cytokines, perforin, and granzymes [41,42,43]. Tregs, which rely primarily on the oxidation of fatty acids for the generation of cellular energy, appear unaffected by this deletion, persisting and functioning in vivo regardless of Glut1 expression [41].
2.3.2 Amino Acid Uptake
T cell activation also results a heightened demand for amino acids, resulting in the upregulation of amino acid transporters following activation. Indeed, as with glucose deficiency, an inadequate supply or uptake of these metabolites can adversely affect the proliferative capacity of activated T cells and can significantly shape their differentiation. Within activated T cells, the amino acid glutamine is particularly critical. Glutamine is absorbed and broken down in a process termed glutaminolysis. Through glutaminolysis, the amino acid is broken down, and its derivatives can be fed into the TCA cycle, restoring levels of intermediates that are consumed by biosynthetic processes. This allows for the de novo synthesis of lipids and NADPH. The activation-induced enhancement of glutamine absorption is accomplished through several well-characterized transporters. The antiporter, ASC amino acid transporter 2 (ASCT2; also called Slc1a5), is upregulated as a direct result of T cell receptor (TCR) ligation [44]. ASCT2 is surprisingly not necessary for the proliferation of T cells. However, even moderate restrictions to glutamine import can have profound effects on the ability of a T cell to differentiate, particularly into the proinflammatory T helper (Th) cell subsets, Th1 and Th17. In murine models of autoimmunity, the in vivo CD4+ T cell-specific deletion of ASCT2 induces a markedly diminished Th1 and Th17 immune response, alleviating the progression of host-reactive disease in murine EAE (a disease driven by Th1 and Th17 immune responders) [44]. This diminished pathology is the result of deficient induction of proinflammatory Th cell subsets alone, without hampering the induction of Tregs or CD8+ T cells [44]. This fact illustrates the potential heterogeneity of metabolite acquisition needs among T cell lineages.
CD8+ T cell function appears to rely primarily on another amino acid transport pathway, preferentially absorbing the amino acid leucine to regulate effector potential. Following exposure to activating signals through TCR ligation and costimulation, both CD4+ and CD8+ T cells upregulate their surface expression of system L neutral amino acid transporter 1 (LAT1; also called Slc7a5). LAT1 is a heterodimeric transporter molecule and is chiefly responsible for the absorption of essential branched-chain (e.g., leucine and isoleucine) and aromatic (e.g., tryptophan and phenylalanine) amino acids while also participating mildly in glutamine transport [45,46,47]. Specifically, the enhanced leucine influx facilitated by LAT1 is critical for the regulation of CD8+ T cell differentiation and migration [17, 48, 49]. As will be discussed in the sections that follow, concentrations of intracellular amino acids are sensed by the key metabolic regulator, mTOR, with a deficiency in amino acids resulting in the inhibition of anabolic processes and cellular proliferation. Accordingly, LAT1-deficient T cells do not proliferate well and display stunted effector differentiation. Specifically, LAT1-deficiency in CD4+ and CD8+ T cells results in a less pronounced induction of Th1/Th17 cells and CTLs, respectively. These observations were linked to an inability to increase amino acid uptake and a decrease in mTOR activity during T cell activation [45]. Moreover, the diminished uptake of leucine resulted in the loss of c-Myc expression. c-Myc is a key regulator of the glycolytic metabolism essential for activated Teff function, and as such, activation-induced upregulation of Glut1 expression was lacking in LAT1-deficient T cells. Interestingly, this deficit in regulator and transporter expression was only apparent at the protein level; levels of c-Myc and Glut1 mRNA were unaffected [45]. A similar loss of proliferation and effector potential can be accomplished through the system L neutral amino acid transporter inhibitors, BCH (2-aminobicylo-(2,2,1)-heptane-2-carboxylic acid), and brasilicardin A [50, 51]. Thus, it is the enhanced uptake of amino acids facilitated by upregulated expression of amino acid transporters, like ASCT2 and LAT1, which allows for the augmented anabolic metabolism that is essential for adequate T cell activation.
Certain immune cell subtypes, namely, dendritic cells (DCs) and macrophages (Mø), can control the availability of the crucial amino acid cysteine to mediate the cellular growth and proliferation of T cells [52]. Due to the typically low extracellular concentrations of the amino acid, DC-/Mø-mediated cysteine release adds an effective level of control to T cell function. Cysteine is processed intracellularly into the tripeptide, glutathione, a principal molecule in mechanisms that protect the DNA replication process and, thus, cellular proliferation. Glutathione has been shown to protect T cells from the effector functions of phagocytic cells by functioning as a key antioxidant, allowing for effective T cell targeting of foreign and transformed cells in harsh microenvironments. Both phagocytic cells (e.g., Mø, neutrophils) and activated T cells can initiate respiratory burst, utilizing NADPH to produce and release reactive oxygen species (ROS) to facilitate the destruction of foreign cells [53]. While ROS have been shown to function as important signaling molecules within T cells, promoting, among other things, the production of IL-2, too much intracellular ROS can be damaging. By releasing ROS into the inflammatory milieu, resident T cells may come under oxidative stress, defined by an overabundance of ROS as compared to neutralizing antioxidants. Prolonged oxidative stress can diminish cellular function or induce apoptosis through chemical reactions that alter protein and lipid conformation/function and damage DNA [54,55,56]. Thus, in order to maintain the functionality of an immune response, mechanisms of protecting T cells against ROS exposure are critical. Therefore, the antioxidant function of glutathione promotes the oxidative balance in conditions of high ROS presence. Interestingly, Tregs too utilize this mechanism to control proinflammatory events. Tregs, while naturally more resistant to oxidative stress-induced apoptosis [57], interact with APCs to facilitate increased cystine uptake by Tregs, thereby competing with local Teff cells for available amino acids and inhibiting the ability of Teff cells to replicate and function [58].
Clearly, an appropriate intracellular concentration of amino acids during T cell activation and expansion is paramount to establish an effective immune response. Indeed, the availability and metabolism of other amino acids such as tryptophan [59, 60] have also been revealed to dictate the effector/regulatory immune axis. The enzyme indoleamine 2,3-dioxygenase (IDO) is responsible for the breakdown of tryptophan, and the metabolites generated by this enzyme as well as the resulting amino acid depletion limit T cell activation and promote suppressive Tregs. IDO is notably produced by tolerogenic dendritic cells and promotes the generation and expansion of Tregs while discouraging the conversion or reprogramming of established Tregs - favoring instead the retention of a traditional, predominantly suppressive phenotype. Importantly abundant IDO levels in the gut and tumor-draining lymph nodes were shown to suppress Treg reprogramming by IL-6 [61,62,63]. IDO activity is also a potent mechanism for immunosuppression capable of mediating maternal tolerance of the fetus and preventing effective antitumor immunity in the cancer setting [64]. Inhibiting IDO in the cancer setting can be an avenue to overcome tumor-induced immunosuppression [65].
Scarcity of extracellular amino acids can lead to the upregulation of several genes involved in amino acid transport and in the de novo synthesis of nonessential amino acids [47, 59, 66]. Artificial induction of these genes, referred to as the amino acid starvation response (AAR), in T cells can suppress the generation of proinflammatory lineages like Th17 cells and reduce effector responses in in vitro and in vivo models of disease [47, 66].
2.4 Key Factors in Metabolic Regulation
An impressive web of receptors, kinases, and transcription factors orchestrate the aforementioned shifting of metabolic lifestyle that is necessary to coordinate the developing immune response (summarized in Fig. 2.3). Several key molecules that govern multiple aspects of cellular metabolism will be described within this chapter. As will be revealed, the precise regulation of these factors and their activities is requisite for appropriate immune function. Their dysregulation, on the other hand, can result in cellular transformation, autoimmunity, or metabolic disorder. How the therapeutic modulation of the expression or function of these metabolic regulators can provide tantalizing avenues for the control of these ailments will also be discussed.

Regulation of metabolic factors and their interplay. Key metabolic factors are highlighted in white. Downstream gene regulation is described in terms of the genes upregulated (up arrow) upon activation of metabolic factors. Acronyms: FAO fatty acid oxidation, OXPHOS oxidative phosphorylation
2.4.1 Cellular Myc (c-Myc)
c-Myc, the cellular homologue to viral Myc (v-Myc; the avian myelocytomatosis viral oncogene), is a proto-oncogene that can independently transform numerous mammalian cell lineages [67]. The gene was first discovered to be the driving transformative factor in Burkitt’s lymphoma, a tumor that arises exclusively from the dysregulation of myc gene expression [68, 69]. In fact, activity-altering mutations or overexpression of c-Myc are such prevalent factors across cancer lineages that Myc is considered to be a central oncogenic determinant [70]. Since its discovery, the complex regulatory role that Myc plays in cellular metabolism and function has been progressively delineated. c-Myc is categorized as an immediate early gene, meaning that it is rapidly upregulated upon TCR- and costimulatory molecule-triggered signaling cascades. A considerable number of pathways (e.g., NF-κB, MAPK/ERK, PI3Kinase/Akt/mTOR) drive the upregulation of c-Myc expression following cellular activation. Stable expression of c-Myc allows for its heterodimerization with the transcription factor myc-associated factor X (MAX) and subsequent genetic influence over a slew of cellular functions key to effector potential, including the regulation of cell cycle progression, growth and proliferation, and cellular differentiation [70, 71]. Indeed, while global c-Myc deficiency is lethal in in vivo models, homologous deletion of c-Myc in vitro significantly prolongs the time between cellular divisions in affected cells and impairs the dominance of glycolytic metabolism [1, 72]. Specifically, pentose phosphate pathway enzymes are downregulated upon c-Myc deficiency, while other related metabolic pathways remain unaffected. Interestingly, c-Myc also appears to promote the uptake of amino acids, as expression of LAT1 and ASCT2 is correspondingly diminished upon T cell-specific deletion of the molecule (knockout of the receptors also reduces the expression of c-Myc) [1, 44]. This would suggest a multifaceted role of c-Myc in mechanisms that follow T cell activation.
2.4.2 c-Myc Regulates Early Response Pathways Downstream of TCR Ligation
Despite the rapid enhancement of c-Myc expression following TCR engagement, this augmentation does not persist throughout the T cell clonal expansion stage [1, 73,74,75]. Indeed, irrespective of the pivotal roles played by c-Myc in the adoption of a glycolysis-dominated, rapidly proliferative profile, the activity of the molecule is limited by its relatively short half-life (15 min) [76, 77] and a rapid depreciation of expression ~48 h postinfection in murine models of T cell activation [74]. These data propose that other (potentially Myc-induced) transcription factors may be responsible for the persistence of an activated T cell metabolic profile following the initial reprogramming. Recent studies have revealed a number of factors that participate in this metabolic maintenance, including activating enhancer-binding protein 4 (AP4), interferon regulatory factor-4 (IRF4), and hypoxia-inducible factor alpha (HIF-1α; to be discussed later in detail) [73, 78,79,80]. Together, through both c-Myc transcriptional activity and persistent TCR signaling, the expression of AP4, IRF4, and HIF-1α increases and resultantly stabilizes the glycolytic profile characteristic of proinflammatory effector lineages. Interestingly, the genetic ablation of these three “second-wave” factors does not impede the initial metabolic reprogramming seen during T cell activation. Rather, deletion of these factors results in a failure of T cells to sustain aerobic glycolysis and, thus, effector cell function and survival [73, 78,79,80,81], as c-Myc levels diminish following T cell activation.
IRF4 has revealed itself to be a rather unique factor in the genetic reprogramming of effector T cells. Several studies have revealed that IRF4 levels in T cells correspond to the relative affinity of TCR-antigen/MHC complex interactions [78, 79, 82]. Specifically, lower-affinity ligands diminished the longevity and intensity of TCR signaling through reduced IRF4, allowing for a fine-tuning of downstream metabolic and effector programs by the strength of T cell activation. Similar to AP4 null experiments, CD8+ T cell-specific deletion of IRF4 results in a reduction of cellular proliferation and effector cytokine production following c-Myc-driven activation [78, 79, 82]. Within CD4+ T cells, IRF4 transcriptional activity appears to coordinate significant pathways in the effector functions of Th lineages and Treg cells. Loss of IRF4 significantly impaired the adoption of effector characteristics upon naïve CD4+ T cell skewing in vitro (revealed by diminished cytokine production and lineage-specific transcription factor expression) and Treg-mediated suppression in vivo [83,84,85,86,87].
c-Myc facilitates an impressive repertoire of mechanisms following T cell activation. Included among these is the upregulation of glucose and amino acid transporters [1] as well as the promotion of glycolysis and glutaminolysis [1, 45]. Indeed, the loss of c-Myc induced a corresponding decrease in amino acid transport expression, inhibiting the central metabolic pathways involved in establishing effector function [1, 44, 45]. Interestingly, LAT1 deletion was revealed to prevent the translation (but not the transcription) of the c-Myc gene in a mechanism not involving the amino acid sensor, mTOR. Destabilization of c-Myc expression resulted in the expected reduction in glycolysis (downregulation of Glut1; decreased lactate output) and glutaminolysis (decreased glutamine and arginine uptake) [45]. The mechanism behind the leucine sensitivity of c-Myc has yet to be fully delineated.
2.4.3 Therapeutic Opportunities in Targeting the c-Myc Signaling Pathway
Compounds that target c-Myc in cancer cells may be of particular value therapeutically. Aside from its role in metabolic control [88,89,90], c-Myc appears to also promote tumor growth through augmented expression of immunomodulatory cell-surface molecules that can facilitate immune evasion [91]. On the other hand, therapeutic downregulation of c-Myc activity within immune cells could allow for the suppression of the activation-/effector-related functions downstream of c-Myc/MAX, thereby dampening unwanted inflammation. The small-molecule inhibitor, 10058-F4, has been shown to obstruct c-Myc/Max heterodimerization, preventing DNA binding/transcriptional activity [92]. Accordingly, 10058-F4 treatment hampers T helper development in in vitro cultures [93].
2.4.4 Mechanistic Target of Rapamycin (mTOR)
The mechanistic target of rapamycin (mTOR; formerly mammalian TOR) is a key regulator of cellular function, integrating intracellular and extracellular signals to coordinate shifting metabolic states with cell growth, proliferation, and longevity. The smooth transition between catabolic and anabolic metabolism in accordance with available extracellular nutrients and intracellular energy storage is paramount to the survival of cells in the dynamic environments that exist within multicellular organisms. mTOR was discovered between 1993 and 1995 to be the molecular target of the cellular proliferation inhibitor, rapamycin [94,95,96,97], and it was so named. Constraining mTOR function through rapamycin has substantial effects on the ability of T cells to mount an effective proinflammatory response. As will be discussed in the subsequent sections, the suppression of mTOR activity has profound effects on the metabolic profile, and thus the function, of T cell subsets.
The mTOR protein is a well-conserved serine-threonine kinase within the phosphoinositide 3-kinase (PI3K)-related kinase family and has been found to nucleate in two multi-protein complexes, termed mTOR complexes 1 and 2, respectively (mTORC1 and mTORC2). mTORC1 operates as an environmental sensor, promoting protein synthesis, and cellular growth (mass accumulation) and proliferation when intracellular energy and amino acid concentrations are sufficiently high [98,99,100]. mTORC2 plays a supporting role by mediating the organization of the actin cytoskeleton and potentially supporting shifting metabolic states [101, 102]. Both mTORC1 and mTORC2 also promote cellular survival (inhibition of autophagy) through phosphorylation of ULK1 and Atg13 by mTORC1 [103,104,105] or AKT (also called protein kinase B) by mTORC2 [106, 107]. In the following sections, we will discuss the regulation, function, and activity of mTOR complexes in T cell subsets.
2.4.5 Fundamentals of mTORC1 Regulation
mTORC1 is a six-protein complex, consisting of regulatory-associated protein of mTOR (Raptor) [108, 109], mammalian lethal with SEC13 protein 8 (mLST8), proline-rich AKT substrate 40 kDa (PRAS40), DEP-domain-containing mTOR-interacting protein (Deptor) [110], and TELO2-interacting protein 1 homologue and telomere maintenance 2, respectively (Tti1/Tel2) [111]. The distinct functions of each protein have yet to be fully delineated, but significant findings have hinted toward the functional roles of the mTORC1-associated proteins. For example, it has been revealed that Raptor interacts with both mTOR and two downstream substrates, ribosomal protein S6 kinase 1 (S6K1) and eukaryotic initiation factor 4E (eIF4E)-binding protein 1 (4E-BP1), effectively providing the scaffolding to facilitate mTOR kinase activity [108, 109, 112,113,114]. Knockout studies of Raptor reveal it to be an obligate binding partner for effective mTORC1 activity. Consequences of S6K1 and 4E-BP1 phosphorylation include enhanced protein synthesis, which facilitates cellular growth and progression through the cell cycle [98, 100, 115, 116]. Tti1/Tel2 constitutively associate with mTOR and appear to coordinate the assembly of the mTORC1 complex. Deletion of either Tti1 or Tel2 results in disassembly of mTORC1 and a loss of mTOR kinase activity [111]. Conversely, PRAS40 and Deptor have been proposed to negatively regulate the complex, as activation of mTORC1 signaling results in the immediate phosphorylation of PRAS40 and Deptor, thereby hindering their ability to bind to the complex and promoting their subsequent degradation. This results in enhanced mTORC1 substrate binding and activity. PRAS40 has been suggested to directly interfere mTORC1 substrate binding [117], while less is known about the exact function of Deptor in mTORC1 inhibition. Indeed, these data and the observation that diminished mTORC1 signaling are associated with PRAS40 and Deptor binding to mTORC1 and support the notion that they act as negative regulators of mTORC1 activity [110, 118, 119].
Pathways downstream of the growth factor receptors, namely, the insulin and Ras signaling pathways, facilitate the activation of mTORC1 activity through the inhibition of the key mTOR regulatory complex, tuberous sclerosis complex (TSC) [120,121,122,123]. TSC is a heterodimer of TSC1 (also called hamartin) and TSC2 (also called tuberin) and functions as a GTPase-activating protein (GAP), converting protein-bound GTP to GDP. TSC1 stabilizes its dimeric partner, TSC2, which contains the GAP domain. TSC actively targets the GTP-bound protein, Ras homologous enriched in brain (Rheb). Kinases downstream of growth factor receptors respond to stimulatory signals by phosphorylating TSC2, inhibiting the GAP activity of the protein. Considering that GTP-bound Rheb stimulates the activity of mTORC1 through direct association [118, 124], TSC, therefore, acts to inhibit mTORC1 signaling by dephosphorylating the GTP bound to Rheb, thereby inactivating the protein’s effect [120, 125]. Specific mutations within TSC inactivates the complex, leading to uncontrollable cellular growth and proliferation through loss of mTORC1 inhibition and gives rise to the proliferative disease that bears its name, tuberous sclerosis [126]. Ligation of the insulin receptor stimulates a negative feedback loop, where activated S6K1 (downstream of mTORC1) inhibits the stimulatory phosphorylation of the proximal substrate of the insulin signaling pathway, destabilizing insulin receptor signaling and preventing deregulated mTOR activity [127]. Loss of this feedback control through inhibitory mutations or deletions of S6K1 can result in cellular transformation (tumorigenesis) or metabolic disorder (insulin desensitization) [128].
As noted in the “Metabolite Acquisition” section, mTOR functions as a key sensor of intracellular amino acid concentrations [129,130,131,132]. While the exact mechanism of amino acid sensing has yet to be determined, it has been well established that the amino acids, glutamine [132,133,134], leucine [118, 135], and arginine [135] facilitate mTORC1 activation. The lysosome appears to be the staging platform of mTORC1 regulation [136, 137] as stimulatory and inhibitory factors (e.g., Rheb and TSC, respectively) tend to localize in proximity of these organelles [138]. One model suggests that when sufficient amino acid concentrations are sensed, Rag protein heterodimers recruit mTORC1 to the lysosome, allowing for Rheb-dependent activation of mTORC1 and subsequent promotion of cellular growth and proliferation [139].
As described, the activation of the mTORC1 pathway is accomplished through various extracellular and intracellular signals such as the ligation of growth factor receptors or the acquisition of certain nutrients. Additionally, other factors can negatively regulate the downstream signaling of mTORC1, including the energy status of the cell and the oxygen tensions found within the cell’s microenvironment. Immune cells carefully coordinate cellular growth and proliferation so that these processes may only occur when sufficient energy stores and biomaterials exist within the cell. mTORC1 is a master regulator of this synchronization, promoting cellular growth only when conditions are ideal. In an example of negative regulation of mTORC1, AMP-activated protein kinase (AMPK) inhibits mTORC1 when cellular energy is lacking (i.e., when AMP/ATP ratios are high); AMPK phosphorylates the Raptor subunit of mTORC1, leading to the recruitment of 14-3-3 protein to Raptor and the inhibition of mTORC1 activity [140]. Additionally, AMPK has been shown to phosphorylate TSC upstream of mTORC1. AMPK phosphorylates TSC2, promoting the activity of TSC and allowing for the control of transcription and cellular growth through mTORC1 inhibition while also protecting against energy-deprivation-induced apoptosis [120]. Low oxygen tension, too, can regulate the activity of mTORC1 [141] by diminishing ATP levels and increasing AMPK activation and by transcriptional activation of regulated in development and DNA damage responses 1 (REDD1), which facilitates TSC2 activation [142, 143].
2.4.6 Fundamentals of mTORC2 Regulation
mTORC2 is a seven-protein complex which shares some protein subunits with mTORC1, including the central protein, mTOR, as well as mLST8, Deptor, and Tti1/Tel2. mTORC2 also includes the proteins rapamycin-insensitive companion of mTOR (Rictor), mammalian stress-activated protein kinase interacting protein (mSIN1), and protein observed with Rictor-1 and Rictor-2, respectively (Protor-1/Protor-2). Like Raptor in mTORC1, Rictor appears to play a central role in stabilizing mTORC2 and is absolutely essential for characteristic mTORC2-mediated phosphorylation of Akt/PKB. Rictor and mSIN1 are stabilized when associated together and maintain the structural integrity of the complex, thus promoting mTORC2 function [144, 145]. Again, Deptor appears to function as a negative regulator of mTORC2 activity, just as it was described with mTORC1 [110]. Uniquely, however, although mLST8 does not appear to be necessary for mTORC1 function, its association with mTORC2 is paramount for the stability and function of the complex. In vitro knockout studies of mLST8 reveal unperturbed phosphorylation of mTORC1 substrates S6K1 and 4E-BP1, while mTORC2-mediated phosphorylation of AKT/PKB was diminished. This inhibition of mTORC2 function yields a phenotype similar to that seen in Rictor knockout studies [146].
Considering that the deletion of mTORC2 activity is lethal in in vivo models and that the complex is insensitive to rapamycin treatment except at prolonged high doses [147], studies in the function of mTORC2 have been met with difficulty. Consequentially, substantially less is known concerning the mTORC2 control mechanisms. What is known is that, like mTORC1, mTORC2 also responds to growth factor signals. Growth factor ligation results in mTORC2-mediated phosphorylation of a unique serine residue on the serine-/threonine-specific kinase, AKT, thereby enhancing the activity of AKT [148, 149]. A second serine residue is phosphorylated by phosphoinositide-dependent kinase-1 (PDK1), coordinating the full activation of AKT. It has been suggested that mSIN1 may coordinate this mTORC2 activity [150]. The complete phosphorylation of AKT results in enhanced proliferation, promotion of a glycolytic metabolism, and cellular migration. Downstream effects of mTORC2 activation will be described in the following section.
2.4.7 Activation of and the Downstream Effects of mTOR Signaling
According to the two-signal model of lymphocyte activation, antigen presentation alone cannot induce an activated state. Instead, the absence of a second costimulatory signal (e.g., CD28 cross-linking or cytokine receptor ligation) results in cellular anergy and an accordingly diminished mTORC1 signaling through S6K1. Similarly, the inhibition of mTOR activity in vivo through rapamycin treatment results in T cell anergy following antigen presentation [151]. It appears that mTOR not only coordinates intracellular nutrient/energy levels and downstream growth factor receptor signaling with cellular activation but that it also senses extracellular environmental cues to determine whether activation or anergy represents the appropriate response.
Increased uptake of amino acids following T cell activation, specifically the absorption of glutamine and leucine, promotes the activity of mTORC1, which in turn, facilitates the adoption of a predominately glycolytic metabolic profile. Genetic deletion of mTOR in CD4+ T cells dampens their proliferative capacity and prevents their differentiation into effector T helper subsets – e.g., Th1, Th2, Th17 – even under the appropriate in vitro skewing conditions. Instead, activating naïve CD4+ T cells in the absence of mTOR activity induces a hypersensitivity to TGF-β signaling and a preferential generation of Foxp3+ Tregs [152]. Interestingly, the inhibition of mTOR activity induces expansion of natural CD4+ Tregs in vivo and the corresponding de novo synthesis of Foxp3 in differentiating naïve CD4+ T cells, supporting the notion that this pathway negatively regulates immune tolerance [153].
Adding complexity to mTOR’s role in determining CD4+ T cell fate was the revelation that the two mTOR complexes have distinct effects on this process. Mice lacking Rheb have been used to study the effects of defective mTORC1 activity. CD4+ T cells from these knockout mice fail to differentiate into proinflammatory Th1 and Th17 effector cells. Treating T cells with rapamycin suppresses rapamycin-sensitive mTORC1 activity and thus inhibits the induction of proinflammatory T helper subsets, Th1 and Th17, but not Th2 [151]. On the other hand, Rictor-deficient CD4+ T cells, which lack mTORC2 activity, display markedly impaired Th2 differentiation [24].
mTOR signaling is also foundational in determining the fate of CD8+ T cells. Upon naïve CD8+ T cell activation, mTOR activity is increased in response to IL-12/STAT4 signaling and costimulation. In these cells, mTOR can upregulate the expression of the transcription factor, T-bet, promoting a cytotoxic CD8+ T cell (CTL) expression profile [48]. While rapamycin treatment of CD4+ T cells leads to cellular anergy and/or the induction of Treg characteristics (including Foxp3 upregulation), CD8+ T cell-specific suppression of mTOR activity results in the generation of memory precursor CD8+ T cells [17, 48, 49]. Indeed, it appears that CD8+ Tmem cells are negatively regulated by mTOR activity. Studies in which TSC1 or TSC2 have been specifically deleted in CD8+ T cells reveal TORC1 to positively enforce effector characteristics in CD8+ cells while inhibiting the formation of Tmem precursors during the early-stage immune response [154,155,156,157]. Indeed, TSC2 null mice, which display constitutive mTORC1 activity, boast a CD8+ T cell compartment with enhanced glycolysis and effector function [154]. Inhibition of mTORC2 can similarly enhance the development of Tmem [154].
The downstream effects of mTORC1 and mTORC2 activation are diverse. mTORC1 largely functions to promote pathways involved with anabolic processes. Through the phosphorylation of downstream S6K1 and 4E-BP1, mTORC1 promotes protein synthesis through enhanced ribosome biogenesis and the release of transcriptional inhibition, respectively [100]. mTORC2, too, appears to positively effect protein synthesis through direct association with ribosomal complexes [158]. Lipid and nucleotide synthesis also appear to be under mTOR control. The principal transcriptional regulators of lipogenesis, sterol regulatory element-binding proteins (SREBPs), are activated by both mTORC1 and mTORC2 [159,160,161]. mTORC1 phosphorylates the SREBP inhibitor, lipin 1, preventing the nuclear translocation of lipin 1 and the subsequent inactivation of SREBP-mediated transcription of nucleotide and lipid synthesis-related genes [161, 162]. mTORC2 has been described to transcriptionally activate SREBP genes through ATK activity, thus also promoting de novo nucleotide and lipid synthesis [160, 163].
2.4.8 Negative Regulation of mTOR Through AMPK Activity
The serine/threonine kinase, adenosine monophosphate-activated protein kinase (AMPK), is often cast as an mTOR antagonist. Each molecule directly inhibits the activity of the other [164,165,166,167], oscillating the preferential utilization of their respective metabolic pathways; AMKP promotes FAO, while mTOR supports a glycolytic profile. A sensor of energy stress, AMPK is activated, in part, in response to high AMP/ATP ratios (i.e., low cellular energy), responding by inhibiting anabolic processes and upregulating specific metabolic pathways in order to restore cellular ATP levels. One such avenue for AMPK-mediated ATP production is the promotion of FAO. FAO is promoted through the upregulation of the rate-limiting enzyme for the pathway known as carnitine palmitoyltransferase 1 (CPT-1; a key component of the CTS) [168]. AMPK can also play a role in the maintenance of CPT-1 activity. By inhibiting acetyl-CoA carboxylase, which is itself a suppresser of CPT-1, AMPK further drives the process of FAO [169]. In addition to promoting the transcription of genes that facilitate lipid metabolism, AMPK activation directly inhibits glycolysis and protein synthesis through the inhibition of mTOR activity. Despite the fact that both mTOR and AMPK are induced by TCR stimulation, the latter kinase is a potent negative regulator of the former [166]. AMPK targets the mTORC1 protein, Raptor, disrupting the protein complex and diminishing mTOR kinase activity [140, 170]. Additionally, AMPK activates the TSC by phosphorylating TSC2, further driving the inhibition of mTOR [171]. Reflecting the inverse relationships between AMPK and mTOR activity, T cells lacking AMPK display enhanced mTORC1 signaling and a correspondingly elevated effector cytokine production [172].
AMPK has also been thought of as an enforcer of quiescence. It is easy to fathom that under conditions of metabolic stress (such as glucose scarcity), cell survival may depend on the resetting of the metabolic lifestyle to adapt to austere conditions. In line with this notion, AMPK was recently suggested to be dispensable for the activation and expansion of effector CD8+ T cells but important for the survival of CD8+ T cells during the so-called contraction phase of the immune response and the mounting of a recall response upon secondary challenge [173]. CD8+ T cell-specific deletion of AMPK in vivo resulted in a dramatic loss of CD8+ Tmem populations following exposure to parasitic infection [173]. Just as effector cells are dependent on a glycolytic metabolism, Tregs and Tmem rely on FAO. Inhibition of FAO by disruption of AMPK signaling curtails the generation of these key immune populations. Both thymically derived natural Tregs and Tregs induced in the periphery display a high degree of AMPK activity. Furthermore, in vivo administration of an AMPK activator (metformin) elevates Treg numbers in a mouse model of asthma while also diminishing cell-surface expression of GLUT1 [23]. Clearly, the reciprocal regulation of mTOR and AMPK represents a principal control mechanism to balance proinflammatory and tolerogenic cues.
2.4.9 Therapeutic Opportunities in Targeting the mTOR Signaling Pathway
Since mTOR is a central player in promoting glycolysis, the metabolic pathway of choice for potently inflammatory effector T cells, inhibiting the kinase has been explored as a means to prevent or down-modulate unwanted immune responses. Indeed, the mTOR agonist, rapamycin, has been administered in the control of the autoimmune disease, systemic lupus erythematosus (SLE) [174, 175]. Rapamycin and sirolimus have been shown to function as immunosuppressants, promoting tolerance in organ and tissue transplants [176].
While mTOR inhibition has the potential to undermine glycolysis and the proinflammatory effector T cells that are heavily dependent on this form of metabolism, somewhat paradoxically, this same strategy can bolster desirable immune responses – particularly through the enhancement of memory T cells. It is important to note that multiple studies have suggested that the memory-like subsets of CD8+ T cells are superior mediators of antitumor immunity [48, 49, 177,178,179]. In light of this, it stands to reason that modulating the metabolic profile of CD8+ T cells may be an effective strategy to improve their cancer-fighting potential. Sirolimus (rapamycin) administration can enhance the memory CD8+ T cell responses of following vaccination of nonhuman primates [180]. Furthermore, suppressing mTOR activity and glycolysis with transient, high-dose rapamycin treatments can enhance the efficacy of antitumor vaccines as well [181]. An aptamer-conjugated RNAi approach intended to specifically knock down mTORC1 activity has also been shown to suppress tumor progression in vivo [182]. Everolimus, another mTOR inhibitor, is an FDA-approved therapeutic option for the treatment of specific brain tumors (subependymal giant-cell astrocytomas) associated with inactivating TSC mutations. Studies are ongoing in proving the efficacy of everolimus in other TSC-associated diseases [126].
The role played by AMPK in modulating T cell immunity appears to lend itself to pharmaceutical intervention as well. The therapeutic benefit of metformin (N,N-dimethylbiguanide) and 5-aminoimidazole-4-carboxamide-ribonucleoside (AICAR) has been explored in their capacity to correct for insulin resistance by the AMPK-mediated upregulation of the glucose transporter, GLUT4. Both compounds are capable of activating AMPK, effectively facilitating the increased uptake of glucose in the absence of insulin signaling [183, 184]. Metformin and AICAR have both shown efficacy in the treatment of the insulin resistance associated with type-2 diabetes while also efficiently suppressing effector T cell activation and the production of IL-2 [51, 185]. Chemical activation of AMPK reduces the effector functions of murine CTLs [164]. In line with these findings, therapeutic intervention of murine EAE with metformin or AICAR dampens inflammatory cytokine production by self-reactive CD4+ T cells and lessens the severity of disease progression [186]. AICAR treatment also ameliorates disease in murine colitis models as evidenced by stunted body weight loss and subdued colon pathology. Moreover, this treatment inhibited macrophage activation and reduced Th1/Th17 cell frequency and cytokine production [187, 188].
While AMPK-activating agents can negatively impact effector T cell biology, they can have opposite effects on memory T cell subsets. Illustrating this, metformin treatment has been found to markedly enhance the memory CD8+ T cell response to Listeria monocytogenes infection in mice [17]. Additionally, others have found that metformin can enhance CD8+ T cell function in tumor sites [189], an effect that likely reflects the more potent antitumor activity of the memory-like CD8+ T cells expected to be bolstered by AMPK activation.
2.4.10 Hypoxia-Inducible Factor 1 (HIF-1)
Great cellular energy demand requires the strict regulation of the byproducts of T cell metabolism. The most efficient method for generating ATP is the catabolism of glucose- or fatty acid-derived pyruvate through oxidative phosphorylation. Hydrogen ions are forced against their concentration and electrochemical gradients into the acidic space between mitochondrial membranes, thereby providing a fuel source to drive the rotary mechanism of ATP synthase [190]. The free electrons generated by this process couple with hydrogen byproducts and free diatomic molecules of oxygen to form water within the mitochondria. This reaction occurs through the action of the terminal enzyme of the electron transport chain, cytochrome c oxidase (COX). COX has been revealed to play a major regulatory role in orchestrating this oxygen-dependent pyruvate metabolism, consuming the majority of the oxygen inspired by the lungs [191]. COX ceases to function under extreme oxygen deprivation (<1 mmHg) [192], a condition termed hypoxia. Specifically, hypoxia refers to tissue oxygen tensions that fall below ~10 mmHg [193]. Low oxygen tensions have a significant impact on immune cell function and proliferation, even when oxygen concentrations have not dropped sufficiently to alter oxidative phosphorylation within the mitochondria. Notably, many tissues that T cells are likely to inhibit – healthy or diseased – can be oxygen scarce. Competition for nutrients (e.g., glucose, fatty acids, amino acids) and oxygen drives characteristic alterations to T cell’s metabolism. Consequentially, T cells must be highly adaptive to hypoxic environments and be able to readily alter their metabolic profile in order to function in sites of varying oxygen tensions.
2.4.11 Control of HIF-1 Activity Through the Regulation of the HIF-1α Subunit
The principal sensor of oxygen deprivation is hypoxia-inducible factor alpha (HIF-1α). HIF-1 complexes form as heterodimers of HIF-1α and HIF-1β (also called the aryl hydrocarbon nuclear translator). These factors are expressed, at least at the mRNA level, in every organ of the body [194]. HIF-1β is constitutively expressed at the protein level and functions in a pathway separate from HIF-1α that is associated with the transcriptional activation of enzymes responsible for the catabolism of xenobiotic toxins, such as the environmental pollutant, dioxin [195]. HIF-1α expression, on the other hand, is tightly regulated and is typically stabilized only under hypoxic conditions. The nuclear translocation of HIF-1α and subsequent binding to HIF-1β only occurs following the stabilization of the α-subunit [196]. Within the nucleus, HIF-1α associates with HIB-1β and functions as a transcription factor, binding to specific promotor sequences of hypoxia response elements (HREs) [197, 198] allowing for a robust cellular response to oxygen deprivation. Both subunits possess helix-loop-helix motifs and a Per-ARNT-Sim (PAS) domain that both facilitate DNA binding and α-/β-subunit dimerization [199,200,201,202]. Another α-subunit, HIF-3α, was initially reported to propagate an inhibitory function by preventing HIF-1α binding to HIF-1β though the sequestering of the β-subunit, yet recent data has suggested an additional transcriptional regulatory role for the protein [203,204,205].
The expression of the α-subunit, HIF-1α, is largely reliant on the local oxygen tension. As you breathe, atmospheric oxygen concentrations (159 mmHg) allow for the rapid diffusion of oxygen into the circulatory system which then pushes arterial blood throughout the body, dispensing oxygen where oxygen tensions are low. In tissues, oxygen concentrations are considerably lower (~15–38 mmHg) than in circulating arterial (75–100 mmHg) and venous blood (≥40 mmHg) [206, 207]. Moreover, within the architecture of tissues, oxygen tension can vary dramatically depending on the proximity of a cell to local capillary beds [208, 209]. Although oxygen tensions are lower within tissues than in circulation, the oxygen and nutrient concentrations therein are sufficiently replete due to elaborate networks of capillary beds. Oxygen tensions are particularly low in lymphoid tissues (~8–35 mmHg) where immune cells are derived and mature. The same is generally true of sites of ongoing inflammation or tumor growth, implicating the importance of HIF-1α in immune cell development and function. During an inflammatory response, competition for available oxygen and nutrients increases as immune cells infiltrate and expand in peripheral tissue sites, resulting in nutrient-/oxygen-destitute microenvironments. Within a tumor, this and the prodigious consumption of glucose and oxygen by tumor cells coupled with the inability of angiogenesis to keep pace with the intratumoral demand result in regions of extreme hypoxia [210, 211]. It is the stabilization of HIF-1α expression, largely consequential of hypoxic conditions (> 10 mmHg) alone, that drives much of the cellular adaptations to oxygen deprivation.
HIF-1α expression is largely controlled posttranslationally by prolyl hydroxylases (PHD), which catalyze the formation of L-hydroxyproline by incorporating oxygen to two specific proline residues (P402 and P564) on HIF-1α [212]. Hydroxylation of proline residues increases the affinity of these sites on HIF-1α for the active site of the von Hippel-Lindau/Elongin-C E3 ligase (VHL) complex by nearly 1,000-fold. The bound VHL transfers ubiquitin molecules to L-hydroxyproline residues, marking HIF-1α for proteome-dependent proteolysis [207, 213]. Thus, the repression of HIF-1α hinges on the presence of free oxygen within the cell, and as such, HIF-1α becomes stabilized under hypoxic conditions [214]. Under normoxic conditions (i.e., non-hypoxic; >10 mmHg), HIF-1α is rapidly degraded through the aforementioned process.
However, this oxygen-dependent degradation has been shown to be superseded in CD4+ and CD8+ T cells where high mTORC1 expression is associated with elevated HIF-1α levels even in normoxic conditions [18, 80, 215, 216]. Indeed, TCR ligation and costimulation stabilizes the expression of HIF-1α, possibly through mTORC1-facilitated enhancement HIF-1α protein synthesis [216]. It should be noted that decreased intracellular oxygen tensions resulting from cellular activation may also contribute to the aforementioned HIF-1α stabilization [207]. Proinflammatory cytokines, too, appear to be capable of augmenting HIF-1α expression in T cells. Specifically, interleukin 6 (IL-6) induces HIF-1α stabilization in vitro through the activation of signal transducer and activator of transcription 3 (STAT3)-driven transcriptional regulation [81]. Additionally, numerous studies have elucidated the role of the receptor of activated protein kinase C (RACK1) in the proteasomal degradation of HIF-1α in an oxygen-/PHD-/VHL-independent manner. RACK1 competes with heat-shock protein 90 (HSP90) for binding to the PAS domain of HIF-1α. Upon RACK1/Elongin-C E3 ubiquitin ligase association, RACK1 facilitates the ubiquitination and subsequent proteome-dependent proteolysis of HIF-1α [217,218,219]. These data suggests various levels of HIF-1α control outside of oxygen tension alone.
2.4.12 Downstream Effects of HIF-1α Stabilization in T Cells
The cellular effects of HIF-1α stabilization are expansive, resulting in the expression of many genes implicated in the control of glucose and pyruvate metabolism in effector T cells. Following TCR ligation and costimulation, T cells undergo clonal expansion and transcriptome reprograming to express effector molecules including cytotoxins, cytokines, and cell-surface molecules. This robust increase in cellular proliferation and protein anabolism demands a matched increase in metabolite uptake to provide components for cellular machinery and free energy to catalyze chemical reactions. HIF-1α expression has been shown to enhance the cell-surface expression of the glucose transporter, Glut1, in CD4+ and CD8+ T cells, supporting the development of proinflammatory T cell lineages [42, 43]. Indeed, within CD4+ T cells, HIF-1 transcriptionally directs the upregulation of the enzymatic machinery required for glycolysis, prioritizing this metabolic pathway which is favored by Teff cells [18]. Interestingly, while c-Myc and mTOR are centrally important for engaging this metabolic reprogramming of recently activated cells, HIF-1 expression appears dispensable at this early juncture. Although HIF-1 controls the transcription of numerous glycolytic genes, HIF-1-deficiency in T cells does not inhibit activation-induced Teff proliferation [80, 81]. It is believed, however, that HIF-1 instead plays an important role in sustaining Teff populations and their glycolytic metabolism following activation.
In CD4+ T cells, HIF-1α reciprocally regulates the key transcriptional regulators of the differentiation programs responsible for Th17 and Treg development, RAR-related orphan receptor gamma (RORγt) and Foxp3, respectively [18, 81]. While Th17 cells are notoriously proinflammatory and functionally share very little with the characteristically immunosuppressive Treg lineage, they nevertheless can be derived in vitro and in peripheral tissues by differentiation pathways that partially overlap, namely, through transforming growth factor beta (TGF-β) receptor signaling. It is the sensing of additional proinflammatory cytokines, like IL-6, that simultaneously promotes the establishment of the Th17 lineage and inhibits the Treg phenotype. Enhanced HIF-1α expression resulting from TCR ligation/IL-6-mediated STAT3 activation can support Th17 differentiation irrespective of local oxygen tensions. Stabilized HIF-1α subunits directly promote the expression of RORγt in differentiating CD4+ T cells, supporting the adoption of a Th17 fate. HIF-1 further cooperates with RORγt in a transcription factor complex to promote transcription/translation of several Th17-associated genes [81]. Another facet of HIF-1’s influence over CD4+ T cell fate decisions appears to involve the downregulation of Foxp3, which can prevent commitment to an induced Treg fate. Indeed, HIF-1 promotes the proteasome-dependent degradation of Foxp3 protein during T cell differentiation. Reflecting these multiple roles of HIF-1, CD4+ T cells from conditional HIF-1α knockout mice are defective in Th17 commitment and are more prone to upregulate Foxp3 in vitro. Moreover, T cell-specific knockout of HIF-1 reduces disease burden in EAE relative to wild type controls. Such protection was seen alongside impaired Th17 immunity and the accumulation of Foxp3+ Tregs when T cells lack HIF-1 expression [18, 81].
The role of HIF-1 in established Tregs appears to be complex. Indeed, HIF-1 deficiency during CD4+ T cell differentiation in vitro results in an accumulation of Foxp3+ T cells [18, 81], suggesting that HIF-1 opposes Treg differentiation. However, other studies suggest that HIF-1α contributes positively to Tregs by promoting transcription at the Foxp3 and Ctla4 genes [220]. Moreover, the suppressive capacity of established Tregs in vivo appears to benefit from HIF-1 expression under some conditions as Tregs from HIF-1-deficient mice were found to be less effective suppressors of colitis than wild-type controls [220]. In contrast, other studies have shown that heightened levels of HIF-1 activity can be detrimental to the normal suppressive functions of Tregs. Specific deletion of key components of the HIF-1α degradation machinery (e.g., Deltex, VHL) in Tregs induces robust expression of HIF-1 that is accompanied by unstable expression of Foxp3 and compromised suppressive function [221, 222]. In one study, these ineffective Tregs were found to have uncharacteristic, Th1 effector-like attributes that resulted from HIF-1-mediated upregulation of Teff cell genes (e.g., Ifng) and metabolic genes responsible for promoting glycolysis [23, 222]. These findings suggest that a precise balance of HIF-1 expression may be required for optimal Treg function.
HIF-1α also influences the development of CD8+ cytotoxic T lymphocytes (CTLs). The cellular activities and functions of activated CD8+ T cells, like those of their CD4+ counterparts, are fueled primarily by a glycolysis-dominated metabolism [18, 81, 223]. Newly activated CD8+ T cells upregulate HIF-1 through the action of mTORC1. This activation begins with TCR signaling/CD28 costimulation, which induces c-Myc expression and subsequent activation of c-Myc-controlled genes that are crucial for efficient cellular activation and expansion. A second wave of augmented gene expression, coordinated by HIF-1 and other transcriptional regulators (e.g., AP4, IRF4), continues to enforce a glycolytic metabolic profile through the upregulation of enzymes involved in glycolysis and glutaminolysis [224].
Additionally, HIF-1α stabilization appears to promote the establishment of effector functions within CTLs. HIF-1 drives the expression of key effector molecules, such as perforin and granzymes, which partially account for the cytotoxic capacity of these cells [80]. Genetic deletion of VHL (the complex responsible for oxygen-dependent HIF-1α degradation) in vivo results in elevated expression of granzyme B and effector cytokines such as IFN-γ and TNF-α. These enhancements, which result from elevated HIF-1 activity, translate into improved CTL-mediated immunity in models of melanoma and viral infection [223]. Boosting HIF-1 levels by exposing cells to hypoxic culture conditions can also enhance the expression of Glut1 and effector molecules [80], further supporting the role of HIF-1 activity in promoting glycolysis and CTL function. Correspondingly, HIF-1 deficiency impairs the acquisition of these effector molecules.
In keeping with the theme of contrasting metabolic lifestyle and requirements for Teff and Tmem lineages, HIF-1 appears to negatively impact memory CD8+ T cells. Chemical inhibition of glycolysis (and HIF-1 expression) promotes a memory CD8+ phenotype instead of a short-lived, effector fate. Indeed, these conditions result in a more memory-like phenotype as evidenced by low levels of KLRG1 and an abundance of surface CD62L. CD62L, also called L-selectin, is expressed most notably in naïve and memory T cells, facilitating the trafficking of these cells to secondary lymphoid organs. CD62L appears a key checkpoint of T cell function as its expression has been shown to control lytic activity of tumor reactive T cells and the memory response to previously encountered antigen [225, 226]. Conversely, forced utilization of glycolysis in CD8+ T cells restricts the generation of memory cells. Interestingly, memory CD8+ T cells that accumulate during inhibition of glycolysis and HIF-1 downregulation appear to be more potent mediators of antitumor immunity than their short-lived Teff counterparts [20].
2.4.13 Therapeutic Opportunities in Targeting the HIF-1 Signaling Pathway
Interest in generating potent HIF inhibitors has been understandably whetted by the possibility of simultaneously disrupting its multiple tumor-abetting functions (e.g., upregulation of glycolytic enzymes, promotion of angiogenesis, facilitation of metastasis). Indeed, HIF-1 is thought to facilitate tumor growth in its characteristically oxygen-deprived microenvironments, and, in keeping, HIF-1 expression trends upward as certain brain tumors increase in grade [227]. Currently, several HIF-1α inhibitors are undergoing clinical or preclinical trials. The small-molecule inhibitor, topotecan, is a well-established anti-neoplastic agent that inhibits HIF-1α expression through a topoisomerase I-mediated mechanism [228, 229]. A daily low-dose regimen of topotecan was revealed to destabilize HIF-1α expression and thus diminish tumor growth and angiogenesis in murine tumor models [228, 230]. Another molecule being explored as a molecular HIF antagonist is PX-478, which showed early promise in treating human tumor xenografts via diminishing HIF-1α and Glut1 expression [231,232,233,234].
In keeping with the notion that HIF-1 stabilization promotes cellular survival, activation of HIF-1α within CD8+ T cells has been shown to prevent immune cell exhaustion, a consequence of chronic viral infection or persistent tumor growth. Loss of VHL derails HIF regulation and thereby enhances effector T cell potential and longevity [223]. Within CD4+ T cells, too, HIF-1 has revealed itself to be targetable. Digoxin, a cardiac glycoside, has been shown to inhibit HIF-1α expression and disrupt HIF-mediated transcriptional activation [235]. Administration of digoxin or its derivatives to naïve CD4+ T cell cultures effectively hinders the skewing of the proinflammatory Th17 subset. Indeed, digoxin treatment in in vivo models of EAE markedly suppressed disease progression [236]. Although digoxin has been shown to directly inhibit the Th17-promoting transcription factor RORγt, digoxin-mediated inhibition of HIF-1α mechanisms, namely, the adoption of a glycolytic profile, is likely another contributing factor to the ineffectual generation of Th17 cells [18, 81].
2.4.14 Peroxisome Proliferator-Activated Receptor (PPAR)
Peroxisome proliferator-activated receptors (PPARs) represent a group of nuclear receptor proteins that are activated by fatty acids and initiate gene transcription to coordinate both lipid and carbohydrate metabolism with cellular proliferation and differentiation. Activated PPARs associate with retinoid x receptors (RXRs) to transcriptionally regulate genes with PPAR response element (PPRE) promotor regions [237, 238]. Three PPAR isoforms exist – α, δ (occasionally defined as β or β/δ), and γ – which possess specific tissue expression patterns and differ in their physiological consequences following ligand binding. Ligation of PPARs by fatty acids can promote the dissociation of corepressors and association of co-activators, facilitating the direct binding of PPAR to DNA and the activation of gene transcription. Furthermore, co-activator complexes have been shown to direct chromatin remodeling through histone acetyltransferase activity, further promoting gene transcription [239,240,241,242,243,244].
While little is known about the direct mechanistic role of PPARδ (also called PPARβ or PPARβ/δ), the functions of the α and γ isoforms have been described at length in both immune and nonimmune cells. PPARα expression is constrained to areas of elevated metabolic activity (e.g., the liver, brown adipose, cardiac and skeletal muscle fibers, intestinal mucosa) and its ligand-dependent activation is associated with augmented fatty acid metabolism. PPARα activation induces the transcriptional upregulation of genes that facilitate intracellular fatty acid transport into sites of β-oxidation as well as the enzymes that are associated with this catabolic pathway [245, 246]. PPARγ, on the other hand, principally promotes anabolic processes, participating in adipogenesis (γ expression remains elevated in both brown and white adipose), lipid biosynthesis, and insulin sensitivity [247]. PPARδ appears to augment the accumulation of lipids within a cell through the repression of metabolic mechanisms and the increased expression of influx-specific transport molecules [248]. Importantly, while other isoforms require corepressor release to facilitate DNA binding, PPARδ has been shown to associate with PPRE promotor regions irrespective of corepressor binding [249]. This unique ability of PPARδ could represent a specific regulatory function, regulating the transcriptional activity of lipid metabolism-associated PPARα and γ.
PPARs are expressed in a variety of immune cell subtypes – e.g., T and B cells, macrophages, and dendritic cells – and have been reported to primarily support anti-inflammatory and tolerogenic activity [239, 246, 247, 250,251,252,253]. Indeed, PPARs have been revealed to transcriptionally downregulate several genes associated with immune cell activation and T cell effector function in inflammation. Notably, PPARs control the activity of T cell activation- and proliferation-related transcription factors that include activator protein-1 (AP-1), nuclear factor of activated T cells (NFAT), nuclear factor-κB (NF-κB), and signal transducers and activators of transcription (STATs) [251, 254,255,256], as well as expression of the stimulatory cytokine, IL-2 [257].
2.4.15 Downstream Effects of PPAR Ligation in Effector T Cells
A consequence of the PPAR-mediated promotion of lipid metabolism (at the expense of glycolytic metabolism) is the downregulation of effector responses, supporting the notion that PPARs function indirectly as immunosuppressors. Indeed, Th1-/Th17-mediated EAE disease is exacerbated in mice with diminished PPARγ (PPARγ+/−) as compared to their wild-type littermates. This was revealed by enhanced neuron demyelination and ex vivo IFNγ-production from CNS-infiltrating Th1 cells in PPARγ+/− mice. Correspondingly, PPARγ antagonists also enhanced the clinical scores of EAE symptoms as well as the induction of cellular activation and IFNγ-production, suggesting an augmented proinflammatory response [258, 259]. PPARγ agonists, on the other hand, ameliorated EAE severity as evidenced by a reduction of leukocyte infiltration into the CNS, a diminishing of proinflammatory cytokine release following in vitro reactivation, and an overall reduced disease duration [260, 261]. Considering the significant Th17 component in EAE disease progression, PPARγ activity was hypothesized to interfere with Th17 differentiation. Interestingly, PPARγ agonists specifically interfered with the induction of RORγt expression in vitro, without directly altering the expression of other pro- and anti-inflammatory transcription factors – e.g., T-bet (Th1), GATA3 (Th2), or Foxp3 (Treg) [262]. While these data suggest that PPARγ does not directly interfere with Th1 skewing in vitro, significant studies have illustrated the PPAR-mediated control of IL-12 production and signaling in vivo, which in turn would effectively diminish Th1 responses in a physiological setting [263, 264]. Indeed, the previous reports of augmented Th1 effector responses upon diminished PPARγ expression [258, 265] have been well supported [263, 266, 267].
Interestingly, while the transcriptional regulation of RORc (the gene encoding RORγt) falls under the control of PPARγ, expression of T-bet (the Th1-associated transcription factor) appears to be mediated by PPARδ. Agonists of the δ isoform result in similar amelioration of CNS inflammation and demyelination in EAE, although not through the reduction of antigen-specific Teff cellular proliferation. Instead, PPARδ agonists hindered the production of effector cytokines, IL-12 and IL-23, resulting in diminished Th1/Th17 differentiation and thus control of T helper-specific cytokine production – e.g., IFNγ, IL-17 – and the expression of the Th1-favoring T-bet [256, 266]. Notably, PPARδ agonists did not altogether downregulate cytokine expression, but propagated the production of anti-inflammatory cytokines, IL-4 and IL-10, further suppressing the effector functions of some T helper subsets. PPARδ knockout results in the converse effect, enhancing Th1/Th17 responses, prolonging the induction phase of EAE, and worsening the disease burden [266].
While PPARδ and γ agonists directly regulate the effector functions of Th17 cells, the loss of PPARα does not appear to alter Th17 effector cytokine (IL-17) production, suggesting that there are differing methods of proinflammatory regulation between PPAR isoforms [256, 262, 266]. Ligand-dependent activation of PPARα effectively protects against autoimmunity, but apparently through the control of a different immune axis. Indeed, PPARα activation prevents the development of severe EAE in mice by reducing the generation of potentially pathologic Th1 cells in favor of an augmented Th2 cell population. This is accomplished through the direct regulation of the Th1- and Th2-specific transcription factors, T-bet and GATA3, respectively, resulting in diminished IFNγ (Th1) and heightened IL-4 and IL5 (Th2) [268, 269]. Also, similar to PPARγ, activation of PPARα also suppresses the production of proinflammatory cytokines by innate cells within the CNS – e.g., microglia and astrocytes [270, 271].
Recently, a role for PPARγ was revealed in a unique subset of Treg cells. Visceral adipose tissues (VATs) house a population of Treg cells that suppress inflammation specifically within this niche. Interestingly, their presence and function are linked to a reversal of insulin resistance. VAT Tregs were found to uniquely express PPARγ, and they display a gene expression profile significantly distinct from their lymphoid tissue-dwelling counterparts. Mice lacking PPARγ specifically within the Treg compartment (Foxp3-cre/Pparg flox/flox) have lower VAT Treg frequencies and are susceptible to organismal metabolic dysfunction [272]. It may be possible that PPAR activation contributes to the heightened lipid-dominated metabolic profile that imparts VAT Tregs with the survival advantage optimal for their unique microenvironment.
2.4.16 Therapeutic Opportunities in Targeting the PPAR Signaling Pathway
Although the complete mechanistic picture of PPAR-mediated immunosuppression remains to be resolved, it is clear that manipulating the activity of any PPAR isoforms alters a cell’s metabolic ability, resulting in support of either pro- or anti-inflammatory conditions. PPAR activation drives enhanced lipid anabolism and curtails the glycolytic profile required for complete Teff function. Inhibition of the PPAR pathway results in the antithesis, namely, a prolonged and more intense inflammatory response. While the involvement of PPAR in lipid metabolism-favoring Treg populations was not directly addressed in many of the aforementioned studies, there is reason to believe that the immune-dampening effects of PPAR activity may also involve these tolerance-promoting cells. For instance, the suppression-inducing effects of the PPARγ agonists 15d-PGJ2 and Ciglitazone can be heightened by the co-administration of retinoic acid, a known promoter of Treg-mediated function [273]. Ciglitazone was revealed to promote the conversion of Teff cells to induced Tregs in vitro through the enhanced secretion of retinoic acid from DCs and a corresponding decrease in pro-effector IL-12 production. Moreover, high-dose Ciglitazone was sufficient to induce this conversion in a DC-independent manner [273]. This synthetic ligand also was found to offer a protective effect in a model of graft versus host disease by enhancing Tregs, further supporting the notion that PPAR activation induces tolerogenic conditions [274]. Moreover, PPARα or γ ligation in human CD4+ T cells treated with TGF-β can induce the stabilized expression of Foxp3 and a Treg phenotype [275]. It is also noteworthy that PPAR agonists can directly activate AMPK [169], a metabolic regulator important for inhibiting the adoption of the glycolytic metabolism that can negatively affect the generation of Tregs.
PPAR agonists are currently being clinically vetted for their ability to suppress unwanted inflammation. Experimental ligation of PPAR has been shown to successfully ameliorate immunopathology in several animal models of autoimmunity [261, 276,277,278,279], supporting the notion that PPAR activation can inhibit proinflammatory immune responses. The PPAR ligands, rosiglitazone and pioglitazone, were found to diminish Th2- and Th17-specific cytokine production and afford protection from asthma. Considering that exogenous IL-17 negates the protective effect of these drugs [280], rosiglitazone and pioglitazone may have the potential to specifically combat Th17-driven pathologies. In one multiple sclerosis trial, pioglitazone (in combination with IFNβ-1α) yielded promising MRI results (i.e., less gray matter atrophy and trends toward decreased lesions) in a 1-year follow-up [281]. Also, encouragingly, in a randomized, placebo-controlled study, rosiglitazone was found to provide a promising strategy to ameliorate ulcerative colitis in patients suffering from the disease [282].
2.5 Concluding Statements
The mechanisms that control the balance of proinflammatory and tolerogenic T cell lineages are closely tied to the activity and the maintenance of specific metabolic pathways. Activating stimuli necessitate increased absorption and utilization of metabolites to facilitate costly cellular proliferation, effector function, and control of apoptosis in many cell types. Yet the particular nutrients absorbed and metabolic enzymes expressed to meet these demands can vary between subsets of T cells. Teff cells, responding to TCR and costimulatory cascades, will principally metabolize glucose through aerobic glycolysis, while shunting off glycolytic intermediates for anabolic processes and maintaining some meager levels of OXPHOS. Tmem and Treg prioritize lipid metabolism, procuring fatty acids for the majority of their energetic needs and facilitating their longevity even in sites of inflammation and tumor growth.
The key regulators of the metabolic programming described here control the expression of genes related to nutrient absorption and metabolism. As such, the manipulating the function of these proteins has proven to be an effective level of control over immune responses in disease settings. In brief, c-Myc expression is dramatically upregulated shortly after cellular activation, promoting a glucose absorption and glycolytic metabolic profile and inducing the expression of transcription factors that maintain this glycolysis-dominant profile (e.g., mTOR, HIF-1α, AP4, IRF4, Bim). Both mTOR and AMPK are upregulated upon TCR and growth hormone receptor ligation and the interplay of their kinase activity promotes either anabolic machinery, cellular growth, and proliferation (mTOR) or the regeneration of energy storages through enhanced catabolism (AMPK). Lipid-mediated ligation of PPARs activate the transcription of genes that directly upregulate FAO, facilitating the distinct functions and longevity associated with Tmem and Treg.
Intense study of the mechanisms that control T cell immunity have revealed pathways that can be manipulated for effective control of immune disease and disorders. Although great bounds have been made in delineating these systems, questions remain concerning how they might be more effectively manipulated to provide therapeutic benefit. As laboratory technology continues to advance, a more intimate understanding of the interplay between c-Myc, mTOR, AMPK, HIF-1α, PPARs, etc. in relation to T cell function will no doubt soon come to light.
References
Wang R et al (2011) The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35(6):871–882
Zhu J, Yamane H, Paul WE (2010) Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol 28:445–489
Littman DR, Rudensky AY (2010) Th17 and regulatory T cells in mediating and restraining inflammation. Cell 140(6):845–858
Rudensky AY (2011) Regulatory T cells and Foxp3. Immunol Rev 241(1):260–268
Vignali DA, Collison LW, Workman CJ (2008) How regulatory T cells work. Nat Rev Immunol 8(7):523–532
Nishikawa H, Sakaguchi S (2010) Regulatory T cells in tumor immunity. Int J Cancer 127(4):759–767
Josefowicz SZ, Lu LF, Rudensky AY (2012) Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol 30:531–564
Schell JC, Rutter J (2013) The long and winding road to the mitochondrial pyruvate carrier. Cancer Metab 1(1):6
Warburg O (1956) On the origin of cancer cells. Science 123(3191):309–314
Fox CJ, Hammerman PS, Thompson CB (2005) Fuel feeds function: energy metabolism and the T-cell response. Nat Rev Immunol 5(11):844–852
Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324(5930):1029–1033
Pearce EL, Pearce EJ (2013) Metabolic pathways in immune cell activation and quiescence. Immunity 38(4):633–643
Pearce EL et al (2013) Fueling immunity: insights into metabolism and lymphocyte function. Science 342(6155):1242454
Segal AW (2008) The function of the NADPH oxidase of phagocytes and its relationship to other NOXs in plants, invertebrates, and mammals. Int J Biochem Cell Biol 40(4):604–618
Wahl DR et al (2012) Distinct metabolic programs in activated T cells: opportunities for selective immunomodulation. Immunol Rev 249(1):104–115
De Boer RJ, Homann D, Perelson AS (2003) Different dynamics of CD4+ and CD8+ T cell responses during and after acute lymphocytic choriomeningitis virus infection. J Immunol 171(8):3928–3935
Pearce EL et al (2009) Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 460(7251):103–107
Shi LZ et al (2011) HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med 208(7):1367–1376
Dardalhon V et al (2008) Role of Th1 and Th17 cells in organ-specific autoimmunity. J Autoimmun 31(3):252–256
Sukumar M et al (2013) Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest 123(10):4479–4488
Schwenk RW et al (2010) Fatty acid transport across the cell membrane: regulation by fatty acid transporters. Prostaglandins Leukot Essent Fat Acids 82(4–6):149–154
Lochner M, Berod L, Sparwasser T (2015) Fatty acid metabolism in the regulation of T cell function. Trends Immunol 36(2):81–91
Michalek RD et al (2011) Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol 186(6):3299–3303
Delgoffe GM et al (2011) The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol 12(4):295–303
Kaech SM, Cui W (2012) Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol 12(11):749–761
van der Windt GJ et al (2012) Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 36(1):68–78
van der Windt GJ et al (2013) CD8 memory T cells have a bioenergetic advantage that underlies their rapid recall ability. Proc Natl Acad Sci U S A 110(35):14336–14341
Fraser KA et al (2013) Preexisting high frequencies of memory CD8+ T cells favor rapid memory differentiation and preservation of proliferative potential upon boosting. Immunity 39(1):171–183
Nicholls DG (2009) Spare respiratory capacity, oxidative stress and excitotoxicity. Biochem Soc Trans 37(Pt 6):1385–1388
Newsholme EA, Crabtree B, Ardawi MS (1985) The role of high rates of glycolysis and glutamine utilization in rapidly dividing cells. Biosci Rep 5(5):393–400
van Stipdonk MJ et al (2003) Dynamic programming of CD8+ T lymphocyte responses. Nat Immunol 4(4):361–365
Rathmell JC et al (2000) In the absence of extrinsic signals, nutrient utilization by lymphocytes is insufficient to maintain either cell size or viability. Mol Cell 6(3):683–692
Newell MK et al (2006) Cellular metabolism as a basis for immune privilege. J Immune Based Ther Vaccines 4:1
Macintyre AN et al (2014) The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab 20(1):61–72
Mueckler M, Thorens B (2013) The SLC2 (GLUT) family of membrane transporters. Mol Asp Med 34(2–3):121–138
Scheepers A, Joost HG, Schurmann A (2004) The glucose transporter families SGLT and GLUT: molecular basis of normal and aberrant function. JPEN J Parenter Enteral Nutr 28(5):364–371
Wood IS, Trayhurn P (2003) Glucose transporters (GLUT and SGLT): expanded families of sugar transport proteins. Br J Nutr 89(1):3–9
Qu Q et al (2016) Fatty acid oxidation and carnitine palmitoyltransferase I: emerging therapeutic targets in cancer. Cell Death Dis 7:e2226
Chakrabarti R et al (1994) Changes in glucose transport and transporter isoforms during the activation of human peripheral blood lymphocytes by phytohemagglutinin. J Immunol 152(6):2660–2668
Frauwirth KA et al (2002) The CD28 signaling pathway regulates glucose metabolism. Immunity 16(6):769–777
Jacobs SR et al (2008) Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol 180(7):4476–4486
Cham CM et al (2008) Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. Eur J Immunol 38(9):2438–2450
Cham CM, Gajewski TF (2005) Glucose availability regulates IFN-gamma production and p70S6 kinase activation in CD8+ effector T cells. J Immunol 174(8):4670–4677
Nakaya M et al (2014) Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity 40(5):692–705
Sinclair LV et al (2013) Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol 14(5):500–508
Verrey F et al (2004) CATs and HATs: the SLC7 family of amino acid transporters. Pflugers Arch 447(5):532–542
Hayashi K et al (2013) LAT1 is a critical transporter of essential amino acids for immune reactions in activated human T cells. J Immunol 191(8):4080–4085
Rao RR et al (2010) The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and Eomesodermin. Immunity 32(1):67–78
Araki K et al (2009) mTOR regulates memory CD8 T-cell differentiation. Nature 460(7251):108–112
Usui T et al (2006) Brasilicardin a, a natural immunosuppressant, targets amino acid transport system L. Chem Biol 13(11):1153–1160
Zheng Y et al (2009) Anergic T cells are metabolically anergic. J Immunol 183(10):6095–6101
Yan Z, Banerjee R (2010) Redox remodeling as an immunoregulatory strategy. Biochemistry 49(6):1059–1066
Dahlgren C, Karlsson A (1999) Respiratory burst in human neutrophils. J Immunol Methods 232(1–2):3–14
Sies H (1997) Oxidative stress: oxidants and antioxidants. Exp Physiol 82(2):291–295
Thoren FB et al (2007) Cutting edge: Antioxidative properties of myeloid dendritic cells: protection of T cells and NK cells from oxygen radical-induced inactivation and apoptosis. J Immunol 179(1):21–25
Cemerski S, van Meerwijk JP, Romagnoli P (2003) Oxidative-stress-induced T lymphocyte hyporesponsiveness is caused by structural modification rather than proteasomal degradation of crucial TCR signaling molecules. Eur J Immunol 33(8):2178–2185
Mougiakakos D, Johansson CC, Kiessling R (2009) Naturally occurring regulatory T cells show reduced sensitivity toward oxidative stress-induced cell death. Blood 113(15):3542–3545
Yan Z, Garg SK, Banerjee R (2010) Regulatory T cells interfere with glutathione metabolism in dendritic cells and T cells. J Biol Chem 285(53):41525–41532
Munn DH, Mellor AL (2013) Indoleamine 2, 3 dioxygenase and metabolic control of immune responses. Trends Immunol 34(3):137–143
Prodinger J et al (2016) The tryptophan metabolite picolinic acid suppresses proliferation and metabolic activity of CD4+ T cells and inhibits c-Myc activation. J Leukoc Biol 99(4):583–594
Baban B et al (2009) IDO activates regulatory T cells and blocks their conversion into Th17-like T cells. J Immunol 183(4):2475–2483
Sharma MD et al (2007) Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J Clin Invest 117(9):2570–2582
Sharma MD et al (2013) An inherently bifunctional subset of Foxp3+ T helper cells is controlled by the transcription factor eos. Immunity 38(5):998–1012
Mellor AL, Munn DH (2004) IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol 4(10):762–774
Qian F et al (2009) Inhibition of tumor cell growth, invasion, and metastasis by EXEL-2880 (XL880, GSK1363089), a novel inhibitor of HGF and VEGF receptor tyrosine kinases. Cancer Res 69(20):8009–8016
Carlson TJ et al (2014) Halofuginone-induced amino acid starvation regulates Stat3-dependent Th17 effector function and reduces established autoimmune inflammation. J Immunol 192(5):2167–2176
Lee CM, Reddy EP (1999) The v-myc oncogene. Oncogene 18(19):2997–3003
Boxer LM, Dang CV (2001) Translocations involving c-myc and c-myc function. Oncogene 20(40):5595–5610
Erikson J et al (1983) Transcriptional activation of the translocated c-myc oncogene in burkitt lymphoma. Proc Natl Acad Sci U S A 80(3):820–824
Dang CV (1999) C-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol 19(1):1–11
Douglas NC et al (2001) Defining the specific physiological requirements for c-Myc in T cell development. Nat Immunol 2(4):307–315
Mateyak MK et al (1997) Phenotypes of c-Myc-deficient rat fibroblasts isolated by targeted homologous recombination. Cell Growth Differ 8(10):1039–1048
Chou C et al (2014) C-Myc-induced transcription factor AP4 is required for host protection mediated by CD8+ T cells. Nat Immunol 15(9):884–893
Best JA et al (2013) Transcriptional insights into the CD8(+) T cell response to infection and memory T cell formation. Nat Immunol 14(4):404–412
Nie Z et al (2012) C-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 151(1):68–79
Hann SR, Eisenman RN (1984) Proteins encoded by the human c-myc oncogene: differential expression in neoplastic cells. Mol Cell Biol 4(11):2486–2497
Hann SR et al (1983) Proteins encoded by v-myc and c-myc oncogenes: identification and localization in acute leukemia virus transformants and bursal lymphoma cell lines. Cell 34(3):789–798
Man K et al (2013) The transcription factor IRF4 is essential for TCR affinity-mediated metabolic programming and clonal expansion of T cells. Nat Immunol 14(11):1155–1165
Yao S et al (2013) Interferon regulatory factor 4 sustains CD8(+) T cell expansion and effector differentiation. Immunity 39(5):833–845
Finlay DK et al (2012) PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J Exp Med 209(13):2441–2453
Dang EV et al (2011) Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell 146(5):772–784
Nayar R et al (2014) Graded levels of IRF4 regulate CD8+ T cell differentiation and expansion, but not attrition, in response to acute virus infection. J Immunol 192(12):5881–5893
Brustle A et al (2007) The development of inflammatory T(H)-17 cells requires interferon-regulatory factor 4. Nat Immunol 8(9):958–966
Cretney E et al (2011) The transcription factors blimp-1 and IRF4 jointly control the differentiation and function of effector regulatory T cells. Nat Immunol 12(4):304–311
Lohoff M et al (2002) Dysregulated T helper cell differentiation in the absence of interferon regulatory factor 4. Proc Natl Acad Sci U S A 99(18):11808–11812
Mittrucker HW et al (1997) Requirement for the transcription factor LSIRF/IRF4 for mature B and T lymphocyte function. Science 275(5299):540–543
Zheng Y et al (2009) Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature 458(7236):351–356
Dang CV et al (2008) The interplay between MYC and HIF in cancer. Nat Rev Cancer 8(1):51–56
Miller DM et al (2012) C-Myc and cancer metabolism. Clin Cancer Res 18(20):5546–5553
Cairns RA, Harris IS, Mak TW (2011) Regulation of cancer cell metabolism. Nat Rev Cancer 11(2):85–95
Casey SC et al (2016) MYC regulates the antitumor immune response through CD47 and PD-L1. Science 352(6282):227–231
Yin X et al (2003) Low molecular weight inhibitors of Myc-max interaction and function. Oncogene 22(40):6151–6159
Bandukwala HS et al (2012) Selective inhibition of CD4+ T-cell cytokine production and autoimmunity by BET protein and c-Myc inhibitors. Proc Natl Acad Sci U S A 109(36):14532–14537
Cafferkey R et al (1993) Dominant missense mutations in a novel yeast protein related to mammalian phosphatidylinositol 3-kinase and VPS34 abrogate rapamycin cytotoxicity. Mol Cell Biol 13(10):6012–6023
Brown EJ et al (1994) A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 369(6483):756–758
Sabatini DM et al (1994) RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell 78(1):35–43
Sabers CJ et al (1995) Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J Biol Chem 270(2):815–822
Fingar DC, Blenis J (2004) Target of rapamycin (TOR): an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene 23(18):3151–3171
Wullschleger S, Loewith R, Hall MN (2006) TOR signaling in growth and metabolism. Cell 124(3):471–484
Ma XM, Blenis J (2009) Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol 10(5):307–318
Sarbassov DD et al (2004) Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol 14(14):1296–1302
Jacinto E et al (2004) Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol 6(11):1122–1128
Hosokawa N et al (2009) Nutrient-dependent mTORC1 association with the ULK1-Atg 13-FIP200 complex required for autophagy. Mol Biol Cell 20(7):1981–1991
Jung CH et al (2009) ULK-Atg 13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 20(7):1992–2003
Ganley IG et al (2009) ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem 284(18):12297–12305
Hresko RC, Mueckler M (2005) mTOR.RICTOR is the Ser 473 kinase for Akt/protein kinase B in 3T3-L1 adipocytes. J Biol Chem 280(49):40406–40416
Sarbassov DD et al (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307(5712):1098–1101
Hara K et al (2002) Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 110(2):177–189
Kim DH et al (2002) mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110(2):163–175
Peterson TR et al (2009) DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 137(5):873–886
Kaizuka T et al (2010) Tti 1 and Tel 2 are critical factors in mammalian target of rapamycin complex assembly. J Biol Chem 285(26):20109–20116
Nojima H et al (2003) The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p 70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J Biol Chem 278(18):15461–15464
Loewith R et al (2002) Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell 10(3):457–468
Foster KG et al (2010) Regulation of mTOR complex 1 (mTORC1) by raptor Ser 863 and multisite phosphorylation. J Biol Chem 285(1):80–94
Fingar DC et al (2004) mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Mol Cell Biol 24(1):200–216
Schalm SS et al (2003) TOS motif-mediated raptor binding regulates 4E-BP1 multisite phosphorylation and function. Curr Biol 13(10):797–806
Wang L et al (2007) PRAS40 regulates mTORC1 kinase activity by functioning as a direct inhibitor of substrate binding. J Biol Chem 282(27):20036–20044
Sancak Y et al (2008) The rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320(5882):1496–1501
Vander Haar E et al (2007) Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol 9(3):316–323
Inoki K, Zhu T, Guan KL (2003) TSC2 mediates cellular energy response to control cell growth and survival. Cell 115(5):577–590
Potter CJ, Pedraza LG, Xu T (2002) Akt regulates growth by directly phosphorylating Tsc 2. Nat Cell Biol 4(9):658–665
Ma L et al (2005) Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 121(2):179–193
Roux PP et al (2004) Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p 90 ribosomal S6 kinase. Proc Natl Acad Sci U S A 101(37):13489–13494
Long X et al (2005) Rheb binds and regulates the mTOR kinase. Curr Biol 15(8):702–713
Tee AR et al (2003) Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr Biol 13(15):1259–1268
Curatolo P, Moavero R (2012) mTOR inhibitors in tuberous sclerosis complex. Curr Neuropharmacol 10(4):404–415
Harrington LE et al (2005) Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol 6(11):1123–1132
Manning BD (2004) Balancing Akt with S6K: implications for both metabolic diseases and tumorigenesis. J Cell Biol 167(3):399–403
Powell JD, Delgoffe GM (2010) The mammalian target of rapamycin: linking T cell differentiation, function, and metabolism. Immunity 33(3):301–311
Beugnet A et al (2003) Regulation of targets of mTOR (mammalian target of rapamycin) signalling by intracellular amino acid availability. Biochem J 372(Pt 2):555–566
Schriever SC et al (2013) Cellular signaling of amino acids towards mTORC1 activation in impaired human leucine catabolism. J Nutr Biochem 24(5):824–831
Duran RV et al (2012) Glutaminolysis activates rag-mTORC1 signaling. Mol Cell 47(3):349–358
Nicklin P et al (2009) Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 136(3):521–534
Kim SG et al (2013) Metabolic stress controls mTORC1 lysosomal localization and dimerization by regulating the TTT-RUVBL1/2 complex. Mol Cell 49(1):172–185
Hara K et al (1998) Amino acid sufficiency and mTOR regulate p 70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J Biol Chem 273(23):14484–14494
Jewell JL, Guan KL (2013) Nutrient signaling to mTOR and cell growth. Trends Biochem Sci 38(5):233–242
Jewell JL et al (2015) Metabolism. Differential regulation of mTORC1 by leucine and glutamine. Science 347(6218):194–198
Zoncu R et al (2011) mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 334(6056):678–683
Sancak Y, Sabatini DM (2009) Rag proteins regulate amino-acid-induced mTORC1 signalling. Biochem Soc Trans 37(Pt 1):289–290
Gwinn DM et al (2008) AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 30(2):214–226
Wouters BG, Koritzinsky M (2008) Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer 8(11):851–864
DeYoung MP et al (2008) Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev 22(2):239–251
Katiyar S et al (2009) REDD1, an inhibitor of mTOR signalling, is regulated by the CUL4A-DDB1 ubiquitin ligase. EMBO Rep 10(8):866–872
Frias MA et al (2006) mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr Biol 16(18):1865–1870
Jacinto E et al (2006) SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 127(1):125–137
Guertin DA et al (2006) Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell 11(6):859–871
Sarbassov DD et al (2006) Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell 22(2):159–168
Guertin DA, Sabatini DM (2007) Defining the role of mTOR in cancer. Cancer Cell 12(1):9–22
Populo H, Lopes JM, Soares P (2012) The mTOR signalling pathway in human cancer. Int J Mol Sci 13(2):1886–1918
Lawlor MA, Alessi DR (2001) PKB/Akt: a key mediator of cell proliferation, survival and insulin responses? J Cell Sci 114(Pt 16):2903–2910
Zheng Y et al (2007) A role for mammalian target of rapamycin in regulating T cell activation versus anergy. J Immunol 178(4):2163–2170
Delgoffe GM et al (2009) The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity 30(6):832–844
Chi H (2012) Regulation and function of mTOR signalling in T cell fate decisions. Nat Rev Immunol 12(5):325–338
Pollizzi KN et al (2015) mTORC1 and mTORC2 selectively regulate CD8(+) T cell differentiation. J Clin Invest 125(5):2090–2108
Kaech SM, Ahmed R (2003) Immunology. CD8 T cells remember with a little help. Science 300(5617):263–265
Kaech SM et al (2003) Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol 4(12):1191–1198
Shrestha S et al (2014) Tsc 1 promotes the differentiation of memory CD8+ T cells via orchestrating the transcriptional and metabolic programs. Proc Natl Acad Sci U S A 111(41):14858–14863
Oh WJ et al (2010) mTORC2 can associate with ribosomes to promote cotranslational phosphorylation and stability of nascent Akt polypeptide. EMBO J 29(23):3939–3951
Porstmann T et al (2008) SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab 8(3):224–236
Hagiwara A et al (2012) Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c. Cell Metab 15(5):725–738
Duvel K et al (2010) Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell 39(2):171–183
Peterson TR et al (2011) mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 146(3):408–420
Yuan M et al (2012) Identification of Akt-independent regulation of hepatic lipogenesis by mammalian target of rapamycin (mTOR) complex 2. J Biol Chem 287(35):29579–29588
Blagih J et al (2015) The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity 42(1):41–54
Faubert B et al (2013) AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab 17(1):113–124
Tamas P et al (2006) Regulation of the energy sensor AMP-activated protein kinase by antigen receptor and Ca2+ in T lymphocytes. J Exp Med 203(7):1665–1670
MacIver NJ et al (2011) The liver kinase B1 is a central regulator of T cell development, activation, and metabolism. J Immunol 187(8):4187–4198
Deberardinis RJ, Lum JJ, Thompson CB (2006) Phosphatidylinositol 3-kinase-dependent modulation of carnitine palmitoyltransferase 1A expression regulates lipid metabolism during hematopoietic cell growth. J Biol Chem 281(49):37372–37380
Lee WH, Kim SG (2010) AMPK-dependent metabolic regulation by PPAR agonists. PPAR Res 2010
Suzuki T et al (2013) Inhibition of AMPK catabolic action by GSK3. Mol Cell 50(3):407–419
Inoki K, Kim J, Guan KL (2012) AMPK and mTOR in cellular energy homeostasis and drug targets. Annu Rev Pharmacol Toxicol 52:381–400
Carretero J et al (2007) Dysfunctional AMPK activity, signalling through mTOR and survival in response to energetic stress in LKB1-deficient lung cancer. Oncogene 26(11):1616–1625
Rolf J et al (2013) AMPKalpha1: a glucose sensor that controls CD8 T-cell memory. Eur J Immunol 43(4):889–896
Fernandez D et al (2006) Rapamycin reduces disease activity and normalizes T cell activation-induced calcium fluxing in patients with systemic lupus erythematosus. Arthritis Rheum 54(9):2983–2988
Warner LM, Adams LM, Sehgal SN (1994) Rapamycin prolongs survival and arrests pathophysiologic changes in murine systemic lupus erythematosus. Arthritis Rheum 37(2):289–297
Augustine JJ, Bodziak KA, Hricik DE (2007) Use of sirolimus in solid organ transplantation. Drugs 67(3):369–391
Gao Y, Whitaker-Dowling P, Bergman I (2015) Memory antitumor T-cells resist inhibition by immune suppressor cells. Anticancer Res 35(9):4593–4597
Klebanoff CA, Gattinoni L, Restifo NP (2006) CD8+ T-cell memory in tumor immunology and immunotherapy. Immunol Rev 211:214–224
Klebanoff CA et al (2005) Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci U S A 102(27):9571–9576
Turner AP et al (2011) Sirolimus enhances the magnitude and quality of viral-specific CD8+ T-cell responses to vaccinia virus vaccination in rhesus macaques. Am J Transplant 11(3):613–618
Li Q et al (2012) Regulating mammalian target of rapamycin to tune vaccination-induced CD8(+) T cell responses for tumor immunity. J Immunol 188(7):3080–3087
Berezhnoy A et al (2014) Aptamer-targeted inhibition of mTOR in T cells enhances antitumor immunity. J Clin Invest 124(1):188–197
Foretz M et al (2014) Metformin: from mechanisms of action to therapies. Cell Metab 20(6):953–966
Sajan MP et al (2010) AICAR and metformin, but not exercise, increase muscle glucose transport through AMPK-, ERK-, and PDK1-dependent activation of atypical PKC. Am J Physiol Endocrinol Metab 298(2):E179–E192
Jhun BS et al (2005) AICAR suppresses IL-2 expression through inhibition of GSK-3 phosphorylation and NF-AT activation in Jurkat T cells. Biochem Biophys Res Commun 332(2):339–346
Nath N et al (2009) Metformin attenuated the autoimmune disease of the central nervous system in animal models of multiple sclerosis. J Immunol 182(12):8005–8014
Bai A et al (2010) AMPK agonist downregulates innate and adaptive immune responses in TNBS-induced murine acute and relapsing colitis. Biochem Pharmacol 80(11):1708–1717
Bai A et al (2010) Novel anti-inflammatory action of 5-aminoimidazole-4-carboxamide ribonucleoside with protective effect in dextran sulfate sodium-induced acute and chronic colitis. J Pharmacol Exp Ther 333(3):717–725
Eikawa S et al (2015) Immune-mediated antitumor effect by type 2 diabetes drug, metformin. Proc Natl Acad Sci U S A 112(6):1809–1814
Walker JE (2013) The ATP synthase: the understood, the uncertain and the unknown. Biochem Soc Trans 41(1):1–16
Li Y et al (2006) Cytochrome c oxidase subunit IV is essential for assembly and respiratory function of the enzyme complex. J Bioenerg Biomembr 38(5–6):283–291
Jimenez-Gutierrez LR et al (2014) The cytochrome c oxidase and its mitochondrial function in the whiteleg shrimp Litopenaeus vannamei during hypoxia. J Bioenerg Biomembr 46(3):189–196
Hockel M, Vaupel P (2001) Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J Natl Cancer Inst 93(4):266–276
Semenza GL et al (1997) Structural and functional analysis of hypoxia-inducible factor 1. Kidney Int 51(2):553–555
Eguchi H et al (1997) A nuclear localization signal of human aryl hydrocarbon receptor nuclear translocator/hypoxia-inducible factor 1beta is a novel bipartite type recognized by the two components of nuclear pore-targeting complex. J Biol Chem 272(28):17640–17647
Freeburg PB, Abrahamson DR (2004) Divergent expression patterns for hypoxia-inducible factor-1beta and aryl hydrocarbon receptor nuclear transporter-2 in developing kidney. J Am Soc Nephrol 15(10):2569–2578
Schodel J et al (2011) High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood 117(23):e207–e217
Wenger RH, Stiehl DP, Camenisch G (2005) Integration of oxygen signaling at the consensus HRE. Sci STKE 2005(306):re12
Wang GL et al (1995) Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A 92(12):5510–5514
Chapman-Smith A, Lutwyche JK, Whitelaw ML (2004) Contribution of the per/Arnt/Sim (PAS) domains to DNA binding by the basic helix-loop-helix PAS transcriptional regulators. J Biol Chem 279(7):5353–5362
Yang J et al (2005) Functions of the per/ARNT/Sim domains of the hypoxia-inducible factor. J Biol Chem 280(43):36047–36054
Ziello JE, Jovin IS, Huang Y (2007) Hypoxia-inducible factor (HIF)-1 regulatory pathway and its potential for therapeutic intervention in malignancy and ischemia. Yale J Biol Med 80(2):51–60
Makino Y et al (2001) Inhibitory PAS domain protein is a negative regulator of hypoxia-inducible gene expression. Nature 414(6863):550–554
Semenza GL (2010) Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 29(5):625–634
Zhang P et al (2014) Hypoxia-inducible factor 3 is an oxygen-dependent transcription activator and regulates a distinct transcriptional response to hypoxia. Cell Rep 6(6):1110–1121
Jiang BH et al (1996) Hypoxia-inducible factor 1 levels vary exponentially over a physiologically relevant range of O2 tension. Am J Phys 271(4 Pt 1):C1172–C1180
McNamee EN et al (2013) Hypoxia and hypoxia-inducible factors as regulators of T cell development, differentiation, and function. Immunol Res 55(1–3):58–70
Vaupel P, Harrison L (2004) Tumor hypoxia: causative factors, compensatory mechanisms, and cellular response. Oncologist 9(Suppl 5):4–9
Tsai AG, Johnson PC, Intaglietta M (2003) Oxygen gradients in the microcirculation. Physiol Rev 83(3):933–963
Chang CH et al (2015) Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 162(6):1229–1241
Gilkes DM, Semenza GL, Wirtz D (2014) Hypoxia and the extracellular matrix: drivers of tumour metastasis. Nat Rev Cancer 14(6):430–439
Gorres KL, Raines RT (2010) Prolyl 4-hydroxylase. Crit Rev Biochem Mol Biol 45(2):106–124
Ohh M et al (2000) Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat Cell Biol 2(7):423–427
Schofield CJ, Ratcliffe PJ (2004) Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol 5(5):343–354
Hudson CC et al (2002) Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol Cell Biol 22(20):7004–7014
Nakamura H et al (2005) TCR engagement increases hypoxia-inducible factor-1 alpha protein synthesis via rapamycin-sensitive pathway under hypoxic conditions in human peripheral T cells. J Immunol 174(12):7592–7599
Baek JH et al (2007) Spermidine/spermine N(1)-acetyltransferase-1 binds to hypoxia-inducible factor-1alpha (HIF-1alpha) and RACK1 and promotes ubiquitination and degradation of HIF-1alpha. J Biol Chem 282(46):33358–33366
Liu YV et al (2007) RACK1 competes with HSP90 for binding to HIF-1alpha and is required for O(2)-independent and HSP90 inhibitor-induced degradation of HIF-1alpha. Mol Cell 25(2):207–217
Liu YV et al (2007) Calcineurin promotes hypoxia-inducible factor 1alpha expression by dephosphorylating RACK1 and blocking RACK1 dimerization. J Biol Chem 282(51):37064–37073
Clambey ET et al (2012) Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc Natl Acad Sci U S A 109(41):E2784–E2793
Hsiao HW et al (2015) Deltex1 antagonizes HIF-1alpha and sustains the stability of regulatory T cells in vivo. Nat Commun 6:6353
Lee JH et al (2015) E3 ubiquitin ligase VHL regulates hypoxia-inducible factor-1alpha to maintain regulatory T cell stability and suppressive capacity. Immunity 42(6):1062–1074
Doedens AL et al (2013) Hypoxia-inducible factors enhance the effector responses of CD8(+) T cells to persistent antigen. Nat Immunol 14(11):1173–1182
Chisolm DA, Weinmann AS (2015) TCR-signaling events in cellular metabolism and specialization. Front Immunol 6:292
Hengel RL et al (2003) Cutting edge: L-selectin (CD62L) expression distinguishes small resting memory CD4+ T cells that preferentially respond to recall antigen. J Immunol 170(1):28–32
Yang S et al (2011) The shedding of CD62L (L-selectin) regulates the acquisition of lytic activity in human tumor reactive T lymphocytes. PLoS One 6(7):e22560
Zagzag D et al (2000) Expression of hypoxia-inducible factor 1alpha in brain tumors: association with angiogenesis, invasion, and progression. Cancer 88(11):2606–2618
Rapisarda A et al (2004) Schedule-dependent inhibition of hypoxia-inducible factor-1alpha protein accumulation, angiogenesis, and tumor growth by topotecan in U251-HRE glioblastoma xenografts. Cancer Res 64(19):6845–6848
Rapisarda A et al (2002) Identification of small molecule inhibitors of hypoxia-inducible factor 1 transcriptional activation pathway. Cancer Res 62(15):4316–4324
Rapisarda A et al (2009) Increased antitumor activity of bevacizumab in combination with hypoxia inducible factor-1 inhibition. Mol Cancer Ther 8(7):1867–1877
Koh MY et al (2008) Molecular mechanisms for the activity of PX-478, an antitumor inhibitor of the hypoxia-inducible factor-1alpha. Mol Cancer Ther 7(1):90–100
Welsh S et al (2004) Antitumor activity and pharmacodynamic properties of PX-478, an inhibitor of hypoxia-inducible factor-1alpha. Mol Cancer Ther 3(3):233–244
Lang M et al (2016) Arsenic trioxide plus PX-478 achieves effective treatment in pancreatic ductal adenocarcinoma. Cancer Lett 378(2):87–96
Zhao T et al (2015) Inhibition of HIF-1alpha by PX-478 enhances the anti-tumor effect of gemcitabine by inducing immunogenic cell death in pancreatic ductal adenocarcinoma. Oncotarget 6(4):2250–2262
Zhang H et al (2008) Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem 283(16):10892–10903
Huh JR et al (2011) Digoxin and its derivatives suppress TH17 cell differentiation by antagonizing RORgammat activity. Nature 472(7344):486–490
Osada S et al (1997) Identification of an extended half-site motif required for the function of peroxisome proliferator-activated receptor alpha. Genes Cells 2(5):315–327
Krey G et al (1993) Xenopus peroxisome proliferator activated receptors: genomic organization, response element recognition, heterodimer formation with retinoid X receptor and activation by fatty acids. J Steroid Biochem Mol Biol 47(1–6):65–73
Alleva DG et al (2002) Regulation of murine macrophage proinflammatory and anti-inflammatory cytokines by ligands for peroxisome proliferator-activated receptor-gamma: counter-regulatory activity by IFN-gamma. J Leukoc Biol 71(4):677–685
DiRenzo J et al (1997) Peroxisome proliferator-activated receptors and retinoic acid receptors differentially control the interactions of retinoid X receptor heterodimers with ligands, coactivators, and corepressors. Mol Cell Biol 17(4):2166–2176
Dowell P et al (1997) p300 functions as a coactivator for the peroxisome proliferator-activated receptor alpha. J Biol Chem 272(52):33435–33443
Yuan CX et al (1998) The TRAP220 component of a thyroid hormone receptor- associated protein (TRAP) coactivator complex interacts directly with nuclear receptors in a ligand-dependent fashion. Proc Natl Acad Sci U S A 95(14):7939–7944
Zhu Y et al (1996) Cloning and identification of mouse steroid receptor coactivator-1 (mSRC-1), as a coactivator of peroxisome proliferator-activated receptor gamma. Gene Expr 6(3):185–195
Zhu Y et al (1997) Isolation and characterization of PBP, a protein that interacts with peroxisome proliferator-activated receptor. J Biol Chem 272(41):25500–25506
Desvergne B, Wahli W (1999) Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev 20(5):649–688
Jones DC, Ding X, Daynes RA (2002) Nuclear receptor peroxisome proliferator-activated receptor alpha (PPARalpha) is expressed in resting murine lymphocytes. The PPARalpha in T and B lymphocytes is both transactivation and transrepression competent. J Biol Chem 277(9):6838–6845
Harris SG, Phipps RP (2001) The nuclear receptor PPAR gamma is expressed by mouse T lymphocytes and PPAR gamma agonists induce apoptosis. Eur J Immunol 31(4):1098–1105
Vosper H et al (2001) The peroxisome proliferator-activated receptor delta promotes lipid accumulation in human macrophages. J Biol Chem 276(47):44258–44265
Shi Y, Hon M, Evans RM (2002) The peroxisome proliferator-activated receptor delta, an integrator of transcriptional repression and nuclear receptor signaling. Proc Natl Acad Sci U S A 99(5):2613–2618
Chinetti G et al (1998) Activation of proliferator-activated receptors alpha and gamma induces apoptosis of human monocyte-derived macrophages. J Biol Chem 273(40):25573–25580
Delerive P et al (1999) Peroxisome proliferator-activated receptor activators inhibit thrombin-induced endothelin-1 production in human vascular endothelial cells by inhibiting the activator protein-1 signaling pathway. Circ Res 85(5):394–402
Gosset P et al (2001) Peroxisome proliferator-activated receptor gamma activators affect the maturation of human monocyte-derived dendritic cells. Eur J Immunol 31(10):2857–2865
Padilla J et al (2000) Peroxisome proliferator activator receptor-gamma agonists and 15-deoxy-Delta(12,14)(12,14)-PGJ(2) induce apoptosis in normal and malignant B-lineage cells. J Immunol 165(12):6941–6948
Delerive P et al (1999) Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-kappaB and AP-1. J Biol Chem 274(45):32048–32054
Delerive P et al (2000) Oxidized phospholipids activate PPARalpha in a phospholipase A2-dependent manner. FEBS Lett 471(1):34–38
Peters JM, Hollingshead HE, Gonzalez FJ (2008) Role of peroxisome-proliferator-activated receptor beta/delta (PPARbeta/delta) in gastrointestinal tract function and disease. Clin Sci (Lond) 115(4):107–127
Yang XY et al (2000) Activation of human T lymphocytes is inhibited by peroxisome proliferator-activated receptor gamma (PPARgamma) agonists. PPARgamma co-association with transcription factor NFAT. J Biol Chem 275(7):4541–4544
Raikwar HP et al (2005) PPARgamma antagonists exacerbate neural antigen-specific Th1 response and experimental allergic encephalomyelitis. J Neuroimmunol 167(1–2):99–107
Raikwar HP et al (2006) PPARgamma antagonists reverse the inhibition of neural antigen-specific Th1 response and experimental allergic encephalomyelitis by Ciglitazone and 15-deoxy-Delta12,14-prostaglandin J2. J Neuroimmunol 178(1–2):76–86
Niino M et al (2001) Amelioration of experimental autoimmune encephalomyelitis in C57BL/6 mice by an agonist of peroxisome proliferator-activated receptor-gamma. J Neuroimmunol 116(1):40–48
Feinstein DL et al (2002) Peroxisome proliferator-activated receptor-gamma agonists prevent experimental autoimmune encephalomyelitis. Ann Neurol 51(6):694–702
Klotz L et al (2009) The nuclear receptor PPAR gamma selectively inhibits Th17 differentiation in a T cell-intrinsic fashion and suppresses CNS autoimmunity. J Exp Med 206(10):2079–2089
Natarajan C, Bright JJ (2002) Peroxisome proliferator-activated receptor-gamma agonists inhibit experimental allergic encephalomyelitis by blocking IL-12 production, IL-12 signaling and Th1 differentiation. Genes Immun 3(2):59–70
Jakobsen MA et al (2006) Peroxisome proliferator-activated receptor alpha, delta, gamma1 and gamma2 expressions are present in human monocyte-derived dendritic cells and modulate dendritic cell maturation by addition of subtype-specific ligands. Scand J Immunol 63(5):330–337
Storer PD et al (2005) Peroxisome proliferator-activated receptor-gamma agonists inhibit the activation of microglia and astrocytes: implications for multiple sclerosis. J Neuroimmunol 161(1–2):113–122
Kanakasabai S et al (2010) Peroxisome proliferator-activated receptor delta agonists inhibit T helper type 1 (Th1) and Th17 responses in experimental allergic encephalomyelitis. Immunology 130(4):572–588
Natarajan C et al (2003) Peroxisome proliferator-activated receptor-gamma-deficient heterozygous mice develop an exacerbated neural antigen-induced Th1 response and experimental allergic encephalomyelitis. J Immunol 171(11):5743–5750
Gocke AR et al (2009) Transcriptional modulation of the immune response by peroxisome proliferator-activated receptor-{alpha} agonists in autoimmune disease. J Immunol 182(7):4479–4487
Cunard R et al (2002) Regulation of cytokine expression by ligands of peroxisome proliferator activated receptors. J Immunol 168(6):2795–2802
Xu J, Racke MK, Drew PD (2007) Peroxisome proliferator-activated receptor-alpha agonist fenofibrate regulates IL-12 family cytokine expression in the CNS: relevance to multiple sclerosis. J Neurochem 103(5):1801–1810
Kapadia R, Yi JH, Vemuganti R (2008) Mechanisms of anti-inflammatory and neuroprotective actions of PPAR-gamma agonists. Front Biosci 13:1813–1826
Cipolletta D et al (2012) PPAR-gamma is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature 486(7404):549–553
Housley WJ et al (2009) PPARgamma regulates retinoic acid-mediated DC induction of Tregs. J Leukoc Biol 86(2):293–301
Wohlfert EA et al (2007) Peroxisome proliferator-activated receptor gamma (PPARgamma) and immunoregulation: enhancement of regulatory T cells through PPARgamma-dependent and -independent mechanisms. J Immunol 178(7):4129–4135
Lei J et al (2010) Peroxisome proliferator-activated receptor alpha and gamma agonists together with TGF-beta convert human CD4+CD25- T cells into functional Foxp3+ regulatory T cells. J Immunol 185(12):7186–7198
Bassaganya-Riera J et al (2004) Activation of PPAR gamma and delta by conjugated linoleic acid mediates protection from experimental inflammatory bowel disease. Gastroenterology 127(3):777–791
Diab A et al (2002) Peroxisome proliferator-activated receptor-gamma agonist 15-deoxy-Delta(12,14)-prostaglandin J(2) ameliorates experimental autoimmune encephalomyelitis. J Immunol 168(5):2508–2515
Mueller C et al (2003) Peroxisome proliferator-activated receptor gamma ligands attenuate immunological symptoms of experimental allergic asthma. Arch Biochem Biophys 418(2):186–196
Beales PE et al (1998) Troglitazone prevents insulin dependent diabetes in the non-obese diabetic mouse. Eur J Pharmacol 357(2–3):221–225
Park SJ et al (2009) Peroxisome proliferator-activated receptor gamma agonist down-regulates IL-17 expression in a murine model of allergic airway inflammation. J Immunol 183(5):3259–3267
Kaiser CC et al (2009) A pilot test of pioglitazone as an add-on in patients with relapsing remitting multiple sclerosis. J Neuroimmunol 211(1–2):124–130
Lewis JD et al (2008) Rosiglitazone for active ulcerative colitis: a randomized placebo-controlled trial. Gastroenterology 134(3):688–695
Acknowledgment
Our research is supported by the grants from the Bloomberg-Kimmel Institute, the Melanoma Research Alliance (Established Investigator Award), the National Institutes of Health (RO1AI099300 and RO1AI089830), Department of Defense (PC130767), “Kelly’s Dream” Foundation, the Janey Fund, the Seraph Foundation, gifts from Bill and Betty Topecer and Dorothy Needle, and the Roswell Park Alliance Foundation. FP is a Stewart Trust Scholar.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Science+Business Media B.V.
About this chapter
Cite this chapter
Vignali, P.D.A., Barbi, J., Pan, F. (2017). Metabolic Regulation of T Cell Immunity. In: Li, B., Pan, F. (eds) Immune Metabolism in Health and Tumor. Advances in Experimental Medicine and Biology, vol 1011. Springer, Dordrecht. https://doi.org/10.1007/978-94-024-1170-6_2
Download citation
DOI: https://doi.org/10.1007/978-94-024-1170-6_2
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-024-1168-3
Online ISBN: 978-94-024-1170-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)