
Abstract
Purpose of Review
Pituitary tumors are undergoing a transformation in histopathologic and molecular classification, coincident with the continued refinement of increasingly powerful methods of genomic annotation and discovery. We highlight novel genomic alterations identified in pituitary adenomas and craniopharyngiomas and discuss their clinical implications.
Recent Findings
Sporadic pituitary adenomas are associated with relatively few recurrent somatic mutations. Recurrent mutations occur largely in subsets of hormone-producing tumors, including GNAS and GPR101 in somatotroph adenomas and USP8 in corticotroph adenomas. Additionally, they manifest with a dichotomous signature of copy number alterations, ranging from almost none to widespread genome instability, while microduplication of chromosome Xq26.3, containing the GNAS gene, defines X-linked acrogigantism. Papillary craniopharyngiomas are defined by BRAFV600E mutations while β-catenin alterations characterize adamantinomatous craniopharyngiomas.
Summary
Genomic annotation of pituitary tumors is defining increasing subsets of neuroendocrine adenohypophyseal tumors and craniopharyngiomas, offering rationale-based pharmacologic targets and potential biomarkers for clinical outcome.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Tumors of the pituitary region are among the most common of the primary brain tumors. Adenomas of the pituitary, while generally non-cancerous, may still cause significant morbidity, through either elaboration of supraphysiologic levels of hormones, compression and subsequent hypofunction of the pituitary gland, or growth and compression of sensitive adjacent neurologic and vascular anatomy. As common as they are—and for as long as they have been described, which dates to nearly the dawn of modern neurological surgery—the sentinel genetic events which drive their origin have been poorly characterized. In an era where genomic annotation of human tumors is revolutionizing our understanding of biological mechanisms and uncloaking new therapeutic approaches, pituitary tumors are just now becoming defined by their molecular features. This is especially timely as pituitary adenomas are undergoing a reappraisal of their pathologic classification, and additional classifiers and predictors of aggressive behavior are being sought.
We herein review the contemporary understanding of the genomic architecture of pituitary adenomas and craniopharyngiomas, highlighting important clinically relevant alterations where possible.
Pituitary Gland
The pituitary gland serves as a nexus of embryologic tissues and cell types, leading to a variety of tumor lineages. The anterior pituitary gland, or adenohypophysis, derives from Rathke’s pouch, which is formed from an upward invagination of oral ectoderm, while the posterior pituitary gland, or neurohypophysis, derives from neuroectodermal floor of the diencephalon. The anterior pituitary contains five primary hormone-secreting cellular lineages that are distinct in structure and function. These corticotroph, lactotroph, somatotroph, thyrotroph, and gonadotroph cells are topologically arranged and secrete adrenocorticotropic hormone (ACTH), prolactin (PRL), growth hormone (GH), thyroid-stimulating hormone (TSH) and gonadotropins (follicle-stimulating hormone [FSH], and luteinizing hormone [LH]), respectively. Additional populations of adenohypophyseal cells secrete no hormones or more than one hormone from the same cell. The posterior pituitary stores and secretes two hormones, vasopressin and oxytocin, that are produced by the supraoptic and paraventricular nuclei of the hypothalamus.
Tumors of the Anterior Pituitary
Classification
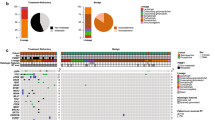
Tumors of the pituitary gland are undergoing a transformation in classification, as codified by updates in the 2017 World Health Organization (WHO) scheme [1•, 2, 3]. Tumors of the adenohypophysis, which account for the majority of pituitary neoplasms, have been traditionally classified by their immunohistochemical hormone profile, histopathologic features, and ultrastructural features. Additionally, renewed focus on the role of transcription factors (TF) in defining adenohypophyseal cell lineage has been incorporated into the formal classification criteria (Fig. 1).

Significant transcription factor expression and hormone staining in adenoma classes and subclasses. ACTH adrenocorticotropic hormone, ERα estrogen receptor α, GH growth hormone, β-LH luteinizing hormone, PIT-1 pituitary-specific POU-class homeodomain transcription factor, PRL prolactin, SF-1 steroidogenic factor 1, TF transcription factor, T-PIT T-box family member TBX19, β-TSH thyroid-stimulating hormone
Specifically, the adenohypophyseal progenitor cell differentiates into three lineages: acidophilic, corticotroph, and gonadotroph. The acidophilic lineage further differentiates into somatotroph, lactotroph, and thyrotroph cells, marked by expression of additional TFs and hormones. The diagnosis of cell lineage-associated adenoma subtypes requires a combination of hormone and transcription factor expression, while a null cell adenoma is defined by absence of adenohypophyseal hormones and transcription factors (Fig. 1).
Within the codified neuroendocrine adenomas, the WHO highlighted five subtypes that have demonstrated increased propensity for clinical aggressiveness in terms of rapid growth, early recurrence, resistance to treatment, and increased proliferative index. These are the sparsely granulated somatotroph adenoma, silent corticotroph adenoma, lactotroph adenoma in men, plurihormonal PIT-1 positive adenoma, and Crooke’s cell adenoma (Table 1).
Sparsely granulated somatotroph adenomas express GH and pituitary-specific POU-class homeodomain transcription factor (PIT-1). The tumors are reactive to cytokeratin staining, which distinguishes them from other somatotroph morphological variants, although consistent interpretation of staining patterns can be challenging in practice at times. Both silent corticotroph and Crooke’s cell adenoma express ACTH and the T-box family member TBX19 transcription factor (T-PIT). Silent corticotrophs are considered non-functional, with absence of clinically apparent sequelae from hormone overproduction, and have variable immunohistochemical expression of ACTH. Crooke’s cell adenomas contain a preponderance of cells with Crooke’s hyaline change, which demonstrates perinuclear reactivity to cytokeratin in a ring-like shape. Plurihormonal PIT-1 positive adenomas express GH, PRL, β-TSH, and PIT-1. These tumors were formally known as silent subtype 3 adenomas but were reclassified because of varying hormone secretion and clinical functional status observed in patients with this tumor.
Additionally, appreciation for variability in the clinical behavior of pituitary tumors has led to a proposal to shift the terminology from adenoma, which implies a benign nature of the lesion, to tumor, and cessation of the “atypical” designation, given its inability to accurately and consistently predict aggressive clinical behavior [1•, 2, 3]. Assessment of tumor proliferation and tumor invasiveness remains recommended for individual cases of clinically aggressiveness tumors. However, more objective markers for tumor behavior and clinical outcome remain to be defined for pituitary adenomas.
Mutations
Given the inconsistent correlation of traditional histopathologic classifications with the natural history of pituitary tumors, increased efforts have centered upon understanding the genetic and cellular origins of pituitary tumors. Hereditary syndromes, including multiple endocrine neoplasia type I (MEN1), MEN4, familial isolated pituitary adenomas (FIPA), McCune-Albright syndrome, and Carney complex, have long suggested a link between discrete genetic causes and pituitary tumors. In addition to germline mutations in AIP [4, 5], MEN1 [6, 7], CDKN1B [8, 9], and PRKAR1A, which have been associated with these familial syndromes, next-generation sequencing of pituitary adenomas has identified several somatic genomic alterations associated with specific tumor subtypes [10, 11•, 12•, 13•].
Observation of recurrent mutations in the catalytic subunit of protein kinase A in cortisol-producing adenomas of the adrenal cortex prompted application of a similar whole-exome sequencing strategy for Cushing’s disease, with identification of mutations in the deubiquitinase USP8 in a third to two-thirds of pituitary corticotroph adenomas [11•, 12•]. USP8 contributes to lysosomal trafficking of ligand-activated epidermal growth factor receptor (EGFR). Gain-of-function mutations in USP8 inhibit EGFR endocytosis, leading to increased EGFR stimulation of Pomc gene transcription and increased ACTH secretion [12•, 14]. Interestingly, while USP8 is exclusively localized to the nucleus in mutant adenomas, non-mutant tumors can manifest with either cytoplasmic or nuclear expression, suggestive of alternative mechanisms other than mutation leading to subcellular compartmentalization and activation of USP8 [12•]. Furthermore, USP8 mutations have been associated with increased expression of somatostatin reception 5 (SSTR5) and O6-methylguanine DNA methyl-transferase (MGMT) [14]. Collectively, these data support a therapeutic role for EGFR inhibition and somatostatin analogues in USP8-mutated corticotroph adenomas.
In somatotroph adenomas, recurrent somatic mutations in the gene encoding the stimulatory G-protein α subunit, GNAS, are identified in a third of tumors [15,16,17,18]. This is concordant with the causative role of GNAS mutations in McCune-Albright syndrome, which is associated with acromegaly, and leads to constitutive activation of the cyclic AMP (cAMP) mitogenic pathway. Other mutations in scattered genes involved in cAMP signaling have also been reported, although not at a significantly recurrent incidence [18].
Additionally, a recurrent somatic mutation in the G-protein coupled receptor, GPR101E308D, has been reported in approximately 4.4% of acromegaly patients [13•]. Introduction of this mutation into a rat GH-secreting cell line produced increased proliferation and GH secretion in vitro. GPR101 resides on chromosome Xq26.3, and microduplication of this locus is implicated as the cause of X-linked acrogigantism in children [13•].
On the whole, recurrent mutations are sparse across the majority of sporadic pituitary adenomas, including the most common subtype, null cell adenomas [10]. This suggests alternative biological mechanisms that contribute to pituitary tumorigenesis, including copy number alterations, rearrangements, and epigenetic changes [10, 19•, 20].
Chromosomal Alterations
Beyond somatic and germline mutations associated with pituitary adenomas, genomic profiling points to two distinct classes of pituitary adenomas based on their level of copy number alterations [19•]. One class harbors almost no chromosomal alterations, while the other is marked by widespread genomic disruption (Fig. 2). Compared to most other tumor types, focal gains and losses are rare relative to broad arm-level chromosomal alterations in pituitary adenomas, the significance of which remains to be elucidated [19•].

Schematic of two genomic categories of pituitary adenomas, stratified by the copy number alteration burden. a One class harbors almost no chromosomal alterations, while b the other is marked by widespread copy number gains and losses in recurrently observed chromosomes. Red indicates amplification, navy blue indicates high frequency of loss, pale blue indicates lower frequency of loss, and gray indicates neutral copy number status
While the genomically quiet cohort is enriched for null cell adenomas and the genomically disrupted cohort has a relatively higher fraction of hormone-expressing adenomas, this association is not consistent [21]. Silent corticotroph adenoma and prolactinoma express a particularly high frequency of genome disruption in small series [21]. However, the threshold for and incidence of genome disruption in adenomas merit closer inspection in larger cohorts to better define possible associations with high-risk adenoma subtypes and clinical outcome [10, 19•, 21].
Mutation Signatures of Craniopharyngioma
The molecular landscape of the craniopharyngioma, like pituitary adenoma, has also been subjected to genomic profiling—with some of the findings bearing immediate therapeutic relevance. Craniopharyngiomas, arising from the pituitary stalk, are among the most challenging of intracranial tumors to manage because of their pattern of growth, associated morbidities, and high recurrence rate. Radiotherapy and chemotherapy provide adjuvant treatment options, but many cases remain refractory despite aggressive treatment and re-treatment. Of the two histological subtypes, adamantinomatous craniopharyngiomas are the most common overall, with almost exclusive representation among pediatric cases, while papillary craniopharyngiomas are predominantly observed in adults.
Strikingly, the BRAFV600E mutations were identified in 81–95% of papillary craniopharyngiomas, illuminating a powerful target for pharmacotherapy [22•, 23]. BRAFV600E mutation leads to overactivity of B-Raf signaling and subsequent mitogenic sequelae, an oncogenic strategy usurped by multiple tumors, including melanoma, where BRAF inhibition has led to promising extension of survival [24,25,26]. This has translated to successful administration of combined BRAF and MEK inhibitors in two patients with multiple recurrent papillary craniopharyngioma [27, 28].
In comparison, the adamantinomatous subtype is characterized by near ubiquitous mutations in the β-catenin gene, CTNNB1, and also demonstrates disruptions in multiple growth factor signaling axes, extracellular matrix regulation, and its immune microenvironment [22•, 23]. Interestingly, co-occurrence of the BRAFV600E mutation and CTNNB1 mutation in two cases of adamantinomatous craniopharyngioma has been reported, suggestive of a potential shared phylogeny in the pathogenesis of select craniopharyngiomas [23].
Conclusions and Future Directions
Genomic characterization of tumor of the pituitary region—pituitary adenomas and craniopharyngiomas in particular—has shed new insights into pharmacologic mutational targets as well as unique copy number alterations. As molecular profiling becomes incorporated into routine clinical practice, elucidation of the epidemiologic associations and prognostic consequences of genomic and epigenomic signatures may yield new insights into the ontology of tumors in this area, the relationship of specific genomic profiles to growth and recurrence, and the prospect of tailored management strategies for patients as the scope of therapeutic targets broadens.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
• Lopes MBS. The 2017 World Health Organization classification of tumors of the pituitary gland: a summary. Acta Neuropathol. 2017;134(4):521–35. https://doi.org/10.1007/s00401-017-1769-8. Summary of updated WHO pituitary tumor classification, with elimination of prior atypical category, and codification of “high risk” adenoma subtypes.
Mete O, Lopes MB. Overview of the 2017 WHO classification of pituitary tumors. Endocr Pathol. 2017;28(3):228–243.
Asa SL, Casar-Borota O, Chanson P, Delgrange E, Earls P, Ezzat S, et al. From pituitary adenoma to pituitary neuroendocrine tumor (PitNET): an International Pituitary Pathology Club proposal. Endocr Relat Cancer. 2017;24(4):C5–8. https://doi.org/10.1530/ERC-17-0004.
Vierimaa O, Georgitsi M, Lehtonen R, Vahteristo P, Kokko A, Raitila A, et al. Pituitary adenoma predisposition caused by germline mutations in the AIP gene. Science. 2006;312(5777):1228–30. https://doi.org/10.1126/science.1126100.
Daly AF, Vanbellinghen JF, Khoo SK, Jaffrain-Rea ML, Naves LA, Guitelman MA, et al. Aryl hydrocarbon receptor-interacting protein gene mutations in familial isolated pituitary adenomas: analysis in 73 families. J Clin Endocrinol Metab. 2007;92(5):1891–6. https://doi.org/10.1210/jc.2006-2513.
Marx SJ. Molecular genetics of multiple endocrine neoplasia types 1 and 2. Nat Rev Cancer. 2005;5(5):367–75. https://doi.org/10.1038/nrc1610.
Wu X, Hua X. Menin, histone h3 methyltransferases, and regulation of cell proliferation: current knowledge and perspective. Curr Mol Med. 2008;8(8):805–15. https://doi.org/10.2174/156652408786733702.
Tichomirowa MA, Lee M, Barlier A, Daly AF, Marinoni I, Jaffrain-Rea ML, et al. Cyclin-dependent kinase inhibitor 1B (CDKN1B) gene variants in AIP mutation-negative familial isolated pituitary adenoma kindreds. Endocr Relat Cancer. 2012;19(3):233–41. https://doi.org/10.1530/ERC-11-0362.
Occhi G, Regazzo D, Trivellin G, Boaretto F, Ciato D, Bobisse S, et al. A novel mutation in the upstream open reading frame of the CDKN1B gene causes a MEN4 phenotype. PLoS Genet. 2013;9(3):e1003350. https://doi.org/10.1371/journal.pgen.1003350.
Song ZJ, Reitman ZJ, Ma ZY, Chen JH, Zhang QL, Shou XF, et al. The genome-wide mutational landscape of pituitary adenomas. Cell Res. 2016;26(11):1255–1259.
• Ma ZY, Song ZJ, Chen JH, Wang YF, Li SQ, Zhou LF, et al. Recurrent gain-of-function USP8 mutations in Cushing’s disease. Cell Res. 2015;25(3):306–17. https://doi.org/10.1038/cr.2015.20. Discovery of novel oncogenic driver mutation underlying a subset of Cushing’s disease, as a possible therapeutic target.
• Reincke M, Sbiera S, Hayakawa A, Theodoropoulou M, Osswald A, Beuschlein F, et al. Mutations in the deubiquitinase gene USP8 cause Cushing’s disease. Nat Genet. 2015;47(1):31–8. https://doi.org/10.1038/ng.3166. Discovery of novel oncogenic driver mutation underlying a subset of Cushing’s disease, with elaboration of mechanism.
• Trivellin G, Daly AF, Faucz FR, Yuan B, Rostomyan L, Larco DO, et al. Gigantism and acromegaly due to Xq26 microduplications and GPR101 mutation. N Engl J Med. 2014;371(25):2363–74. https://doi.org/10.1056/NEJMoa1408028. Report of novel mutation responsible for a subset of acromegaly patients and report of new pediatric syndrome.
Hayashi Y, Mizumoto M, Akutsu H, Takano S, Matsumura A, Okumura T, et al. Hyperfractionated high-dose proton beam radiotherapy for clival chordomas after surgical removal. Br J Radiol. 2016;89(1063):20151051. https://doi.org/10.1259/bjr.20151051.
Vallar L, Spada A, Giannattasio G. Altered Gs and adenylate cyclase activity in human GH-secreting pituitary adenomas. Nature. 1987;330(6148):566–8. https://doi.org/10.1038/330566a0.
Landis CA, Masters SB, Spada A, Pace AM, Bourne HR, Vallar L. GTPase inhibiting mutations activate the alpha chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature. 1989;340(6236):692–6. https://doi.org/10.1038/340692a0.
Clementi E, Malgaretti N, Meldolesi J, Taramelli R. A new constitutively activating mutation of the Gs protein alpha subunit-gsp oncogene is found in human pituitary tumours. Oncogene. 1990;5(7):1059–61.
Ronchi CL, Peverelli E, Herterich S, Weigand I, Mantovani G, Schwarzmayr T, et al. Landscape of somatic mutations in sporadic GH-secreting pituitary adenomas. Eur J Endocrinol. 2016;174(3):363–72. https://doi.org/10.1530/EJE-15-1064.
• Bi WL, Horowtiz P, Greenwald N, Abedalthagafi M, Agarwalla PK, Gibson WJ, et al. Landscape of genomic alterations in pituitary adenomas. Clin Cancer Res. 2016;23(7):1841–51. https://doi.org/10.1158/1078-0432.CCR-16-0790. This study highlights a dichotomous pattern among pituitary adenomas, as stratified by the presence or absence of genome disruption.
Yacqub-Usman K, Richardson A, Duong CV, Clayton RN, Farrell WE. The pituitary tumour epigenome: aberrations and prospects for targeted therapy. Nat Rev Endocrinol. 2012;8(8):486–94. https://doi.org/10.1038/nrendo.2012.54.
Bi WL, Greenwald NF, Ramkissoon SH, Abedalthagafi M, Coy SM, Ligon KL, et al. Clinical identification of oncogenic drivers and copy number alterations in pituitary tumors. Endocrinology. 2017;158(7):2284–2291.
• Brastianos PK, Taylor-Weiner A, Manley PE, Jones RT, Dias-Santagata D, Thorner AR, et al. Exome sequencing identifies BRAF mutations in papillary craniopharyngiomas. Nat Genet. 2014;46(2):161–5. https://doi.org/10.1038/ng.2868. Discovery of targetable oncogenic mutation in majority of papillary craniopharyngiomas.
Larkin SJ, Preda V, Karavitaki N, Grossman A, Ansorge O. BRAF V600E mutations are characteristic for papillary craniopharyngioma and may coexist with CTNNB1-mutated adamantinomatous craniopharyngioma. Acta Neuropathol. 2014;127(6):927–9. https://doi.org/10.1007/s00401-014-1270-6.
Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363(9):809–19. https://doi.org/10.1056/NEJMoa1002011.
Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367(18):1694–703. https://doi.org/10.1056/NEJMoa1210093.
Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366(8):707–14. https://doi.org/10.1056/NEJMoa1112302.
Brastianos PK, Shankar GM, Gill CM, Taylor-Weiner A, Nayyar N, Panka DJ, et al. Dramatic response of BRAF V600E mutant papillary craniopharyngioma to targeted therapy. J Natl Cancer Inst. 2015;108(2).
Aylwin SJ, Bodi I, Beaney R. Pronounced response of papillary craniopharyngioma to treatment with vemurafenib, a BRAF inhibitor. Pituitary. 2016;19(5):544–6. https://doi.org/10.1007/s11102-015-0663-4.
Acknowledgments
The authors express gratitude to Winona W. Wu for illustration assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Wenya Linda Bi, Alexandra Giantini Larsen, and Ian F. Dunn declare no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Neuro-Oncology
Rights and permissions
About this article

Cite this article
Bi, W.L., Larsen, A.G. & Dunn, I.F. Genomic Alterations in Sporadic Pituitary Tumors. Curr Neurol Neurosci Rep 18, 4 (2018). https://doi.org/10.1007/s11910-018-0811-0
Published:
DOI: https://doi.org/10.1007/s11910-018-0811-0