
Abstract
The molecular mechanism of neuronal loss and synaptic damage in Alzheimer’s disease (AD), Parkinson’s disease dementia (PDD), frontotemporal dementia (FTD) and Lewy body dementia (LBD) is poorly understood and could differ among different types of neurodegenerative processes. However, the presence of neuroinflammation is a common feature of dementia. In this setting, reactive microgliosis, oxidative damage and mitochondrial dysfunction are associated with the pathogenesis of all types of neurodegenerative dementia. Moreover, an increased body of evidence suggests that microglia may play a central role in AD progression. In this paper, we review the scientific literature on neuroinflammation related to the most common neurodegenerative dementias (AD, PDD, FTD and LBD) focussing on the possible molecular mechanisms and the available clinical evidence. Furthermore, we discuss the neuroimaging techniques that are currently used for the study of neuroinflammation in human brain.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
It was estimated that 35.6 million people lived with dementia worldwide in 2010, with numbers expected to almost double every 20 years, to 65.7 million in 2030 and 115.4 million in 2050 [1]. Apart from Alzheimer’s disease (AD), which accounts for 60 % of dementia, other causes of neurodegenerative dementia are Parkinson’s disease dementia (PDD), frontotemporal dementia (FTD) and Lewy body dementia (LBD) [2]. Mild cognitive impairment (MCI) can represent a transitional stage between health and AD and was originally defined by the Petersen criteria [3]. The estimated prevalence of MCI in older adults is around 7–20 % depending on the population studied [4]. Despite the large number of people affected by these diseases, the molecular mechanisms accounting for neuronal loss and synaptic damage in AD, PDD, FTD and LBD are still poorly understood. Neuroinflammation in the form of glial activation is a component of all these diseases, and oxidative damage, mitochondrial dysfunction and reactive microgliosis are all factors in the pathogenesis and progression of the neurodegenerative dementias [5–7].
Postmortem studies in neurodegenerative dementias have shown activated microglia targeting different areas of the brain depending on the syndrome. The exact role of inflammation in the pathological process of neurodegeneration has led to both primary and secondary hypotheses in neurodegenerative diseases [8]. Some of the first evidence was shown by McGeer et al., who detected large numbers of human leukocyte antigen DR (HLA-DR) positive reactive microglia along with Lewy bodies in the substantia nigra of all cases studied with Parkinson’s disease. They also found that dementia cases with a pre-mortem diagnosis of AD or PD showed large numbers of HLA-DR-positive reactive microglia and significant plaque and tangle counts in the hippocampus, with reduced cortical choline acetyltransferase activity [8]. Although it was previously thought that the central nervous system (CNS) was an immune-privileged site, it is now well established that inflammatory processes occur in the CNS in response to injury, infection or neurodegenerative disease [9]. It has also become evident that glial cells can be activated by the presence of systemic inflammation and that this can act as a trigger for clinical deterioration of Alzheimer’s and Parkinson’s diseases [9].
All current evidence suggests that neuroinflammation is a significant component of neurodegenerative disease development. One hypothesis of its role is that the initial pathogenic insult (e.g. aberrant protein aggregation) induces an ongoing inflammatory/cytotoxic response and the secondary neuroinflammation is a diffuse process, leading to alterations in neuronal function with a predilection for selected regions depending on the nature of the neurodegenerative disease associated with the dementia [10–12]. Growing preclinical and clinical evidence supports this hypothesis, and studies suggest that systemic inflammation can worsen, or possibly trigger, neurodegenerative diseases as cytokines cross the blood–brain barrier. In this paper, we reviewed the scientific English literature on neuroinflammation and dementia related to most common neurodegenerative diseases (AD, PDD, FTD and LBD) focussing on possible molecular mechanisms and clinical evidence. Moreover, we discuss the neuroimaging techniques that are currently available or under investigation for the study of neuroinflammation in humans.
Neuroinflammation in AD, PDD, FTD and LBD Patients
The term “neuroinflammation” as used here refers to the intrinsic cellular response in the central nervous system associated with cell neurodegeneration. Microglial cells and astrocytes are mainly involved in the inflammatory response in the CNS [13]. Microglial cells are the resident macrophages of the CNS; they represent around the 10–12 % of the CNS population [14] and play a crucial role not only in neurogenesis, neuronal plasticity and regeneration but also serve as a first line of immune defence in any type of brain insult. They have the unique capability to phagocytose toxic products, release cytotoxic factors and behave as antigen presenting cells [13, 15]. In the absence of foreign stimuli, microglial cells are in a “resting” state but have spidery processes constantly scanning the local environment for changes in brain milieu without interfering with neurons and neuronal function,[15]. When activated by any insult to the brain, they undergo morphological changes, with the ramified processes becoming ameoboid [16], and may move toward the site of injury [15]. The cells can stay in an activated phase for weeks after an acute insult, releasing cytokines and neurotoxic agents that further exacerbate CNS damage [17]. This cidal activation pattern of behaviour is generally exhibited by microglia expressing an M1 or classical phenotype. However, activated microglial can also behave in a restorative fashion as an M2, or alternatively activated phenotype, releasing growth factors and remodelling synapses [18]. In cell culture, microglia develop an M1 phenotype in the presence of lipopolysaccharide (LPS) and/or interferon (IFN) and tumor necrosis factor (TNF) and produce a massive inflammatory response releasing interleukin-1β (IL-1β), IL-12, TNF-α and inducible nitric oxide synthase (iNOS). The M2 phenotype is generated in cell cultures in the presence of IL-4 and IL-13 and has an anti-inflammatory profile. The M2 phenotype can also exist as subtypes—M2b is stimulated by immune complex formation and toll-like receptor TLR or IL-1β activation; M2c represents deactivated macrophages, which contribute to the suppression of pro-inflammatory cytokines [19•, 20]. The switch of macrophages between M1 and M2 states is a dynamic process in peripheral inflammation [20] and, although peripheral macrophages and microglia have differences, it is possible to adopt a similar classification [7]. Microglial activation can switch from M2 to M1 phenotype during the course of disease [7]; in the hippocampus of aged rodents, Jimenez et al. demonstrated a distinctive shift from M2 to M1 phenotype of microglial activation [7, 21] .
Tanaka et al. [22] have demonstrated that sub-acute administration of LPS activated microglia and increased production of IL-1β and TNF-α, resulting in learning and memory deficits in the animals. This did not occur in IL-1 knockout [23] mice proving that IL-1 plays an important role in microglial LPS-induced impairment of learning and memory [22, 24]. Given that, in microglia cells, amyloid activation of the NOD-like receptor family, pyrin domain containing 3 (NLRP3) inflammasome is a crucial event for IL-1β production and subsequent neuroinflammatory response, Heneka et al. [25] evaluated active caspase-1 expression in human MCI and AD patients, showing a strongly enhanced activity in neurodegeneration. Indeed, they found that NLRP3 (−/−) or Casp1 (−/−) mice carrying mutations associated with familial AD were largely protected from loss of spatial memory and other AD-related features. Moreover, NLRP3 inflammasome deficiency skewed microglial cells to M2 phenotype, resulting in the decreased deposition of amyloid in the APP/PS1 model of AD. Overall, these data suggest an important role of NLRP3/caspase-1 axis in the pathogenesis of AD and indicate new possible therapeutic interventions for the disease [25]. Interestingly, in neurodegenerative diseases, activated microglial cells seem to increase the release of membrane-type 1 (metalloproteinase MMP-1) in the interstitial space. Some evidence suggests that microglial/macrophage MMP-14 expression is upregulated in AD tissue and also in mouse models of human AD [26]. It has also been shown that, in animals with prion disease, microglial cells switch from an anti-inflammatory phenotype to the pro-inflammatory aggressive phenotype when stimulated with LPS, starting an inflammatory response characterised by the increasing of IL-1β, TNF and IL-6 and leading to behavioural changes, neuronal death and more rapid progression of the disease [27, 28]. On this basis, Lunnon et al. [29] studied the molecular mechanisms underlying the microglial phenotype switching, concluding that, during chronic neurodegeneration, microglia show an increased expression levels of activating FcγR and a lower signalling threshold for Ab-mediated cell activation.
In chronic neuroinflammation, microglial cells can remain activated for extended periods, releasing quantities of cytokines and neurotoxic molecules that may contribute to long-term neurodegeneration [30]. In AD, activated microglia surround amyloid plaques, and resting microglia are activated by amyloid oligomers, fibrils and amyloid precursor protein (APP) [31, 32]. Interestingly, knockout mice without the APP gene show decreased microglial activation [33]. In PDD or LBD, microglial cells can be activated by α-synuclein fibrils [34]. Moreover, alterations in inflammatory cells/molecules before the aggregation of tau in several different mouse models of FTD have been observed [35].
Neuroinflammation results in synaptic impairment and neuronal death and contributes to neurodegeneration within the brain [19•]. In this regard, specific markers of neuroinflammation have been found in CNS areas of AD-affected patients, while subjects with high plaque burden without dementia show little evidence of neuroinflammation [36]. In neurodegenerative disorders such as AD, PD, Huntington’s disease and multiple sclerosis, inflammation plays a crucial role, substantiated by high levels of pro-inflammatory cytokines [13, 31, 32, 37]. Wright et al. [38] found that loss of neurons from the hippocampal CA1 region begins as early as 12 weeks of age in hAPP-J20 AD mice and that the extent of neuronal loss increases with age and correlates with the number of activated microglia. In a PD model, it has been demonstrated that chronic stress enhances susceptibility to inflammation in the substantia nigra. Indeed, the induction of inflammatory process in stressed rats resulted in higher inflammatory response and was associated with higher rate of death of dopaminergic neurons in the substantia nigra, suggesting that stress may be an important risk factor in the degenerative processes and symptoms of PD [39].
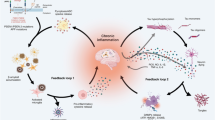
At a molecular level, separate from classical pathways involved in inflammation such as cyclooxygenase (COX), several intracellular pathways have been discovered in the macrophage activation processes and which are also relevant to microglial cells [40]. One of the most important pathways is related to p38 mitogen-activated protein kinase (MAPK) and was observed by Hensley et al. [41]. Activation of the p38 MAPK pathway has been observed at different stages of AD pathogenesis, which is related to microglial activation and inflammatory mediator production [41, 42]. Another pathway potentially involved in neuroinflammation involves peroxisome proliferator-activated receptor gamma (PPAR-γ), a nuclear receptor protein that functions as a transcriptional factor regulating gene expression involved in energy metabolism, adipocyte differentiation, insulin sensitisation and tumour suppression. Interestingly, it has been found that PPAR-γ agonists interfere with inflammatory gene expression and microglia-mediated inflammatory responses in the brain [43]. Recently, the insulin signalling pathway has been implicated in control of neuronal excitability and metabolism and is altered in microglial cells of mouse models of AD. Glucagon-like peptide-1 (GLP-1), an insulinotropic hormone, has similar functions and growth-like properties as insulin/insulin-like growth factor. Alterated function of these pathways can contribute to the progressive loss of neurons in Alzheimer’s disease and Parkinson’s disease [44]. GLP-1 analogues reduce microglial activation in transgenic mice, suggesting a possible role for them as neuroprotectants. A preclinical study on the interrelation between AD and diabetes using a high-fat diet (HFD) in a mouse model of genetically induced AD-like neuropathology (3xTg-AD) showed that that cerebral expression of human AD transgenes led to peripheral glucose intolerance, associated with pancreatic human amyloid accumulation. Moreover, HFD enhanced glucose intolerance, increased brain inflammation, brain soluble amyloid and memory impairment in 3xTg-AD mice. Surprisingly, a single insulin injection reversed the deleterious effects of HFD on memory and soluble amyloid levels. These findings, taken together, strongly support a hypothesis of a common relationship between peripheral metabolic disturbances such as glucose intolerance and an increased risk of neurodegeneration mediated through inflammatory processes [45] Figure 1 details the underlying common mechanism of neurodegenerative diseases.

The common process of neurodegenerative diseases in Alzheimer’s disease (AD), Parkinson’s disease dementia (PDD), and fronto-tempotal dementia (FTD). The inflammatory signals induce microglia to an activated state leading to morphological changes and secretion of pro-inflammatory factors [i.e. interleukin 1, 6 (IL-1–6) and tumor necrosis factor (TNF)], which in turn maintain the inflammatory status and favour the production of reactive oxygen species (ROS). This inflammatory cycle may trigger neurofibrillary tangle (NFT) deposition and/or neuronal death
Preclinical and Pathological Evidence of Neuroinflammation in Neurodegenerative Diseases
Alzheimer’s Disease
An increasing body of evidence suggests that IL-1 released by inflammatory cells into the CNS is an important driving force in the transformation of diffuse amyloid deposits into neuritic amyloid plaques as well as in the spread of these plaques and neuronal degeneration across regions of cerebral cortex in patients with AD [12]. In order to understand the role of IL-1β in AD pathogenesis, Ghosh et al. [46] used an inducible model of sustained IL-1β overexpression and a transgenic mouse model of AD, which develops amyloid deposition and tau phosphorylation. They observed a marked increase in tau phosphorylation and a reduction in amyloid burden along with a huge increase in plaque-associated microglia and evidence of microglial activation at the site of inflammation. They have also demonstrated that increased p38 MAPK and glycogen synthase kinase-3β activity contribute to tau phosphorylation. Interestingly, this finding suggests that neuroinflammation may act in opposite manner in the amyloid and tau pathology associated with mouse transgenic models [46]. TNF-α has also been proposed to contribute to the activation of microglial cells and thus to the onset and progression of AD. Detrait et al. [47] showed that systemic administration of an anti-TNF medication counteracts amyloid-induced memory impairment and normalises increased TNF-α levels in the hippocampus in a non-transgenic mouse model. Moreover, Sun et al. [48] have shown that TNF-α converting enzyme (TACE/ADAM-17) in the cerebrospinal fluid (CSF) of subjects with MCI and in AD patients is significantly higher than that of cognitively healthy controls. They also found a significant correlation between plasma TACE activity and CSF t-tau and Aβ42 levels and CSF Aβ42/tau ratios in AD patients. The levels of plasma TACE activity correlated significantly and negatively with cognition. These findings strongly support a role of TNF-α in AD-related neuroinflammation and the possible use of TNF-α blockers as a protectant from the disease [48]. There is some evidence suggesting that endogenous anti-inflammatory pathways are involved in Aβ-induced inflammation. Interestingly, Woodling et al. showed that signalling through the prostaglandin-E2 (PGE2) EP4 receptor reduces microglial inflammatory responses to Aβ42 peptides. In particular, they documented that, in cultured microglial cells, EP4 stimulation attenuated levels of Aβ42-induced inflammatory factors and potentiated phagocytosis of Aβ42, while deletion of microglial EP4 in APPSwe-PS1ΔE9 (APP-PS1) mice increased inflammatory gene expression, oxidative protein modification and Aβ deposition in the brain at early stages of pathology, but not at later stages. In this regard, EP4 receptor levels decreased significantly in human cortex with progression from normal to AD states. These findings suggest that an early loss of anti-inflammatory signalling system in AD development may contribute to subsequent progression of pathology [49].
Parkinson’s Disease Dementia
In a post-mortem study, Imamura et al. [50] showed that major histocompatibility complex (MHC) class II-positive activated microglia are widely distributed in the affected regions, frequently in association with α-synuclein and monoaminergic neuritis in PD brains. PDD is characterised at a molecular level by abnormal aggregation of α-synuclein into intra-neuronal fibrils [8]. The immune system plays a significant role in the pathophysiology of PDD as evidenced by the activation of both innate and adaptive immunity [51]. In human post-mortem studies and in animal models of PD or PDD, α-synuclein aggregation is associated with inflammation and immune response including reactive microgliosis, increased pro-inflammatory cytokine expression, lymphocyte infiltration [52] and immunoglobulin deposition [53]. In PDD, α-synuclein is not only present in neuronal cells, but it can be released from them and transferred to neighbouring glial cells. Lee et al. [54] demonstrated that neuron-released α-synuclein can be transferred into astrocytes using both tissue cultures and transgenic mouse models, thus explaining how astrocytes of patients with PD and dementia with Lewy bodies, which normally express low levels of α-synuclein, present α-synuclein deposits [55]. Interestingly, a microarray study showed that astrocytes, after the exposition of neuron-released α-synuclein, increased the expression of specific genes that are involved in pro-inflammatory responses, such as cytokines and chemokines [54], which in turn could act as mediators of more extensive inflammation with the recruitment and activation of microglial cells [56]. It has been shown that MHCII has a central role for the activation of both the innate and adaptive immune responses to α-synuclein. Using an in vivo mouse model, induced by viral overexpression of α-synuclein and in vitro systems to study the role of the MHCII complex in α-synuclein-induced neuroinflammation and neurodegeneration, Harms et al. [53] demonstrated that overexpression of full-length human α-synuclein induces MHCII expression in microglia, activation of antigen processing and presentation of antigen leading to CD4 T cell proliferation and subsequent cytokine release. This study also demonstrated that MHCII knockout mice prevented α-synuclein-induced microglial activation, IgG deposition and the degeneration of dopaminergic neurons [53].
Frontotemporal Dementia
Progranulin (PGRN) is 70-kDa protein involved in biological processes such as inflammation and wound healing [57], which acts as a TNF regulator in inflammation [58] and also as a growth factor. Interestingly, in humans, mutations of the PGRN gene are causally linked to FTD [59]. However, how loss of PGRN function targets frontotemporal neurodegeneration is still poorly understood. Interestingly, when mice lacking PGRN (Gr−/−) are treated with 1-methyl-4-(2′-methylphenyl)-1,2,3,6-tetrahydrophine (MPTP), they present greater neuron loss and increased microgliosis compared to controls. The marked neuronal loss in PGRN (Gr−/−) mice seems not to be due to selective vulnerability of neurons to MPTP, but rather to an increased microglial activation response, thus suggesting that PGRN deficiency leads to neuroinflammation and neuronal loss via microglia activation [60]. Numerous mutations of the tau gene in chromosome 17 have been linked to FTD [61]. Changes in CNS of a patient affected by FTD tau P301S mutation are consistent with a strong neuroinflammatory reaction. In this setting, activated microglia expressing MHCII receptors were detected in the cortex and hippocampus. In addition, IL-1β and COX-2 expression were induced in neuronal and glial cells of these patients [62].
Evidence from Genetic Studies
Recent genome-wide association studies (GWAS) have identified significant correlations between components of the innate immune system and the incidence of sporadic AD, supporting a link between the immune system (neuroinflammation) and dementia pathophysiology [63, 64]. In this setting, the identification of a novel variant in the gene encoding the triggering receptor expressed on myeloid cells 2 (TREM2) in AD patients reinforces the hypothesis of the causative link between inflammatory cells and degeneration. This is substantiated by a recent study from Jiang et al. [65] who demonstrated that TREM2 levels in the brains of 3- month old senescence-accelerated mice were increased during the aging process. By knocking down TREM2 expression in brains of the same mice by non-viral RNA interference, they found a significant increase in pro-inflammatory cytokines including TNF-α and IL-6, associated with a reduction in IL-10. Neuronal and synaptic losses along with cognitive impairment have also been observed [65]. In humans, it has been found not only that variants in TREM2 triple the risk of developing late-onset AD but also that its mutation is associated with an increased risk of developing PD and PDD [66–68]. TREM2 is expressed on microglial cells and acts as stimulator of phagocytosis on the one hand and as suppressor of cytokine production and inflammation on the other hand [66–68]. Recent GWAS studies were able to identify several other genetic risk factors: CLU, PICALM and CR1 [69]. Additionally several other loci have been demonstrated including BIN1, EPHA1, MS4A, CD33, CD2AP and ABCA7 [70]. Apart from demonstrating increased risk, these findings shed light on the different pathways, which could be involved in the disease pathogenesis. Even though the role of inflammation has been proposed for some time, genetic data suggest a primary role of inflammation in the development of AD, in contrast with the hypothesis that immune and inflammatory related processes are secondary to AD occurrence. The two genes that code for proteins acting as regulators of the complement system (CLU and CR1) are risk factors for the development of the most common form of AD [71]. Clusterin is a lipoprotein expressed in most mammalian tissues, and it interacts with a variety of molecules. It is involved in a number of physiological processes, including inhibition of the complement system. Clusterin is able to modulate the membrane attack complex (MAC) and can act by preventing the inflammatory response associated with complement activation after protein aggregation. CR1 is a polymorphic protein that also acts as a negative regulator of the complement system (inhibiting both the classical and alternative pathways). CR1 on erythrocytes acts as a vehicle for clearance of C3b-coated immune complexes, being involved in immune adherence and phagocytosis. BIN1, members of the MS4A gene family, CD33, ABCA7, CD2AP and EPHA1 may also potentially be related to the immune and inflammatory responses [72]. BIN1 knockout mosaic mice have been reported to show reduced inflammation with aging. MS4A1 (CD20) has been demonstrated to have a function in regulating calcium influx downstream of the activated B-cell antigen receptor [72]. Furthermore, mutations in toll-like receptors (TLRs), as major innate immune mediators, may be have a role in the clearance amyloid-β (Aβ) deposits. The TLR9 signalling pathway is associated with the immune inflammatory response, and it has been associated with the reduction of Aβ burden in AD mice. A recent publication documented that TLR9 polymorphism may modify LOAD risk in the Han Chinese population [73].
Role of Systemic Inflammation in Central Inflammation and Neurodegeneration
Studies in animal models show that systemic inflammation can lead to exaggerated acute symptoms of brain function (sickness syndrome), accelerate neurodegeneration and increase neuronal loss [74, 75]. Interestingly, systemic inflammation generates an exaggerated immune response in the CNS of AD and PD animals that is probably mediated by the local innate immune system with a priming effect [19•]. Likewise, the influence of peripheral inflammation on microglial cells is thought to be present in the human where it might contribute to neurodegenerative processes. Although the blood–brain barrier (BBB) is not permeable to pro-inflammatory mediators, they can enter via the choroid plexus and peripheral inflammation could trigger a neuroinflammatory response in glial cells and neurons stimulating both to release and transmit inflammatory mediators and allow leucocyte migration into the CNS. Several preclinical animal models have been developed for evaluating the effects of peripheral inflammation and brain inflammatory response [76, 77]. Most of them showed a direct link between specific peripheral inflammation/oxidative stress and AD. Some studies focussed on the protective effect of specific cytokines on AD progression [77, 78••].
Other clinical studies have evaluated the relationship between metabolic inflammatory status and cognition [79–81]. Apart from the classical systemic inflammatory diseases, it is well known that some metabolic alterations such as metabolic syndrome or obesity are characterised by an increased release of pro-inflammatory cytokines resulting in a chronic peripheral inflammatory status [79, 81]. In this regard, it has been found that obesity and metabolic alterations could be associated with development of MCI and AD [79–81]. Moreover, other studies showed a link between specific peripheral chronic inflammatory diseases, such periodontitis, and cognitive deficit [82, 83]. Metabolic diseases as well as chronic inflammatory conditions could contribute to the AD neurodegenerative process in different ways inducing both CNS oxidative stress and/or a chronic neuroinflammation (triggered by peripheral inflammation) [68]. Interestingly, rheumatoid arthritis (RA) seems negatively associated with AD development [84].
One possible explanation of the differential effect of peripheral inflammatory processes on central inflammation in CNS in the AD process could be reflected in the function of microglial cells. The CNS cells could be influenced by peripheral inflammatory/anti-inflammatory mediator switching in one or other activated phenotype (M1 or M2) in neurodegenerative diseases [85]. This hypothesis might explain why obese patients, who show a significant release of IL-6 in the portal circulation, may have an increased risk of developing AD. By contrast, the inverse relationship between AD and RA could be explained by the influence of differential inflammatory mediators produced by the chronic joint inflammation. A large meta-analysis of 17 epidemiological studies suggested that non-steroidal anti-inflammatory drugs (NSAIDs) decrease the risk of developing AD [84], although subsequent randomised trials failed to show a beneficial effect of NSAID on established AD [86, 87]. Otherwise, a recent revision of the AD Anti-inflammatory Prevention Trial [88••] supports a beneficial role of NSAID only in the early, asymptomatic, phases of the disease [89], suggesting that neuroinflammation could be crucial in the initial events of the neurodegeneration process.
In PD patients, an increased body of evidence suggests a possible role of peripheral inflammation, especially in gastrointestinal apparatus, on neurodegeneration [90]. In this setting, increased expression of TNF-α, IFN-γ, IL-6 and IL-1β (similar to that observed in inflammatory bowel diseases) has been found [91, 92]. Devos et al. [92] provided evidence that PD enteric inflammation is tightly associated with glial dysregulation showing that the expression of glial markers in colonic biopsies from PD patients are elevated and correlate with the expression of pro-inflammatory cytokines. Several epidemiological studies have shown that NSAIDs are also associated with a reduced risk of developing PD [93, 94].
Evidence for Neuroinflammation in Neurodegenerative Diseases from Neuroimaging Studies
Considering the central role of microglial cells in neurodegeneration, the evaluation of activated microglia in vivo is an important strategy to shed more light into the pathophysiology of neurodegenerative dementia, to evaluate possible treatment targets and also as a marker of disease progression. Quantification and localisation of CNS inflammation may offer a potential tool for evaluating novel therapeutic targets. Positron emission tomography (PET) is the most widely used in vivo method for detecting microglial activation.
Activated microglial cells overexpress mitochondrial translocator protein (TSPO), which is found only at low levels in healthy CNS [95]. In vivo animal studies of neurodegenerative diseases demonstrated that TSPO can also be overexpressed in reactive astrocytes [96]. However, an animal model of focal ischaemia suggested a temporal relationship between TSPO expression in microglia and reactive astrocytes. Some experimental models have shown that TSPO co-localises better with the temporospatial profile of microglial activation than with that of reactive astrocytes [97]. Moreover, a post-mortem study on AD brains showed that TSPO is mainly overexpressed in microglial cells of several areas of the cortex and limbic lobe and is localised within or surrounding senile plaques [98]. Even though there could be a degree of overlap in TSPO expression between activated microglia and reactive astrocytes, TSPO expression and neuroimaging of TSPO using PET is a good marker of neuroinflammation.
The most commonly used PET radioligand to detect TSPO protein in humans is [11C] (R) PK11195 [1-(2-chlorophenyl)-N-methyl-N-(1-methylpropyl)-3-isoquinoline carboxamide. [11C] PK11195 has a half-life of 20 min and has been extensively used to image neurological diseases in humans [99, 100]. [11C] PK11195 PET may detect in vivo microglial activation in the brain of AD mouse models and in AD patients [101, 102]. In the latter, [11C] PK11195 PET studies confirmed that activated microglial cells throughout the association cortex have a distribution pattern similar to that of amyloid plaque [97, 103]. Increased cortical [11C] PK11195 binding can be detected in around 60 % of mild to moderate AD patients and around 40 % of subjects with MCI [103, 104]. Figure 1 demonstrates [11C] PK11195 PET in different types of dementia. Levels of cortical [11C] PK11195 binding show an inverse correlation with mini-mental state examination (MMSE) ratings, suggesting involvement of microglial activation in neuronal dysfunction and cognitive impairment. However, not all studies have detected increased [11C] PK11195 binding in MCI and mild to moderate AD, which may reflect different sensitivities of the cameras and analytical approaches used. A recent [11C] PK11195 PET study found significant binding in the substantia nigra and putamen of PD cases, and those with demenita had higher microglial activation in association cortex compared to non-demented subjects. This finding is consistent with the pathological studies demonstrating involvement of association cortex in PD and LBD [6, 50, 105] (Fig. 2).

[11C]-(R)-PK11195 binding potential of patients with Alzheimer’s disease (AD), mild cognitive impairment (MCI), Parkinson’s disease dementia (PDD) and Parkinson’s disease (PD) compared to control by positron-emission tomography (PET). The presence of diffuse microglial activation in patients with AD, PDD, PD and MCI suggests a common neuroinflammatory pathway for the neurodegenerative diseases
PET studies have also detected increased [11C] PK11195 binding in FTD [104, 106]. A recent study showed that high levels of microglial activation in the temporal lobe of FTD patients might serve as a marker of inherited FTD associated with intronic mutations in MAPT, with a relatively intense signal in this region in PET studies using [11C] PK11195 [106]. Moreover, a significant increase in CSF carbonylation was shown in FTD patient groups, and [11C] PK11195 PET studies have shown increased uptake in frontotemporal regions and basal ganglia. It is important to note that the pathology of FTD does not involve amyloid plaques, suggesting that microglial activation is not specifically related to the amyloid pathology and may reflect part of a common neurodegenerative process.
Although [11C] PK11195 has been the most commonly used PET radioligand for detecting microglial activation, a second generation of TSPO ligands has been developed, which have a higher affinity for TSPO with lower non-specific signals in preclinical models, and are undergoing clinical evaluation with encouraging results. Compounds with improved BBB permeability and affinity for TSPO, including [11C] DAA1106 [107] and [18F] fluoroethoxy-DAA1106 ([18F] FEDAA1106) [108], were developed and applied to neuroimaging of AD patients [109]. Other promising high affinity TSPO tracers are [18F] PBR111, [11C] PBR28, [18F] GE180 and [11C] DPA713 [110•, 111•]. However, the affinity of these newer tracers for TSPO is dependent on which genetic polymorphism of the protein is present, and it is now clear that the presence of a non-synonymous polymorphism of TSPO, coded by the rs6971 single nucleotide polymorphism (SNP) lowers the binding potential of these tracers [112].
Kreisl et al. [113] found that AD cases without the rs6971 polymorphism showed increased cortical [11C] PBR28 binding to TSPO, which worsens with disease severity; however, they were unable to detect a raised signal in their MCI cases. PET scans with [11C] DAA1106, a potent and selective ligand for TSPO, were performed on ten non-genotyped patients with AD and ten age-matched control subjects by Yasuno et al. [109] again showing that the mean binding potential was increased in the brain of AD patients compared with control subjects in all measured regions. Statistical significance has been reached across many of the regions examined, including dorsal and medial prefrontal cortex, lateral temporal cortex, parietal cortex, occipital cortex, anterior cingulate cortex, striatum and cerebellum [109]. Neuroimaging thus represents a powerful tool to detect the presence of neuroinflammation in neurodegenerative dementia, also in the pre-symptomatic phase of neurodegenerative diseases. If novel PET techniques using 18F-labelled tracers are validated, they may have potential for prognostic purposes and to follow up the clinical outcome in the early phases of the disease in multicentre studies.
Recently, we have demonstrated that temporoparietal cortical regions show a significant inverse correlation between levels of microglial activation measured using [11C] PK11195 PET and cerebral glucose metabolic rate measured by [18F] FDG PET in the immediate vicinity, suggesting a deleterious effect of microglia on neuronal function in AD and PDD [114]. In other brain regions, raised cortical microglial activation was also associated with reduced glucose metabolism in the medial temporal lobe, suggesting microglial activation in the regions of cortical projections from medial temporal lobe could lead to neuronal damage and reduction in glucose metabolism at a distance. This study suggests that microglial activation is a common process in neurodegenerative diseases such as AD, MCI and PDD. This study also suggested that microglial activation, along with amyloid deposition, could cause neuronal damage and reduced glucose metabolism, while microglial activation and neuroinflammation could occur independently of amyloid deposition.
Conclusion
Neuroinflammation in the form of miocroglial activation plays a significant role in the pathogenesis of neurodegenerative dementias. There is compelling evidence to suggest that neuroinflammation is a common process and is associated with neuronal damage and cognitive impairment. It can be detected with TSPO markers such as [11C] PK11195 PET and also newer higher affinity ligands, but brain uptake of the latter is influenced by the TSPO polymorphism expressed by individuals. Chronic M1 phenotype microglial activation leads to the release of inflammatory mediators, which result in neurotoxic responses that could drive the neurodegenerative processes. The M2 microglial phenotype, however, is potentially protective and aids adaptive recovery. There is evidence to suggest that peripheral inflammation can exacerbate or trigger central inflammation. For an effective treatment for neurodegenerative diseases, it may be necessary to intervene in both central and peripheral inflammatory pathways, such as by giving cytokine antagonists. It is likely that anti-inflammatory approaches to halting dementias will dominate the field in the years to come.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Prince M et al. The global prevalence of dementia: a systematic review and metaanalysis. Alzheimers Dement. 2013;9(1):63–75. e2.
Emre M, Cummings JL, Lane RM. Rivastigmine in dementia associated with Parkinson’s disease and Alzheimer’s disease: similarities and differences. J Alzheimers Dis. 2007;11(4):509–19.
Petersen RC et al. Current concepts in mild cognitive impairment. Arch Neurol. 2001;58(12):1985–92.
Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med. 2004;256(3):183–94.
Schapira AH, Jenner P. Etiology and pathogenesis of Parkinson’s disease. Mov Disord. 2011;26(6):1049–55.
Iannaccone S et al. In vivo microglia activation in very early dementia with Lewy bodies, comparison with Parkinson’s disease. Parkinsonism Relat Disord. 2013;19(1):47–52.
Varnum MM, Ikezu T. The classification of microglial activation phenotypes on neurodegeneration and regeneration in Alzheimer’s disease brain. Arch Immunol Ther Exp (Warsz). 2012;60(4):251–66.
McGeer PL et al. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology. 1988;38(8):1285–91.
Sastre M et al. Inflammatory risk factors and pathologies associated with Alzheimer’s disease. Curr Alzheimer Res. 2011;8(2):132–41.
McGeer PL, Itagaki S, McGeer EG. Expression of the histocompatibility glycoprotein HLA-DR in neurological disease. Acta Neuropathol. 1988;76(6):550–7.
Imamura K et al. Cytokine production of activated microglia and decrease in neurotrophic factors of neurons in the hippocampus of Lewy body disease brains. Acta Neuropathol. 2005;109(2):141–50.
Mrak RE, Griffin WS. Common inflammatory mechanisms in Lewy body disease and Alzheimer disease. J Neuropathol Exp Neurol. 2007;66(8):683–6.
Morales I et al. Neuroinflammation in the pathogenesis of Alzheimer’s disease. A rational framework for the search of novel therapeutic approaches. Front Cell Neurosci. 2014;8:112.
Heneka MT, O’Banion MK. Inflammatory processes in Alzheimer’s disease. J Neuroimmunol. 2007;184(1–2):69–91.
Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10(11):1387–94.
Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19(8):312–8.
Liu X et al. Age-dependent neuroinflammatory responses and deficits in long-term potentiation in the hippocampus during systemic inflammation. Neuroscience. 2012;216:133–42.
Mills CD et al. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. 2000;164(12):6166–73.
Cunningham C. Microglia and neurodegeneration: the role of systemic inflammation. Glia. 2013;61(1):71–90. doi:10.1002/glia.22350. This article is an evidence-based review that reported the most significant articles related to role of neuroinflammation and the progression of chronic neurodegenerative disease.
Lyman M et al. Neuroinflammation: the role and consequences. Neurosci Res. 2014;79:1–12.
Jimenez S et al. Inflammatory response in the hippocampus of PS1M146L/APP751SL mouse model of Alzheimer’s disease: age-dependent switch in the microglial phenotype from alternative to classic. J Neurosci. 2008;28(45):11650–61.
Tanaka S et al. Involvement of interleukin-1 in lipopolysaccaride-induced microglial activation and learning and memory deficits. J Neurosci Res. 2011;89(4):506–14.
[Abstracts of the 9th Conference on Alzheimer Disease, Kecskemet, Hungary, September 21–23, 2005]. Ideggyogy Sz, 2005. 58(9–10): p. 344–57.
Tanaka S et al. Activation of microglia induces symptoms of Parkinson’s disease in wild-type, but not in IL-1 knockout mice. J Neuroinflammation. 2013;10:143.
Heneka MT et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493(7434):674–8.
Langenfurth A et al. Membrane-type 1 metalloproteinase is upregulated in microglia/brain macrophages in neurodegenerative and neuroinflammatory diseases. J Neurosci Res. 2014;92(3):275–86.
Cunningham C et al. Systemic inflammation induces acute behavioral and cognitive changes and accelerates neurodegenerative disease. Biol Psychiatry. 2009;65(4):304–12.
Cunningham C et al. Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J Neurosci. 2005;25(40):9275–84.
Lunnon K et al. Systemic inflammation modulates Fc receptor expression on microglia during chronic neurodegeneration. J Immunol. 2011;186(12):7215–24.
Liu B, Hong JS. Role of microglia in inflammation-mediated neurodegenerative diseases: mechanisms and strategies for therapeutic intervention. J Pharmacol Exp Ther. 2003;304(1):1–7.
Dickson DW et al. Microglia and cytokines in neurological disease, with special reference to AIDS and Alzheimer’s disease. Glia. 1993;7(1):75–83.
Barger SW, Harmon AD. Microglial activation by Alzheimer amyloid precursor protein and modulation by apolipoprotein E. Nature. 1997;388(6645):878–81.
DeGiorgio LA et al. Amyloid precursor protein gene disruption attenuates degeneration of substantia nigra compacta neurons following axotomy. Brain Res. 2002;938(1–2):38–44.
McGeer PL, McGeer EG. The alpha-synuclein burden hypothesis of Parkinson disease and its relationship to Alzheimer disease. Exp Neurol. 2008;212(2):235–8.
Yoshiyama Y et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53(3):337–51.
Lue LF et al. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am J Pathol. 1999;155(3):853–62.
Ferrari CC et al. Progressive neurodegeneration and motor disabilities induced by chronic expression of IL-1beta in the substantia nigra. Neurobiol Dis. 2006;24(1):183–93.
Wright AL et al. Neuroinflammation and neuronal loss precede Abeta plaque deposition in the hAPP-J20 mouse model of Alzheimer’s disease. PLoS ONE. 2013;8(4):e59586.
de Pablos RM et al. Chronic stress enhances microglia activation and exacerbates death of nigral dopaminergic neurons under conditions of inflammation. J Neuroinflammation. 2014;11:34.
Griffin EW et al. Cyclooxygenase-1-dependent prostaglandins mediate susceptibility to systemic inflammation-induced acute cognitive dysfunction. J Neurosci. 2013;33(38):15248–58.
Hensley K et al. p38 kinase is activated in the Alzheimer’s disease brain. J Neurochem. 1999;72(5):2053–8.
Kim SH, Smith CJ, Van Eldik LJ. Importance of MAPK pathways for microglial pro-inflammatory cytokine IL-1 beta production. Neurobiol Aging. 2004;25(4):431–9.
Kielian T, Drew PD. Effects of peroxisome proliferator-activated receptor-gamma agonists on central nervous system inflammation. J Neurosci Res. 2003;71(3):315–25.
Bassil F, et al. Insulin, IGF-1 and GLP-1 signaling in neurodegenerative disorders: targets for disease modification? Prog Neurobiol. 2014;118:1–18.
Vandal M, et al. Insulin reverses the high-fat diet-induced increase in brain Aβ and improves memory in an animal model of Alzheimer disease. Diabetes. 2014;63:4291–301
Ghosh S et al. Sustained interleukin-1beta overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer’s mouse model. J Neurosci. 2013;33(11):5053–64.
Detrait ER et al. Peripheral administration of an anti-TNF-alpha receptor fusion protein counteracts the amyloid induced elevation of hippocampal TNF-alpha levels and memory deficits in mice. Neurochem Int. 2014;72:10–3.
Sun Q et al. Increased plasma TACE activity in subjects with mild cognitive impairment and patients with Alzheimer’s disease. J Alzheimers Dis. 2014;41(3):877–86.
Woodling NS et al. Suppression of Alzheimer-associated inflammation by microglial prostaglandin-E2 EP4 receptor signaling. J Neurosci. 2014;34(17):5882–94.
Imamura K et al. Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol. 2003;106(6):518–26.
Hirsch EC, Hunot S. Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol. 2009;8(4):382–97.
Brochard V et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest. 2009;119(1):182–92.
Harms AS et al. MHCII is required for alpha-synuclein-induced activation of microglia, CD4 T cell proliferation, and dopaminergic neurodegeneration. J Neurosci. 2013;33(23):9592–600.
Lee HJ et al. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J Biol Chem. 2010;285(12):9262–72.
Halliday GM, Stevens CH. Glia: initiators and progressors of pathology in Parkinson’s disease. Mov Disord. 2011;26(1):6–17.
Lee HJ, Kim C, Lee SJ. Alpha-synuclein stimulation of astrocytes: potential role for neuroinflammation and neuroprotection. Oxidative Med Cell Longev. 2010;3(4):283–7.
He Z et al. Progranulin is a mediator of the wound response. Nat Med. 2003;9(2):225–9.
Tang W et al. The growth factor progranulin binds to TNF receptors and is therapeutic against inflammatory arthritis in mice. Science. 2011;332(6028):478–84.
Baker M et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442(7105):916–9.
Martens LH et al. Progranulin deficiency promotes neuroinflammation and neuron loss following toxin-induced injury. J Clin Invest. 2012;122(11):3955–9.
Bugiani O. FTDP-17: phenotypical heterogeneity within P301S. Ann Neurol. 2000;48(1):126.
Bellucci A et al. Presence of reactive microglia and neuroinflammatory mediators in a case of frontotemporal dementia with P301S mutation. Neurodegener Dis. 2011;8(4):221–9.
Schmidt R et al. Early inflammation and dementia: a 25-year follow-up of the Honolulu-asia aging study. Ann Neurol. 2002;52(2):168–74.
Engelhart MJ et al. Inflammatory proteins in plasma and the risk of dementia: the rotterdam study. Arch Neurol. 2004;61(5):668–72.
Jiang T et al. Triggering receptor expressed on myeloid cells 2 knockdown exacerbates aging-related neuroinflammation and cognitive deficiency in senescence-accelerated mouse prone 8 mice. Neurobiol Aging. 2014;35(6):1243–51.
Guerreiro R et al. TREM2 variants in Alzheimer’s disease. N Engl J Med. 2013;368(2):117–27.
Rayaprolu S et al. TREM2 in neurodegeneration: evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson’s disease. Mol Neurodegener. 2013;8:19.
Rohn TT. The triggering receptor expressed on myeloid cells 2: “TREM-ming” the inflammatory component associated with Alzheimer’s disease. Oxidative Med Cell Longev. 2013;2013:860959.
Harold D et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41(10):1088–93.
Hollingworth P et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet. 2011;43(5):429–35.
Hollingworth P et al. Genome-wide association study of Alzheimer’s disease with psychotic symptoms. Mol Psychiatry. 2012;17(12):1316–27.
Karch CM et al. Expression of novel Alzheimer’s disease risk genes in control and Alzheimer’s disease brains. PLoS ONE. 2012;7(11):e50976.
Wang YL et al. Toll-like receptor 9 promoter polymorphism is associated with decreased risk of Alzheimer’s disease in Han Chinese. J Neuroinflammation. 2013;10(1):101.
Block ML, Hong JS. Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol. 2005;76(2):77–98.
Wynne AM, Henry CJ, Godbout JP. Immune and behavioral consequences of microglial reactivity in the aged brain. Integr Comp Biol. 2009;49(3):254–66.
Tucsek Z, et al. Obesity in aging exacerbates blood–brain barrier disruption, neuroinflammation, and oxidative stress in the mouse hippocampus: effects on expression of genes involved in beta-amyloid generation and Alzheimer’s disease. J Gerontol A Biol Sci Med Sci. 2014;69:1212–26.
Park SM et al. Effects of collagen-induced rheumatoid arthritis on amyloidosis and microvascular pathology in APP/PS1 mice. BMC Neurosci. 2011;12:106.
Boyd TD et al. GM-CSF upregulated in rheumatoid arthritis reverses cognitive impairment and amyloidosis in Alzheimer mice. J Alzheimers Dis. 2010;21(2):507–18. doi:10.3233/JAD-2010-091471. These authors showed that subcutaneous GM-CSF administration significantly reduced brain amyloidosis and completely reversed the cognitive impairment, while increasing hippocampal synaptic area and microglial density in mice model.
Misiak B, Leszek J, Kiejna A. Metabolic syndrome, mild cognitive impairment and Alzheimer’s disease—the emerging role of systemic low-grade inflammation and adiposity. Brain Res Bull. 2012;89(3–4):144–9.
Doruk H et al. The relationship between body mass index and incidental mild cognitive impairment, Alzheimer’s disease and vascular dementia in elderly. J Nutr Health Aging. 2010;14(10):834–8.
Ho AJ et al. Obesity is linked with lower brain volume in 700 AD and MCI patients. Neurobiol Aging. 2010;31(8):1326–39.
Naorungroj S et al. Cognitive decline and oral health in middle-aged adults in the ARIC study. J Dent Res. 2013;92(9):795–801.
Noble JM et al. Periodontitis is associated with cognitive impairment among older adults: analysis of NHANES-III. J Neurol Neurosurg Psychiatry. 2009;80(11):1206–11.
McGeer PL, Schulzer M, McGeer EG. Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer’s disease: a review of 17 epidemiologic studies. Neurology. 1996;47(2):425–32.
Ferraccioli G et al. Rheumatoid arthritis and Alzheimer’s disease: genetic and epigenetic links in inflammatory regulation. Discov Med. 2012;14(79):379–88.
Aisen PS et al. Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. JAMA. 2003;289(21):2819–26.
Soininen H et al. Long-term efficacy and safety of celecoxib in Alzheimer’s disease. Dement Geriatr Cogn Disord. 2007;23(1):8–21.
Leoutsakos JM et al. Effects of non-steroidal anti-inflammatory drug treatments on cognitive decline vary by phase of pre-clinical Alzheimer disease: findings from the randomized controlled Alzheimer’s disease anti-inflammatory prevention trial. Int J Geriatr Psychiatr. 2012;27(4):364–74. doi:10.1002/gps.2723. This recent revision of the AD Anti-inflammatory Prevention Trial supports a beneficial role of NSAID only in the early, asymptomatic, phases of Alzheimer disease suggesting that neuroinflammation could be a crucial events in the neurodegeneration process.
Breitner JC et al. Extended results of the Alzheimer’s disease anti-inflammatory prevention trial. Alzheimers Dement. 2011;7(4):402–11.
Hirsch EC, Vyas S, Hunot S. Neuroinflammation in Parkinson’s disease. Parkinsonism Relat Disord. 2012;18 Suppl 1:S210–2.
Matsuda R et al. Quantitive cytokine mRNA expression profiles in the colonic mucosa of patients with steroid naive ulcerative colitis during active and quiescent disease. Inflamm Bowel Dis. 2009;15(3):328–34.
Devos D et al. Colonic inflammation in Parkinson’s disease. Neurobiol Dis. 2013;50:42–8.
Chen H et al. Nonsteroidal antiinflammatory drug use and the risk for Parkinson’s disease. Ann Neurol. 2005;58(6):963–7.
Wahner AD et al. Nonsteroidal anti-inflammatory drugs may protect against Parkinson disease. Neurology. 2007;69(19):1836–42.
Leung E et al. Microglia activation mediates fibrillar amyloid-beta toxicity in the aged primate cortex. Neurobiol Aging. 2011;32(3):387–97.
Frautschy SA et al. Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol. 1998;152(1):307–17.
Cagnin A, Gerhard A, Banati RB. In vivo imaging of neuroinflammation. Eur Neuropsychopharmacol. 2002;12(6):581–6.
Cosenza-Nashat M et al. Expression of the translocator protein of 18 kDa by microglia, macrophages and astrocytes based on immunohistochemical localization in abnormal human brain. Neuropathol Appl Neurobiol. 2009;35(3):306–28.
Shah F et al. Synthesis of the enantiomers of [N-methyl-11C]PK 11195 and comparison of their behaviours as radioligands for PK binding sites in rats. Nucl Med Biol. 1994;21(4):573–81.
Chauveau F et al. Nuclear imaging of neuroinflammation: a comprehensive review of [11C]PK11195 challengers. Eur J Nucl Med Mol Imaging. 2008;35(12):2304–19.
Politis M, Su P, Piccini P. Imaging of microglia in patients with neurodegenerative disorders. Front Pharmacol. 2012;3:96.
Yasuno F et al. Increased binding of peripheral benzodiazepine receptor in mild cognitive impairment-dementia converters measured by positron emission tomography with [(1)(1)C]DAA1106. Psychiatry Res. 2012;203(1):67–74.
Edison P et al. Microglia, amyloid, and cognition in Alzheimer’s disease: an [11C](R)PK11195-PET and [11C]PIB-PET study. Neurobiol Dis. 2008;32(3):412–9.
Cagnin A et al. In vivo detection of microglial activation in frontotemporal dementia. Ann Neurol. 2004;56(6):894–7.
McGeer PL et al. Microglia in degenerative neurological disease. Glia. 1993;7(1):84–92.
Lant SB RA, Thompson JC, Rollinson S, Pickering-Brown S, Snowden JS, Davidson YS, Gerhard A, Mann DM. Patterns of microglial cell activation in frontotemporal lobar degeneration. Neuropathol Appl Neurobiol. 2014;40:686–96.
Maeda J et al. Novel peripheral benzodiazepine receptor ligand [11C]DAA1106 for PET: an imaging tool for glial cells in the brain. Synapse. 2004;52(4):283–91.
Wang M, Gao M, Zheng QH. Fully automated synthesis of PET TSPO radioligands [11C]DAA1106 and [18 F]FEDAA1106. Appl Radiat Isot. 2012;70(6):965–73.
Yasuno F et al. Increased binding of peripheral benzodiazepine receptor in Alzheimer’s disease measured by positron emission tomography with [11C]DAA1106. Biol Psychiatry. 2008;64(10):835–41.
Venneti S, Lopresti BJ, Wiley CA. Molecular imaging of microglia/macrophages in the brain. Glia. 2013;61(1):10–23. doi:10.1002/glia.22357. This review provides an interesting and updated overview of positron emission tomography (PET) and magnetic resonance (MR) imaging of microglia/macrophages in the brain.
Yoder KK et al. Influence of TSPO genotype on 11C-PBR28 standardized uptake values. J Nucl Med. 2013;54(8):1320–2. doi:10.2967/jnumed.112.118885. This was one of the first study showing the effect of TSPO genotype on the binding potential of new microglial cell PET tracers.
Kreisl WC et al. A genetic polymorphism for translocator protein 18 kDa affects both in vitro and in vivo radioligand binding in human brain to this putative biomarker of neuroinflammation. J Cereb Blood Flow Metab. 2013;33(1):53–8.
Kreisl WC et al. In vivo radioligand binding to translocator protein correlates with severity of Alzheimer’s disease. Brain. 2013;136(Pt 7):2228–38.
Fan Z, et al. Influence of microglial activation on neuronal function in Alzheimer’s and Parkinson’s disease dementia. Alzheimers Dement. 2014. doi:10.1016/j.jalz.2014.06.016.
Compliance with Ethics Guidelines
Conflict of Interest
Giuseppe Pasqualetti received funding from University of Pisa and from PAIM (Pisa, Italy) and Italian Society of Pharmacology for research travel cost.
David J. Brooks was the chief medical officer for GE healthcare, and he has received consultancy fees/honoraria from the following: Acadia Pharmaceuticals Inc, Amsterdam Molecular Therapeuctics BV, AstraZeneca, BiogeIdec, NeuroNova AB, Eli Lilly and Company, Medtronic Inc, Shire Pharmaceuticals Inc, Synosia Therapeutics AG, GlaxoSmith Kline, UBC Biosciences Inc, Veralis (R&D) limited, Genentech Inc, Navidea.
Paul Edison received grant funding from Medical Research Council, UK; Alzheimer’s Research, UK; Alzheimer’s Society, UK; Novo Nordisk; GE Healthcare; and Piramal Life Science.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Neuroimaging
Rights and permissions
About this article

Cite this article
Pasqualetti, G., Brooks, D.J. & Edison, P. The Role of Neuroinflammation in Dementias. Curr Neurol Neurosci Rep 15, 17 (2015). https://doi.org/10.1007/s11910-015-0531-7
Published:
DOI: https://doi.org/10.1007/s11910-015-0531-7