
Abstract
Purpose of Review
Skeletal adaptation to mechanical loading plays a critical role in bone growth and the maintenance of bone homeostasis. Osteocytes are postulated to serve as a hub orchestrating bone remodeling. The recent findings on the molecular mechanisms by which osteocytes sense mechanical loads and the downstream bone-forming factors are reviewed.
Recent Findings
Calcium channels have been implicated in mechanotransduction in bone cells for a long time. Efforts have been made to identify a specific calcium channel mediating the skeletal response to mechanical loads. Recent studies have revealed that Piezo1, a mechanosensitive ion channel, is critical for normal bone growth and is essential for the skeletal response to mechanical loading.
Summary
Identification of mechanosensors and their downstream effectors in mechanosensing bone cells is essential for new strategies to modulate regenerative responses and develop therapies to treat the bone loss related to disuse or advanced age.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Skeletal Adaptation
Bones adapt to changes in mechanical forces by changing their mass. Specifically, mechanical forces increase osteoblast number, stimulate bone formation, and increase bone mass [1, 2]. A classic example of anabolic bone adaptation is the higher bone mass that baseball players have in their throwing forearm compared with their non-throwing forearm [3]. In contrast, loss of mechanical stimuli decreases bone mass by inhibiting bone formation [4, 5] and promoting osteoclastogenesis and bone resorption [4, 6, 7]. Bone mass is rapidly lost under unloading conditions such as extreme lack of physical activity, being bedridden, or microgravity [8]. The identity of the cells responsible for directly sensing changes in mechanical loading of the skeleton is unclear.
As the most abundant resident cells in bone, osteocytes have been postulated to sense and respond to mechanical cues. They are derived from osteoblasts that become buried in the bone matrix. They are connected to one another by the lacuna-canalicular network, which enables chemical and fluid transport between osteocytes and cells on the bone surface [9]. Changes in the mechanical forces experienced by the bone cause the rate of fluid flow inside the lacuna-canalicular system to change, and this can be detected by osteocytes [10]. In vitro evidence indicates that osteocytes are highly responsive to fluid shear stress [11, 12], and that they sense mechanical signals mainly through their long cellular processes rather than their cell bodies [13]. Being in a rigid microenvironment, osteocytes may also respond to matrix strains directly in addition to fluid shear stress [14]. The idea that osteocytes are involved in mechanosensing is partially supported by in vivo osteocyte ablation models. Tatsumi et al. showed that ablation of osteocytes in mice prevents the cancellous bone loss induced by mechanical unloading [6]. However, in the same study, when the hindlimbs of these mice are reloaded following a short period of unloading, the skeletal response is not affected by osteocyte ablation [6]. These studies indicate that osteocytes are required for the skeletal response to loss of mechanical stimulation, but not for the increase of mechanical stimulation. Other studies with loss or gain of function in osteocytes have revealed the important roles of a set of genes, such as β-catenin [15, 16], Lrp5 [17], Sost [1, 18, 19], and Piezo1 [20], in the skeletal response to mechanical load. The mouse genetic tools used in these studies to delete or over-express genes of interest in osteocytes are mainly Dmp1-Cre transgenic mice, including 8 kb Dmp1-Cre, 10 kb Dmp1-Cre, and 10 kb Dmp1-CreERT2 mice. Although osteocytes are targeted by these Cre driver strains, other cell types, including osteoblasts in particular, are also targeted [7, 21, 22]. Therefore, it is possible that the skeletal phenotype observed in these studies could be attributed to osteoblasts, osteocytes, or both. Consistent with this, in vitro evidence suggests that osteoblasts are also sensitive to mechanical stimulation and produce the same molecules as osteocytes in response, such as prostaglandins [23]. Thus, it is still an open question which cell type(s) in the bone senses changes in mechanical stimulation. The newly developed Sost-Cre and Sost-CreERT2 mouse models may help to answer that question since they target osteocytes but not osteoblasts [21, 24]. Studies using reporter mice showed that Dmp1-Cre transgene causes gene deletion in osteoblasts and osteocytes while the Sost-Cre transgene targets osteocytes as well as hematopoietic cells [21]. Although neither Dmp1-Cre nor Sost-Cre transgene is specific to osteocytes, they are still a common target of both transgenes. Therefore, comparison of results obtained from Sost-Cre and Dmp1-Cre models could help determine the importance of genes of interest expressed in osteocytes.
During physical activity, the skeleton encounters different types of mechanical forces, including gravitational loading and muscle contraction. While it is clear that these mechanical stimuli cause bone deformation or strain, the exact mechanisms by which strain at a whole tissue level is transduced to osteocytes and the nature of the mechanical forces that activate osteocytes in vivo remain unclear. Fluid shear stress has been postulated to be the major driving force for load-induced bone formation. The idea is that loading causes deformation of bone and results in interstitial pressure gradients in the bone marrow and the lacuna-canalicular system of osteocytes. These pressure gradients drive the movement of interstitial fluid in the marrow and through the lacuna-canalicular network, generating shear stress. Mechanosensitive cells in bone, such as osteocytes, perceive the change of shear stress and then orchestrate bone remodeling [25]. Based on this model, fluid shear stress has become the most widely used in vitro approach to study mechanotransduction in osteocytes. A more recently modified model for this transduction suggests that the fluid flow in the lacuna-canalicular system generates drag forces directly on the osteocyte tethering sites of the lacunar wall, which results in an amplified hoop strain on cell processes [26]. The magnitude of this strain is comparable with that used in vitro to activate osteoblastic cells. Therefore, this hoop strain has been proposed to be the driving force that activates osteocytes in vivo. However, an in vitro culture system that models this effect has not been developed.
Mechanosensors in Bone
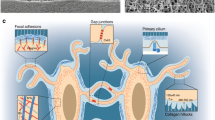
Mechanical signals contribute to normal bone growth and skeletal homeostasis during adulthood. However, the mechanisms by which the skeleton responds to mechanical stimulation are not fully understood. For decades, efforts have been made to identify specific mechanosensors in bone cells to pin down those mechanisms. Although a variety of cell surface proteins and structures has been proposed to facilitate perception of mechanical signals by bone cells (Fig. 1; for a revision on these mechanisms involved in osteocyte mechanotransduction, see Ref. [27,28,29,30]) [27,28,29,30], an integrated pathway explaining how osteocytes perceive and transduce mechanical signals has yet to be elucidated. Integrins are one of the surface proteins implicated in mechanosensation in osteocytes. Deletion of the β1 integrin in osteoblasts and osteocytes using the Col1a1-cre transgene leads to a significant reduction in bone formation induced by mechanical loading compared with control mice [31]. Integrin αV/β3 is found to be expressed in osteocytes and is localized in close proximity to the purinergic channel pannexin1, the ATP-gated purinergic receptor P2X7R, and the low voltage–gated T-type calcium channel CaV3.2 in a specialized structure through which osteocyte cell processes tether to the lacunar wall [32]. These structures potentially facilitate the hoop strain amplification leading to activation of osteocytes. Recently, integrin αV/β5 has been identified as the receptor for the so-called exercise hormone Irisin [33]. Irisin levels increase dramatically with physical activity and decrease with unloading [34]. Importantly, administration of Irisin prevents the bone loss caused by hindlimb unloading [35]. In vitro assays show that Irisin binds to integrin αV/β5 and chemical blocking of β5 integrin diminishes the effects of Irisin on osteocytes [33]. Therefore, integrins not only respond to physical cues directly but also respond to chemical myokines induced by mechanical stimuli.

Cell surface proteins and structures involved in osteocyte mechanotransduction. Wnt receptors including Lrp5, integrin-containing focal adhesions, primary cilia, voltage-gated calcium channels, and connexin-based gap junctions are the major mechanosensors being implicated in bone cells. Upon stimulation by mechanical loading, osteocytes promote osteoblast formation by increasing Wnt ligand expression and decreasing SOST expression. Mechanical stimulation also enhances osteocyte energy production by promoting mTOR signaling
Calcium channels are also involved in the sensing of mechanical signals by osteocytes as indicated by multiple lines of evidence [36,37,38,39]. For instance, calcium influx is an early event following mechanical stimulus in osteocytes both in vitro and in vivo [37, 38]. Although the magnitude of the calcium influx within responding osteocytes does not change with increasing loading, the number of responding osteocytes does [37, 38]. This increase in intracellular calcium concentration is required for the production of Nos2 and Cox-2 induced by mechanical stimulation [40, 41]. In addition, chemical blocking of L-type voltage-sensitive calcium channels (VSCC) suppresses the typical loading-induced increase in bone formation in rats [42]. Similar to many other cell types, osteocytes exhibit different types of calcium channels, including transient receptor potential channels (TRPV), L-type VSCC, and T-type VSCC [39, 43, 44]. To identify a specific calcium channel involved in the response of the skeleton to mechanical loading, loss of function studies for some of these calcium channels has been implemented in the past, and some of these mutant mice have been tested for their skeletal response to loading. For the TRPV channels that expressed in osteocytes, mice lacking TRPV 1, 4, or 6 have been analyzed for their basal skeletal phenotype. TRPV 1 and TRPV4 knockout mice have high bone mass associated with decreased osteoclast number and normal bone formation rate [45,46,47]. This argues against a role for TRPV1 and 4 as mechanosensors in bone, and this is consistent with other observations that TRPV4 does not respond to mechanical stimulation such as membrane stretch [48]. TRPV6 is expressed in osteoblasts and osteocytes at low levels [49]. Moreover, TRPV6 knockout mice exhibit reduced intestinal calcium absorption but maintain a normal bone formation rate indicating that TRPV6 does not play a role in the osteoblast lineage [50, 51]. The role of voltage-gated calcium channels in bone has also been explored. Mice with germline deletion of the L-type VSCC CaV 1.3 have a reduced cross-sectional area in long bones. However, these mice respond normally to mechanical loading [52].
With recent advances in identification of mechanosensitive ion channels in neurons, Piezo1 has emerged as a critical mechanosensor in many cell types [53]. Studies in epithelial cells have shown that Piezo1 responds to various forms of mechanical forces, including membrane stretch, static pressure, and fluid shear stress [54,55,56]. Moreover, Piezo1 can be directly activated by mechanical perturbations of the lipid bilayer alone, demonstrating its role in mechanosensing [57]. Piezo1 is also highly expressed in osteocytes and can be upregulated by mechanical stimulation in vitro as well as in vivo [20]. Recent studies from several different laboratories have found that deletion of the Piezo1 gene in different stages of the osteoblast lineage using the Prx1-Cre, OCN-Cre, Col1a1-Cre, and Dmp1-Cre transgene dramatically reduced both cancellous bone mass and cortical thickness [20, 58,59,60]. The outer and inner circumferences of cortical bone in the midshaft of the femur in Piezo1 knockout mice are also decreased and are associated with decreased bone formation [20]. This skeletal phenotype is consistent with a reduced ability to respond to mechanical stimulation. Direct testing of this idea by performing an anabolic loading regime confirmed that the bones of the conditional knockout mice are less responsive to mechanical signals than controls [20]. This decrease was not due to an overall decrease in cell health since cell survival was not affected by Piezo1 deletion [20]. In addition, deletion of Piezo1 in osteoblast lineage cells also diminished the skeletal response to mechanical unloading induced by both hindlimb suspension [58, 59] and Botox injection [60]. Thus, these studies strongly suggest that Piezo1 plays a critical role in sensing mechanical signals in bone. The findings that the basal skeletal phenotype is similar between mice with Piezo1 deletion in different stages of osteoblast lineage as well as the evidence that the skeletal response to changes in mechanical loading is blunted in mice with Piezo1 deletion in Dmp1-Cre targeted cells suggest that Piezo1 expression in more mature cells including osteocytes is critical for sensing mechanical stimulation in bone.
Since the skeletal response to mechanical loading is not completely abolished in mice lacking Piezo1 in mature bone cells [20], it is most likely that there are other mechanosensors that compensate for the loss of Piezo1 in these mice. In addition, Piezo1 has been reported to be associated with integrin αV activation through Gαq/Gα11 signaling pathways in epithelial cells [61]. Therefore, it will also be important to understand how Piezo1 interacts with other mechanosensors in osteocytes.
Downstream Effectors Mediate Loading-Induced Bone Formation
Mechanical loading stimulates bone formation by rapidly increasing osteoblast formation and function [2]. Osteocytes have been recognized as a hub to control bone formation by communicating with osteoblasts and their progenitors. To date, several effector proteins have been implicated in the bone formation induced by mechanical signals. These include sclerostin, a canonical Wnt signaling inhibitor. Sclerostin expressed in osteocytes has been shown to be critical for osteoblast formation, and its production is governed by mechanical signals [1, 18, 19]. TRPV4 calcium channel has been shown to be required for the suppression of sclerostin by mechanical stimulation in osteocytes in vitro [62], but calcium oscillation is not required for this suppression [63]. Deletion of Sost, the sclerostin coding gene, prevented the bone loss caused by tail suspension [64] and botulinum toxin [65]. Overexpression of Sost in osteocytes blunted the anabolic effects of loading on bone [66]. In contrast, loss of sclerostin in mice did not prevent the increase in bone formation induced by anabolic loading [65], suggesting that sclerostin is a permissive agent for bone formation and that there are additional effector proteins mediating loading-induced bone formation.
In the Wnt/β-catenin pathway, mechanical loading also increases the expression of several Wnt ligands including Wnt1 and Wnt7b in murine bone [67, 68]. Mechanosensitive ion channel Piezo1 has been shown to be essential for load-induced Wnt1 expression [20]. By binding to various receptors on the cell surface, Wnts, such as Wnt1, activate the Wnt signaling pathway and play critical roles in osteoblastogenesis and bone formation [69]. Importantly, earlier studies have demonstrated that Lrp5, a co-receptor for Wnt ligands, is required for loading-induced anabolic effects [17]. Consistent with this, blocking Wnt ligands’ secretion using a pharmacological inhibitor diminished the anabolic effect of loading in adult mice [70]. Moreover, postnatal deletion of Wntless, a conserved transmembrane protein required for Wnts’ secretion in osteoblast lineage cells in adult mice, significantly reduced the skeletal response to mechanical loading [70]. These studies suggest that Wnt ligands play an important role in mediating loading-induced bone formation. Further mouse genetic studies will be required to determine which Wnts are critical for the osteogenic response to mechanical loading.
Besides changing the expression of secreted regulatory proteins in osteocytes, mechanical loading also promotes overall health and energy production of osteocytes. Osteocyte viability is influenced by mechanical signals both in vitro and in vivo [71,72,73]. In addition, mechanical stimulation increases ATP production and mitochondrial function [74,75,76,77]. Although these phenomena have been observed for a long time, it is not clear whether increases in cellular metabolism contribute to anabolic effects of mechanical loading. Recent evidence from studies on the role of the mammalian target of the rapamycin (mTOR) pathway in osteoblast lineage cells has shed light on this matter. The mTOR pathway is a highly conserved central regulator for cellular metabolism [78]. Mechanical loading can stimulate mTOR activity in osteocytes in vitro [79], which in turn promotes glycolysis and energy production in bone cells [80]. Deletion of Rictor, an mTOR complex 2 subunit, in osteoblast lineage cells using Prx1-Cre blunted the anabolic response to mechanical stimuli in adult mice [81], suggesting that cell metabolism plays an important role in the response. More recent studies with deletion of Rictor in Dmp1-cre-expressing cells showed a comparable cancellous and cortical bone phenotype to that observed in mice with Rictor deletion in Prx1-Cre expressing cells [82]. Importantly, the skeletal response to mechanical loading is also prevented in mice lacking Rictor in Dmp1-cre expressing cells [82]. These results demonstrate that the positive impact of the mTOR signaling on the anabolic response to loading occurs in mature cells, possibly osteocytes.
Future Directions
Even though much progress has been made, many questions remain unanswered. For instance, are osteocytes the major mechanosensing cells in bone? If not, what are the other cell types that respond directly to mechanical stimulation in bone? With advances in generating more osteocyte specific Cre driver strains and the development of cell type–specific CRISPR interference technology, the role that particular cell types in the different stages of osteoblast lineage play in mechanosensation could be determined. Other questions, such as whether mechanosensors are specific to certain forms of mechanical stimulation and how different mechanosensors and downstream signaling pathways interact with each other upon activation, are also critical to elucidate the complex system of mechanotransduction in bone. As the exact pathways of mechanosensation and the mediating factors that promote bone formation come to light, it is feasible that bone anabolism could be achieved by pharmacological agents targeting the activation of those pathways.
References
Robling AG, Niziolek PJ, Baldridge LA, Condon KW, Allen MR, Alam I, et al. Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J Biol Chem. 2008;283(9):5866–75.
Turner CH, Owan I, Alvey T, Hulman J, Hock JM. Recruitment and proliferative responses of osteoblasts after mechanical loading in vivo determined using sustained-release bromodeoxyuridine. Bone. 1998;22(5):463–9.
Warden SJ, Mantila Roosa SM, Kersh ME, Hurd AL, Fleisig GS, Pandy MG, et al. Physical activity when young provides lifelong benefits to cortical bone size and strength in men. Proc Natl Acad Sci U S A. 2014;111(14):5337–42.
Nakamura H, Aoki K, Masuda W, Alles N, Nagano K, Fukushima H, et al. Disruption of NF-kappaB1 prevents bone loss caused by mechanical unloading. J Bone Miner Res. 2013;28(6):1457–67.
Kondo H, Nifuji A, Takeda S, Ezura Y, Rittling SR, Denhardt DT, et al. Unloading induces osteoblastic cell suppression and osteoclastic cell activation to lead to bone loss via sympathetic nervous system. J Biol Chem. 2005;280(34):30192–200.
Tatsumi S, Ishii K, Amizuka N, Li M, Kobayashi T, Kohno K, et al. Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab. 2007;5(6):464–75.
Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O'Brien CA. Matrix-embedded cells control osteoclast formation. Nat Med. 2011;17(10):1235–41.
Uda Y, Azab E, Sun N, Shi C, Pajevic PD. Osteocyte Mechanobiology. Curr Osteoporos Rep. 2017;15(4):318–25.
Robling AG, Bonewald LF. The osteocyte: new insights. Annu Rev Physiol. 2020;82:485–506.
Fritton SP, Weinbaum S. Fluid and solute transport in bone: flow-induced mechanotransduction. Annu Rev Fluid Mech. 2009;41:347–74.
Kamel MA, Picconi JL, Lara-Castillo N, Johnson ML. Activation of beta-catenin signaling in MLO-Y4 osteocytic cells versus 2T3 osteoblastic cells by fluid flow shear stress and PGE2: implications for the study of mechanosensation in bone. Bone. 2010;47(5):872–81.
Lu XL, Huo B, Chiang V, Guo XE. Osteocytic network is more responsive in calcium signaling than osteoblastic network under fluid flow. J Bone Miner Res. 2012;27(3):563–74.
Burra S, Nicolella DP, Francis WL, Freitas CJ, Mueschke NJ, Poole K, et al. Dendritic processes of osteocytes are mechanotransducers that induce the opening of hemichannels. Proc Natl Acad Sci U S A. 2010;107(31):13648–53.
Klein-Nulend J, van der Plas A, Semeins CM, Ajubi NE, Frangos JA, Nijweide PJ, et al. Sensitivity of osteocytes to biomechanical stress in vitro. FASEB J. 1995;9(5):441–5.
Javaheri B, Stern AR, Lara N, Dallas M, Zhao H, Liu Y, et al. Deletion of a single beta-catenin allele in osteocytes abolishes the bone anabolic response to loading. J Bone Miner Res. 2014;29(3):705–15.
Kang KS, Hong JM, Robling AG. Postnatal beta-catenin deletion from Dmp1-expressing osteocytes/osteoblasts reduces structural adaptation to loading, but not periosteal load-induced bone formation. Bone. 2016;88:138–45.
Zhao L, Shim JW, Dodge TR, Robling AG, Yokota H. Inactivation of Lrp5 in osteocytes reduces young's modulus and responsiveness to the mechanical loading. Bone. 2013;54(1):35–43.
Robling AG, Bellido T, Turner CH. Mechanical stimulation in vivo reduces osteocyte expression of sclerostin. J Musculoskelet Neuronal Interact. 2006;6(4):354.
Li X, Ominsky MS, Niu QT, Sun N, Daugherty B, D'Agostin D, et al. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res. 2008;23(6):860–9.
Li X, Han L, Nookaew I, Mannen E, Silva MJ, Almeida M, et al. Stimulation of Piezo1 by mechanical signals promotes bone anabolism. Elife. 2019;8. https://doi.org/10.7554/eLife.49631.
Xiong J, Piemontese M, Onal M, Campbell J, Goellner JJ, Dusevich V, et al. Osteocytes, not osteoblasts or lining cells, are the main source of the RANKL required for osteoclast formation in remodeling bone. PLoS One. 2015;10(9):e0138189.
Dallas SL, Xie Y, Shiflett LA, Ueki Y. Mouse Cre models for the study of bone diseases. Curr Osteoporos Rep. 2018;16(4):466–77.
Klein-Nulend J, Bacabac RG, Bakker AD. Mechanical loading and how it affects bone cells: the role of the osteocyte cytoskeleton in maintaining our skeleton. Eur Cell Mater. 2012;24:278–91.
Maurel DB, Matsumoto T, Vallejo JA, Johnson ML, Dallas SL, Kitase Y, et al. Characterization of a novel murine Sost ER(T2) Cre model targeting osteocytes. Bone Res. 2019;7:6.
Weinbaum S, Cowin SC, Zeng Y. A model for the excitation of osteocytes by mechanical loading-induced bone fluid shear stresses. J Biomech. 1994;27(3):339–60.
Han Y, Cowin SC, Schaffler MB, Weinbaum S. Mechanotransduction and strain amplification in osteocyte cell processes. Proc Natl Acad Sci U S A. 2004;101(47):16689–94.
Litzenberger JB, Kim JB, Tummala P, Jacobs CR. Beta1 integrins mediate mechanosensitive signaling pathways in osteocytes. Calcif Tissue Int. 2010;86(4):325–32.
Nguyen AM, Jacobs CR. Emerging role of primary cilia as mechanosensors in osteocytes. Bone. 2013;54(2):196–204.
Thompson WR, Rubin CT, Rubin J. Mechanical regulation of signaling pathways in bone. Gene. 2012;503(2):179–93.
Klein-Nulend J, Bakker AD, Bacabac RG, Vatsa A, Weinbaum S. Mechanosensation and transduction in osteocytes. Bone. 2013;54(2):182–90.
Litzenberger JB, Tang WJ, Castillo AB, Jacobs CR. Deletion of β1 Integrins from cortical osteocytes reduces load-induced bone formation. Cell Mol Bioeng. 2009;2(3):416–24.
Cabahug-Zuckerman P, Stout RF Jr, Majeska RJ, Thi MM, Spray DC, Weinbaum S, et al. Potential role for a specialized beta3 integrin-based structure on osteocyte processes in bone mechanosensation. J Orthop Res. 2018;36(2):642–52.
Kim H, Wrann CD, Jedrychowski M, Vidoni S, Kitase Y, Nagano K, et al. Irisin mediates effects on bone and fat via alphaV integrin receptors. Cell. 2018;175(7):1756–68 e17.
Kawao N, Moritake A, Tatsumi K, Kaji H. Roles of Irisin in the linkage from muscle to bone during mechanical unloading in mice. Calcif Tissue Int. 2018;103(1):24–34.
Colaianni G, Mongelli T, Cuscito C, Pignataro P, Lippo L, Spiro G, et al. Irisin prevents and restores bone loss and muscle atrophy in hind-limb suspended mice. Sci Rep. 2017;7(1):2811.
Hung CT, Pollack SR, Reilly TM, Brighton CT. Real-time calcium response of cultured bone cells to fluid flow. Clin Orthop Relat Res. 1995;313:256–69.
Lu XL, Huo B, Park M, Guo XE. Calcium response in osteocytic networks under steady and oscillatory fluid flow. Bone. 2012;51(3):466–73.
Lewis KJ, Frikha-Benayed D, Louie J, Stephen S, Spray DC, Thi MM, et al. Osteocyte calcium signals encode strain magnitude and loading frequency in vivo. Proc Natl Acad Sci U S A. 2017;114(44):11775–80.
Li J, Duncan RL, Burr DB, Turner CH. L-type calcium channels mediate mechanically induced bone formation in vivo. J Bone Miner Res. 2002;17(10):1795–800.
Ajubi NE, Klein-Nulend J, Alblas MJ, Burger EH, Nijweide PJ. Signal transduction pathways involved in fluid flow-induced PGE2 production by cultured osteocytes. Am J Phys. 1999;276(1):E171–8.
Bakker AD, Silva VC, Krishnan R, Bacabac RG, Blaauboer ME, Lin YC, et al. Tumor necrosis factor alpha and interleukin-1beta modulate calcium and nitric oxide signaling in mechanically stimulated osteocytes. Arthritis Rheum. 2009;60(11):3336–45.
Li J, Duncan RL, Burr DB, Turner CH. L-type calcium channels mediate mechanically induced bone formation in vivo. J Bone Miner Res. 2002;17(10):1795–800.
Abed E, Labelle D, Martineau C, Loghin A, Moreau R. Expression of transient receptor potential (TRP) channels in human and murine osteoblast-like cells. Mol Membr Biol. 2009;26(3):146–58.
Shao Y, Alicknavitch M, Farach-Carson MC. Expression of voltage sensitive calcium channel (VSCC) L-type Cav1.2 (alpha1C) and T-type Cav3.2 (alpha1H) subunits during mouse bone development. Dev Dyn. 2005;234(1):54–62.
Masuyama R, Vriens J, Voets T, Karashima Y, Owsianik G, Vennekens R, et al. TRPV4-mediated calcium influx regulates terminal differentiation of osteoclasts. Cell Metab. 2008;8(3):257–65.
van der Eerden BC, Oei L, Roschger P, Fratzl-Zelman N, Hoenderop JG, van Schoor NM, et al. TRPV4 deficiency causes sexual dimorphism in bone metabolism and osteoporotic fracture risk. Bone. 2013;57(2):443–54.
Nishimura H, Kawasaki M, Tsukamoto M, Menuki K, Suzuki H, Matsuura T, et al. Transient receptor potential vanilloid 1 and 4 double knockout leads to increased bone mass in mice. Bone Rep. 2020;12:100268.
Nikolaev YA, Cox CD, Ridone P, Rohde PR, Cordero-Morales JF, Vasquez V, et al. Mammalian TRP ion channels are insensitive to membrane stretch. J Cell Sci. 2019;132(23). https://doi.org/10.1242/jcs.238360.
Little R, Muimo R, Robson L, Harris K, Grabowski PS. The transient receptor potential ion channel TRPV6 is expressed at low levels in osteoblasts and has little role in osteoblast calcium uptake. PLoS One. 2011;6(11):e28166.
Bianco SD, Peng JB, Takanaga H, Suzuki Y, Crescenzi A, Kos CH, et al. Marked disturbance of calcium homeostasis in mice with targeted disruption of the Trpv6 calcium channel gene. J Bone Miner Res. 2007;22(2):274–85.
Chen F, Ni B, Yang YO, Ye T, Chen A. Knockout of TRPV6 causes osteopenia in mice by increasing osteoclastic differentiation and activity. Cell Physiol Biochem. 2014;33(3):796–809.
Li J, Zhao L, Ferries IK, Jiang L, Desta MZ, Yu X, et al. Skeletal phenotype of mice with a null mutation in Cav 1.3 L-type calcium channel. J Musculoskelet Neuronal Interact. 2010;10(2):180–7.
Murthy SE, Dubin AE, Patapoutian A. Piezos thrive under pressure: mechanically activated ion channels in health and disease. Nat Rev Mol Cell Biol. 2017;18(12):771–83.
Li J, Hou B, Tumova S, Muraki K, Bruns A, Ludlow MJ, et al. Piezo1 integration of vascular architecture with physiological force. Nature. 2014;515(7526):279–82.
Gudipaty SA, Lindblom J, Loftus PD, Redd MJ, Edes K, Davey CF, et al. Mechanical stretch triggers rapid epithelial cell division through Piezo1. Nature. 2017;543(7643):118–21.
Miyamoto T, Mochizuki T, Nakagomi H, Kira S, Watanabe M, Takayama Y, et al. Functional role for Piezo1 in stretch-evoked Ca(2)(+) influx and ATP release in urothelial cell cultures. J Biol Chem. 2014;289(23):16565–75.
Syeda R, Florendo MN, Cox CD, Kefauver JM, Santos JS, Martinac B, et al. Piezo1 channels are inherently mechanosensitive. Cell Rep. 2016;17(7):1739–46.
Sun W, Chi S, Li Y, Ling S, Tan Y, Xu Y, et al. The mechanosensitive Piezo1 channel is required for bone formation. Elife. 2019;8. https://doi.org/10.7554/eLife.47454.
Wang L, You X, Lotinun S, Zhang L, Wu N, Zou W. Mechanical sensing protein PIEZO1 regulates bone homeostasis via osteoblast-osteoclast crosstalk. Nat Commun. 2020;11(1):282.
Zhou T, Gao B, Fan Y, Liu Y, Feng S, Cong Q, et al. Piezo1/2 mediate mechanotransduction essential for bone formation through concerted activation of NFAT-YAP1-ss-catenin. Elife. 2020;9. https://doi.org/10.7554/eLife.52779.
Albarran-Juarez J, Iring A, Wang S, Joseph S, Grimm M, Strilic B, et al. Piezo1 and Gq/G11 promote endothelial inflammation depending on flow pattern and integrin activation. J Exp Med. 2018;215(10):2655–72.
Lyons JS, Joca HC, Law RA, Williams KM, Kerr JP, Shi G, et al. Microtubules tune mechanotransduction through NOX2 and TRPV4 to decrease sclerostin abundance in osteocytes. Sci Signal. 2017;10(506). https://doi.org/10.1126/scisignal.aan5748.
Williams KM, Leser JM, Gould NR, Joca HC, Lyons JS, Khairallah RJ, et al. TRPV4 calcium influx controls sclerostin protein loss independent of purinergic calcium oscillations. Bone. 2020;136:115356.
Lin C, Jiang X, Dai Z, Guo X, Weng T, Wang J, et al. Sclerostin mediates bone response to mechanical unloading through antagonizing Wnt/beta-catenin signaling. J Bone Miner Res. 2009;24(10):1651–61.
Morse A, McDonald MM, Kelly NH, Melville KM, Schindeler A, Kramer I, et al. Mechanical load increases in bone formation via a sclerostin-independent pathway. J Bone Miner Res. 2014;29(11):2456–67.
Tu X, Rhee Y, Condon KW, Bivi N, Allen MR, Dwyer D, et al. Sost downregulation and local Wnt signaling are required for the osteogenic response to mechanical loading. Bone. 2012;50(1):209–17.
Holguin N, Brodt MD, Silva MJ. Activation of Wnt signaling by mechanical loading is impaired in the bone of old mice. J Bone Miner Res. 2016;31(12):2215–26.
Kelly NH, Schimenti JC, Ross FP, van der Meulen MC. Transcriptional profiling of cortical versus cancellous bone from mechanically-loaded murine tibiae reveals differential gene expression. Bone. 2016;86:22–9.
Joeng KS, Lee YC, Lim J, Chen Y, Jiang MM, Munivez E, et al. Osteocyte-specific WNT1 regulates osteoblast function during bone homeostasis. J Clin Invest. 2017;127(7):2678–88.
Lawson L, Brodt M, Silva M. Wnts produced by osteo-lineage cells are required for loading-induced bone formation in mice. JBMR. 2019;32(Suppl 1). https://www.asbmr.org/meetings/annualmeeting/AbstractDetail?aid=f47bcec8-41a2-43ca-b1a9-7334c2f1c43a. Accessed 23 Sep 2019.
Cabahug-Zuckerman P, Frikha-Benayed D, Majeska RJ, Tuthill A, Yakar S, Judex S, et al. Osteocyte apoptosis caused by hindlimb unloading is required to trigger osteocyte RANKL production and subsequent resorption of cortical and trabecular bone in mice femurs. J Bone Miner Res. 2016;31(7):1356–65.
Aguirre JI, Plotkin LI, Stewart SA, Weinstein RS, Parfitt AM, Manolagas SC, et al. Osteocyte apoptosis is induced by weightlessness in mice and precedes osteoclast recruitment and bone loss. J Bone Miner Res. 2006;21(4):605–15.
Kelly T, Suva LJ, Nicks KM, MacLeod V, Sanderson RD. Tumor-derived syndecan-1 mediates distal cross-talk with bone that enhances osteoclastogenesis. J Bone Miner Res. 2010;25(6):1295–304.
Bartolak-Suki E, Imsirovic J, Parameswaran H, Wellman TJ, Martinez N, Allen PG, et al. Fluctuation-driven mechanotransduction regulates mitochondrial-network structure and function. Nat Mater. 2015;14(10):1049–57.
Helle SCJ, Feng Q, Aebersold MJ, Hirt L, Gruter RR, Vahid A, et al. Mechanical force induces mitochondrial fission. Elife. 2017;6. https://doi.org/10.7554/eLife.30292.
Yamamoto K, Imamura H, Ando J. Shear stress augments mitochondrial ATP generation that triggers ATP release and Ca(2+) signaling in vascular endothelial cells. Am J Physiol Heart Circ Physiol. 2018;315(5):H1477–H85.
Genetos DC, Kephart CJ, Zhang Y, Yellowley CE, Donahue HJ. Oscillating fluid flow activation of gap junction hemichannels induces ATP release from MLO-Y4 osteocytes. J Cell Physiol. 2007;212(1):207–14.
Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168(6):960–76.
Liu W, Wang Z, Yang J, Wang Y, Li K, Huang B, et al. Osteocyte TSC1 promotes sclerostin secretion to restrain osteogenesis in mice. Open Biol. 2019;9(5):180262.
Esen E, Chen J, Karner CM, Okunade AL, Patterson BW, Long F. WNT-LRP5 signaling induces Warburg effect through mTORC2 activation during osteoblast differentiation. Cell Metab. 2013;17(5):745–55.
Chen J, Holguin N, Shi Y, Silva MJ, Long F. mTORC2 signaling promotes skeletal growth and bone formation in mice. J Bone Miner Res. 2015;30(2):369–78.
Lewis KJ, Yi X, Wright CS, Pemberton EZ, Bullock WA, Thompson WR, et al. The mTORC2 component Rictor is required for load-induced bone formation in late-stage skeletal cells. JBMR Plus. 2020. https://doi.org/10.1002/jbm4.10366.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Jinhu Xiong, Xuehua Li, and Jacob Kordsmeier declare no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Osteocytes
Rights and permissions
About this article

Cite this article
Li, X., Kordsmeier, J. & Xiong, J. New Advances in Osteocyte Mechanotransduction. Curr Osteoporos Rep 19, 101–106 (2021). https://doi.org/10.1007/s11914-020-00650-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11914-020-00650-y