
Abstract
Purpose of Review
The underlining goal of this review is to offer a concise, detailed look into current knowledge surrounding transient receptor potential canonical channel 6 (TRPC6) in the progression of diabetic kidney disease (DKD).
Recent Findings
Mutations and over-activation in TRPC6 channel activity lead to the development of glomeruli injury. Angiotensin II, reactive oxygen species, and other factors in the setting of DKD stimulate drastic increases in calcium influx through the TRPC6 channel, causing podocyte hypertrophy and foot process effacement. Loss of the podocytes further promote deterioration of the glomerular filtration barrier and play a major role in the development of both albuminuria and the renal injury in DKD. Recent genetic manipulation with TRPC6 channels in various rodent models provide additional knowledge about the role of TRPC6 in DKD and are reviewed here.
Summary
The TRPC6 channel has a pronounced role in the progression of DKD, with deviations in activity yielding detrimental outcomes. The benefits of targeting TRPC6 or its upstream or downstream signaling pathways in DKD are prominent.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Diabetic kidney disease (DKD) is the leading risk factor for end-stage renal disease (ESRD). In 2014, 44% of the diagnosed cases of ESRD were as a result of diabetes. The Center for Disease Control and Prevention predicts that as many as 1 in 3 US adults could have diabetes by 2050 if current trends continue. The prevalence of diabetes with associated renal injury highlights the growing urgency to understand the disease pathophysiology better and to develop effective therapies. Identification of novel therapeutic strategies that specifically target the development of renal damage is desired for patients with DKD. During its progression, loss of the glomerular filtration barrier (GFB) cells and resulting albuminuria are major characteristics of the disease. Glomeruli injury (glomerulopathy) is considered a primary contributor to DKD. Glomerular visceral epithelial cells, or podocytes, are important for normal glomerular permselectivity, and their dysfunction or loss is implicated in the pathogenesis of glomerulonephritis. The podocyte is an integral member of the GFB, and its damage has been linked to the development of the detrimental characteristics of DKD. In the current mini-review, we discuss recent work identifying some novel mechanisms in DKD mediated by activation of transient receptor potential canonical 6 (TRPC6) channels in glomeruli. For details about other members of the TRPC family in the kidney, please see a recent review by Dryer et al. [1•].
Diabetic Kidney Disease and Glomerulopathy
DKD is a disease in which hyperglycemia leading to progressive damage to the glomerular cells, tubules, and capillaries in the kidneys. DKD is a common complication of both type 1 and type 2 diabetes mellitus. Comorbidity of this disease is hypertension associated with untreated hyperglycemia. The combination of diabetes and hypertension is correlated with cardiovascular and kidney diseases [2, 3]. Renin-angiotensin system (RAS) inhibitors and glycemic control with insulin treatment are the current therapies for treating complications associated with DKD. Recent studies also identified sodium glucose co-transporter 2 (SGLT2), dipeptidyl peptidase-4 (DPP-4) inhibitors, and glucagon-like peptide-1 receptor agonists (GLP-1) as potential effective drugs in the treatment or prevention of DKD [4,5,6,7]. Despite these treatment efforts, millions of patients worldwide still have diabetes with a large cohort of these patients being expected to develop chronic kidney disease (CKD) progressing to ESRD.
Reduction in glomerular filtration rate (GFR) along with albuminuria are pivotal markers in predicting the risk for ESRD. Development of DKD is associated with many alterations in the structure of GFB that contribute to the reduction of the GFR [8]. The glomerulus is composed of various cell types and podocytes as a key target for injury leading to glomerulosclerosis, proteinuric kidney disease, and the eventual development of DKD. Key factors determining the pathological changes of glomerular morphology and permeability are linked to an elevation of podocyte intracellular calcium ([Ca2+]i). The alterations in [Ca2+]i signaling in podocytes are precisely controlled by intracellular and extracellular sources. The spatial and temporal patterns of the [Ca2+]i signal due to Ca2+ entry from different routes vary. Podocyte depletion may result from improper calcium handling due to abnormal activation under diseased conditions when pathological factors are elevated. Transient receptor potential (TRP) channels, along with other sources such as store-operated calcium entry (SOCE) and depletion of the internal calcium stores, are responsible for the specific control of [Ca2+]i. SOCE is activated by depletion of the internal calcium stores in the endoplasmic reticulum (ER) and sarcoplasmic reticulum (SR) of the cell [9]. It was shown that inhibition of ER stress attenuates kidney dysfunction and glomeruli injury induced by diabetes mellitus and hypertension [10]. Two families of proteins, in particular, have been identified as essential for the function of SOCE, stromal interaction molecules 1–2 (STIM) as the calcium sensor and Orai 1–3 as the pore former [11]. STIM1, especially, is a protein located predominantly in the ER membrane and monitors the calcium concentrations within the lumen of the ER. When STIM1 senses calcium depletion in the ER, it aggregates and transcolates into plasma membrane junctions where it can interact with Orai1 and allows Ca2+ influx through the STIM1-Orai1 complex. Through the newly formed Orai1 pore, calcium enters the cytosol [12]. Some isoforms of Orai and STIM have also been found to interact with TRPCs, including TRPC1, TRPC3, and TRPC6 [13, 14]. Over-activation of this system and other SOCE mechanisms might also have implications in DKD.
TRPC6 and Its Regulation by GPCR
Members of the TRPC family, including TRPC6 channels, are shown as key contributors in the pathogenesis of renal and cardiovascular diseases [1•, 15,16,17]. TRPC belongs to the superfamily of TRP cation channels that are non-selectively permeable to calcium [15, 17]. Biophysical properties of TRPC6, including details about its permeation and rectification in renal and heterologous systems, have been recently discussed [18]. Briefly, TRPC channels can be activated by stimulating tyrosine phosphorylation-dependent cascade through phospholipase C (PLC). It has been proposed that TRPC6 (and TRPC7 but not TRPC5 or TRPC4) [19, 20] channels can be directly activated by DAG and TRPC6 activation can be prevented by the inhibition of protein kinase C (PKC) [21]. It was also reported that TRPC6 channel is not primarily activated by mechanical stimuli and becomes mechanosensitive once receptor-activated. The simultaneous operation of receptor and mechanical stimulations in podocyte may be similar to smooth muscle cells mechanism, where synergistically amplification of transmembrane Ca2+ mobilization through TRPC6 activation enhanced the [Ca2+]i via both PLC/DAG and phospholipase A2/ω-hydroxylase/20-HETE pathways [22].
Over-activation of the TRPC6 channel was shown to cause glomerular damage due to their role in podocytopenia [17, 23]. Under normal physiological conditions, the channel remains relatively dormant until various stimuli trigger its activation [17]. Both gain-of-function (GOF) and loss-of-function (LOF) mutations of the TRPC6 channel have been identified (see Table 1) as genetic impetus for the development of renal diseases such as focal segmental glomerulosclerosis (FSGS), which displays similar renal injury like loss of the GFB cells and progressive albuminuria seen in DKD [23, 25, 37].
TRPC channels are components of complex pathways often responding to activation of G-protein-coupled receptor (GPCR) cascades and triggering large amounts of Ca2+ influx in the cell. TRPC6 is highly expressed in the soma of the podocyte as shown by electrophysiological recordings [38], and it is also present within the slit diaphragm domains of the foot processes where it interacts with podocin and nephrin [24, 39]. Thus, TRPC6 activation may directly correlate with mechanical stretch and actin reorganization in the podocyte, playing a significant role in the physiological regulation of the permeability barrier during normal fluctuations in glomeruli filtration. Stimulation of TRPC6 channels results in an influx of Ca2 + ions into the cells triggering an intracellular signaling cascade and leading to enhanced gene transcription important for podocyte function and maintenance. Interestingly, the reported genetic presence of GOF mutations in TRPC6 promotes apoptotic elevation in intracellular Ca2+ and rapid progression in renal pathology. The activation of TRPC6 can be mediated by direct pathophysiological stimuli like an increase in oxidative stress, cytoskeleton remodeling, by the chronic activation of GPCR pathways or combination of all these factors [1•, 16, 40]. As we and others previously have shown, TRPC6 activation through the GPCR-signaling mechanisms, such as angiotensin II (Ang II), adenosine triphosphate (ATP), and protease thrombin, is the most common and pharmacologically relevant stimulus [38, 41,42,43,44]. Direct evidence of the contribution of GPCRs in TRPC6 activation and consecutive glomerular diseases was shown by Spurney and colleagues who used the mouse model in which a constitutively active Gq α-subunit was specifically expressed in podocytes [45]. It was reported that Gq-dependent effects on podocyte injury were generalizable to DKD given that the expression of Gq-promoted albuminuria, mesangial expansion, and increased glomerular basement membrane width in diabetic mice. The authors proposed that targeting Gq-TRPC6 signaling may have therapeutic benefits for the treatment of glomerular diseases [45].
It is established that hyperglycemia in combination with Ang II causes an increase in TRPC6 promoter activity and overexpression of the TRPC6 channel accompanied by an increase in Ca2+ influx within podocytes. Multiple studies revealed that Ang II-mediated activation of the TRPC6 channel induces a drastic increase in the calcium influx into the podocytes, leading to its dysfunction and breakdown of the GFB with an increase in albuminuria also occurring [38, 46,47,48,49,50,51,52]. Specifically, Ang II directly and reversibly activates TRPC6 through the AT1 receptor as was shown by the cell-attached single channel recordings in co-transfected CHO cells, isolated glomeruli preparations, and TRPC6 knockout animals [38]. Shown in Fig. 1a is an effect of Ang II in freshly isolated glomeruli of wild-type mice, which was completely absent in TRPC6−/− mice (Fig. 1b) [38].

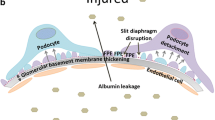
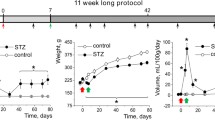
Contribution of TRPC6 and its upstream signaling pathways towards the development of diabetic kidney disease. a Angiotensin II (Ang II; 1 μM) activates TRPC6 channels in the podocytes of freshly isolated mouse glomeruli. Representative current traces of a TRPC6 channel from a cell-attached patch of a podocyte from a wild-type mouse glomerulus. A continuous current trace (upper row) and addition of Ang II (1 μM) to the external bath solution are shown. Membrane potential was − 60 mV. The c and oi denote closed and open current levels, respectively. b A representative recording made on the podocytes of the freshly isolated glomerulus of a TRPC6 knockout mouse. No TRPC6 channel activity was recorded in any of the patches before or after application of Ang II. Effect of Ang II was observed in 11 out of 11 wild-type and 0 out 7 TRPC6−/− mice, respectively. Modified from [38] with permission. c Expression levels of TRPC6 channels in type 1 DKD. A representative Western blot and a summary graph for the densitometry values of TRPC6 from the cortical kidney lysates of the vehicle- and STZ-treated rats (11 weeks after STZ injection); P < 0.05. Modified from [47] with permission. d Schematic depicting role of TRPC6 in DKD. Elevation during diabetes of signaling factors, such as Ang II, serine proteases, ATP, and H2O2, results in enhanced expression and activity of TRPC6 channels, which subsequently leads to substantial increases in [Ca2+]i and further elevation of albuminuria and cell apoptosis
There is a growing evidence that serine proteases like thrombin, kallikrein, and urokinase with corresponding protease-activated receptors (PARs) and soluble urokinase-type plasminogen activator receptor (suPAR) signaling mechanisms may play a significant role in the development of DKD. These receptors are expressed in podocytes, and their activation may promote a substantial elevation in [Ca2+]i [42, 53, 54]. For example, suPAR increased mobilization and stretch-activation of TRPC6 through a pathway that entails Nox2-dependent generation of reactive oxygen species (ROS) [55, 56]. suPAR along with urokinase receptor (uPAR) are also found to be significantly activated in glomeruli and sera of DKD patients [57,58,59]. It was further reported that antagonism of PAR1, which is the most common PAR receptor in the kidney, protects against podocyte injury in a mouse model of nephropathy via TRPC-mediated elevation of intracellular Ca2+ [60]. Recent studies revealed that PAR1 [61, 62] deficiency protects against streptozotocin (STZ)-induced DKD in mice and plays an essential role in the development of type 1 diabetes. Clinical studies also demonstrated that the concentration of urinary thrombin is elevated in patients with glomerulonephritis and is associated with the severity of glomerular injury in type 2 diabetes and DKD [63]. The patients with type 2 diabetes and DKD have disturbances in thrombin generation and associations with enhanced urinary thrombin levels, microalbuminuria, and formation of denser fibrin clots [64, 65]. Therefore, the contribution to the development of podocyte and glomerular injury in DKD can be mediated by PARs along with other GPCRs-TRPC6 signaling cascades (Fig. 1d).
ATP and its derivatives are other important players targeting TRPC6 in the kidney. ATP-evoked currents mediated through P2Y metabotropic receptors are positively coupled to TRPC6 activity [43]. As we have shown previously, P2Y1 receptor signaling cascade is the predominant P2Y purinergic pathway in the glomeruli podocytes [41]; however, contributions of other P2Y receptors were also proposed [43]. Furthermore, we cannot exclude that expression and function of P2 receptors could be modulated in the progression of DKD, similarly to a reported remodeling in the P2 receptors profile in polycystic kidney disease [66]. Overall, mechanisms of P2Y-GPCR-TRPC6 cascade in the progression of renal pathology, specifically DKD, are poorly understood and require further investigations.
Reactive Oxygen Stress and TRPC6 Activation
It has been found that formation of ROS, and in particular, hydrogen peroxide (H2O2) production from NADPH oxidases, is needed for TRPC6 activation [16]. TRPC6 is a redox-sensitive channel, and modulation of TRPC6-mediated Ca2+ signaling by overproduction of ROS may trigger injury in multiple cells including podocyte [67]. In rodents, activation of podocyte TRPC6 channel by GPCRs often correlates with the involvement of ATP-NADPH oxidase 2 (Nox2) [43] or Ang II-NADPH oxidase 4 (Nox4)-mediated elevation of ROS [68••]. In the absence of Nox4, podocytes had stabilized basal calcium concentrations, and H2O2 application in the absence of TRPC6 had blunted pathological elevation in basal calcium level. The data suggest that TRPC6 blockade protects podocyte and whole glomerulus from the oxidative stress similarly to the knockout of Nox4 in the STZ-induced diabetic animals [68••]. Interestingly, human mesangial cells in response to hyperglycemia have increased expression of TRPC6 and promote bidirectional potentiation between TRPC6 and another NADPH subunit, Nox5, that is normally not expressed in rodents. Overall, inhibition of both Nox5 and TRPC6 could represent a new approach to target the deleterious effects of the pathological intracellular signaling cascades in the diabetic kidney [69].
Moreover, Graham et al. found that the protein content of TRPC6 in the mesangial cells of the glomerulus is in part regulated by ROS and protein kinase C (PKC) in the diabetic setting [70]. Hyperglycemia leads to a downregulation of TRPC6 in the mesangial cells and the glomerulus as a whole. H2O2 stifled the expression of TRPC6 protein in cultured mesangial cells which mirrored the hyperglycemic response these cells had, but under the knockdown of Nox4, there was an elevation in the mesangial cell TRPC6 protein content. Also, interestingly, activation of PKC repressed increases in TRPC6 protein in the cells [70].
Role of TRPC6 in the Development of FSGS
Several studies have reported the identification of TRPC6 mutations associated with the FSGS. These genetic findings highlighted the critical importance of the podocyte and the TRPC6 channel in the maintenance of GFB. The majority of identified mutations in the TRPC6 gene are GOF mutations yielding excessive [Ca2+]i levels and podocyte cell death (leading to the development of FSGS). Chiliuza et al. revealed that GOF mutations in TRPC6 lead to extracellular signal-regulated kinases (ERK) activation via at least two distinct pathways [71]. There are also genetic variations which lead to LOF phenotypes [27]. These mutations demonstrate that not only activation but also inhibition of TRPC6 activity might lead to FSGS (see Table 1). This discrepancy could also be due to the fact that a functional analysis in heterologous systems may not always provide definitive proof to discern between mutations and polymorphisms for amino acid substitutions in a particular gene. Recently published, the cryo-EM structure of the cytoplasmic domain of murine TRPC6 [72] and human TRPC6 homotetramer in complex with a high-affinity inhibitor BTDM [73••], both at 3.8 Å resolutions, will provide essential information required to uncover the molecular architecture of TRPC channels and offer a structural basis for understanding the mechanism of the TRPC6 channels regulation. Modeling of known mutations can also reveal a mystery why both GOF and LOF mutations of the same protein result in the same disease phenotype. Interestingly, there is an observed correlation between gain- or loss-of-function mutations and age of patients. It was proposed that loss of TRPC6 function might be implicated in the pathophysiology of juvenile forms of hereditary FSGS [27].
Recent Studies of Trpc6 in DKD
An association between TRPC6 channels with the development of diabetic complications was reported in several studies. Elevated expression and activity of TRPC6 channels, in various in vivo and in vitro diabetic models, were described. As an example, Sonneveld et al. demonstrated that expression of TRPC6 protein in podocyte was elevated in proteinuric STZ-induced diabetic rats [50]. In vitro studies further revealed that high glucose levels increased TRPC6 mRNA and protein expression, and that Ang II also specifically increased TRPC6 expression in cultured podocytes.
Recent genetic studies further provided important information about the role of TRPC6 in the progression of DKD. Both positive and negative findings were reported [74]. To test the contribution of TRPC6 towards the development of type 1 DKD, we used the Dahl salt-sensitive (SS) rat injected with a low-dose of streptozotocin (STZ-SS). The STZ model is well-established and offers the advantage of quickly and reproducibly inducing hyperglycemia and renal hypertrophy in rodents. Other STZ-treated mouse or rat models typically do not develop the glomerular disease unless hypertension is induced, unlike the Dahl SS where only STZ treatment is required. The Dahl SS rat is a salt-dependent model of hypertension that is highly susceptible to the development of renal disease when fed a high-salt diet [75,76,77]. As reported by Dr. Williams’s laboratory [78] and us [47, 68], SS rats treated with STZ develop hyperfiltration and progressive proteinuria and display renal histological lesions characteristic to those seen in patients with DKD. Therefore, while this model also has some limitations, it mimics the development of human DKD. Eleven weeks post-treatment was sufficient for the animals to develop hyperglycemia, diuresis, weight loss, microalbuminuria, nephrinuria, and display renal histological lesions typical for patients with DKD. Western blotting and immunohistochemistry revealed increased expression of TRPC6 in the kidney homogenate and podocytes [47, 79]. Western blot analysis of TRPC6 expression in the cortex homogenate of STZ- or vehicle-injected SS rats is shown in Fig. 1c. Patch-clamp electrophysiology performed on podocytes of the freshly isolated glomeruli revealed enhanced basal TRPC channel activity in the STZ-SS rats, and an increased response to Ang II. Total calcium influx triggered by Ang II application was also augmented in podocytes of these rats [47]. In the experiments that followed, we tested the role of Ang II in glomerular volume dynamics and podocyte Ca2+ handling. Using a novel 3D whole-glomerulus imaging ex vivo assay, we revealed the involvement of both AT1 and AT2 receptors in controlling glomerular permeability. Ang II-induced Ca2+ flux was dramatically diminished upon inhibition of TRPC channels with SAR7334, SKF 96365, clemizole hydrochloride, and La3+, but not ML204. Similarly, specific inhibitors and activators of TRPC channels revealed that TRPC6 channels are implicated in the regulation of glomerular volume dynamics [46]. In our recent studies using a TRPC6 knockout created on the Dahl SS rat background (SSTrpc6−/−), we further tested the contribution of TRPC6 during the development of type 1 DKD [79]. SSTrpc6−/− or control SS rats were injected with STZ or vehicle and simultaneously given an insulin subcutaneous implant to maintain blood glucose at a stable level. There was an increase in albuminuria and glomerular injury following STZ treatment in SSTrpc6−/− and control groups similar to previously published data [17, 78]. Western blot analysis revealed elevated levels of nephrin in urine samples of STZ-SS rats, which was higher compared with STZ-SSTrpc6−/− rats. Furthermore, pathological increases in basal [Ca2+]i levels and foot process damage of podocytes during the development of DKD was attenuated in the STZ-SSTrpc6−/− compared with STZ-treated SS rats. Overall, our data indicate that TRPC6 channel inhibition may have at least partial renoprotective effects [79]. Dryer and colleagues recently presented [80] a similar approach when they used STZ-treated Sprague-Dawley (SD) rats with a deletion of the TRPC6 gene that renders the channels non-functional [81•]. In contrast to our data and that of the authors’ chronic puromycin aminonucleoside nephrosis (PAN) model [81•], they did not observe the protective effects of TRPC6 inactivation on glomerular pathology [80]. As discussed above, this might be due to the difference between rat strains used for the creation of the TRPC6 knockouts. The Dahl SS rats develop more profound renal abnormalities, specifically glomerular injury, compared to SD rats.
In another study, Wang et al. assessed the role of TRPC6 in type 1 diabetes setting by breeding Akita mice with TRPC6 KO mice. It should be noted that this model, as well as many other animal models of DKD, also has a number of limitations [74] [82]. The authors reported that knockout of TRPC6 decreased proteinuria and attenuated tubule injury but promoted mesangial expansion in the Akita mice [83]. Knockout of TRPC6 in Akita mice inhibited albuminuria at 12 and 16 weeks of age, but this difference disappeared by 20 weeks. Hyperglycemia and blood pressure were similar between TRPC6 knockout and wild-type Akita mice, but knockout mice were more insulin resistant. This study provides critical insight into the contribution of TRPC6 channels in DKD since additional attention should be paid to the timeline of the development of podocyte and renal damage.
Additional Potential Mechanisms Contributing to TRPC6 Regulation in DKD
Recently, evidence has arisen linking TRPC6 activity with other pathways to renal immune responses. Nuclear factor of activated T cells (NFAT) is a family of transcription factors with a role in the immune system. In the podocyte, Wang et al. found that mutations in TRPC6 can activate NFAT-dependent transcription that leads to activation of calcineurin, a Ca2+-dependent serine/threonine phosphatase with a role in transcription regulation in renal cells [84]. Conditional activation of NFAT showed the progression of proteinuria in mice. Furthermore, TRPC6 GOF mutants associated with FSGS enhance NFAT activation and determine the susceptibility to the development of glomerulosclerosis [84, 85]. Also, Ang II has been found to increase TRPC6 expression through an NFAT positive feedback pathway [51]. In a Ren2 transgenic rat, a model of elevated tissue RAS activity, the authors showed increased TRPC6 glomerular expression with a positive correlation and calcineurin/NFAT activity. Inhibiting calcineurin/NFAT activity prevented both doxorubicin and Ang II-mediated increases in TRPC6 expression [51].
Ma et al. further provided evidence of the Ang II/TRPC6/NFAT pathway involvement in the development of characteristics of DKD, such as podocyte injury, in particular, for type 2 diabetes. In a rat model of type 2 diabetes, induced by high-caloric diets and STZ, they found a direct correlation between the increase in Ang II-mediated TRPC6 expression, podocyte process effacement, and the development of microalbuminuria. When these animals were given an AT1 receptor blocker valsartan, there was a reduction in expression of both TRPC6 and NFAT. Inhibition of TPRC6 also mediated decreases in hyperglycemia-induced podocyte injury by decreasing NFAT mRNA expression [86]. In line with this, the immunosuppression with tacrolimus (FK506) caused attenuation of elevated expression of both TRPC6 and NFAT. Also, FK506 reduced the extent of podocyte morphology damage related to DKD with improvements in podocytes levels of nephrin expression [87]. In a human tubular epithelial cell line treated with high glucose to simulate DKD, overexpression of TRPC6 resulted in an increase in the NFAT that was deactivated by a TRPC6 knockdown [88]. These findings implicate the importance of the Ang II/TRPC6/NFAT axis in the progression of DKD through its involvement in the podocyte and glomerular injury associated with DKD.
Along with the few other pathways mentioned above, TRPC6 is involved in multiple other signaling mechanisms regulated by calcium such as the AMP-activated protein kinase (AMPK) metabolic pathway. AMPK is one of the key regulators of glucose and fatty acids; thus, its potential in DKD is of no surprise [89]. AMPK has implications in a variety of renal-related pathologies, including podocyte dysfunction and renal hypertrophy. In the diabetes-induced renal hypertrophy setting, AMPK activity is reduced. This indicates that hyperglycemia may have inhibitory effects on the pathway. It was reported that TRPC6 plays an important role in the insulin-dependent reorganization of the podocyte cytoskeleton and activation of AMPK [90]. Both AMPK and TRPC6 activation were shown to be required for the stimulation of the Rac1 pathway, which has connections to podocyte injury in DKD. Ji et al. using immortalized human podocytes proved the involvement of TRPC6-AMPK crosstalk showing that hypoxia-induced autophagy, which is a form of AMPK Ca2+-dependent autophagy initiated by hypoxia, is correlated with TRPC6 activity [91].
Another calcium-dependent pathway that involves TRPC6 is the transforming growth factor beta (TGF-β) signaling pathway. It was reported that inhibition of TRPC6 alleviated glomerulosclerosis induced by TGF-β1 in podocytes and reduced cell apoptosis [92, 93]. Contribution of TGF-β1 was further reported in TRPC3/6/7−/− mice under diabetic conditions. It was reported in this study that TRPC6 protein was increased in the renal cortex in wild-type diabetic mice. Furthermore, the diabetic wild-type and TRPC3/6/7−/− mice had similar levels of hyperglycemia; however, the triple knockout diabetic mice showed less polyuria, kidney hypertrophy, glomerular enlargement, and albuminuria compared with control mice [94]. Therefore, the data suggest a strong correlation of TRPC6 activation with TGF-β signaling, including DKD conditions.
Role of Trpc6 in Other CKDs
The protective role of TRPC6, in various other types of CKD, was also shown. Furthermore, therapeutic efficacy of a selective pharmacological TRPC6 inhibition was recently demonstrated. As an example, TRPC6 inhibitor (BI 749327, acting through the suppression of NFAT activation) treatment reduced renal fibrosis and inhibited renal injury of mice after UUO; this compound also improved cardiac function in pressure overload mice model [95•]. Furthermore, Kim et al. recently reported that TRPC6 inactivation (deletion of essential portion of TRPC6, which caused disruption of the ankyrin-repeat domain and non-functional TRPC6 channels; TRPCdel/del in SD rat background) attenuated glomerulosclerosis, ultrastructural changes, interstitial changes, and renal fibrosis in the progression of chronic PAN, a classic model for acquired nephrotic syndromes [81•]. The authors reported that TRPC6 inactivation reduced albuminuria in the chronic phase of the disease. Using the same genetic model (TRPCdel/del), the authors examined the role of TRPC6 in the progression of inflammation and fibrosis in the nephrotoxic serum (NTS) model of crescentic glomerulonephritis and reported that TRPC6 inactivation reduces glomerulosclerosis and podocyte loss but does not reduce renal fibrosis or tubulointerstitial disease [96]. Li et al. assessed GFR in conscious wild-type and TRPC6 knockout mice, and in anesthetized rats with and without in vivo knockdown of TRPC6 in the kidneys and found that GFR was significantly greater in TRPC6 knockout mice and rats with knockdown of TRPC6. Serum creatinine level was significantly lower in TRPC6−/− mice as well. Consistently, knockdown of TRPC6 in the kidney using a TRPC6 specific shRNA construct significantly attenuated Ang II-induced GFR decline in rats. The authors contributed these effects to the function of TRPC6 in the glomerular mesangial cells [97]. Wu et al. found that in a model of renal interstitial fibrosis, loss of TRPC6 prevented the development of fibrosis as well as attenuated increases in expression of fibrotic genes such as connective tissue growth factor (CTGF) and TGF-β1, a fibrotic factor. Also, interestingly, they showed that a double knockout of both TRPC3 and TRPC6 did not have the same renoprotective benefits as knocking out TRPC6 alone [98]. There are also implications of TRPC6 involvement in idiopathic membranous nephropathy (iMN), a proteinuric disease [51, 99, 100]. It was found that RAS inhibitors and calcineurin blockers commonly used to treat iMN were able to attenuate these intracellular pathways that involve TRPC6 [51, 85, 99], which was shown to be overexpressed in iMN [99]. Huang et al. showed that in a model of basic nephropathy, when podocytes were treated with a TRPC6 inhibitor following TGF-β1 podocyte, apoptosis was attenuated [92]. Recent studies revealed progressive aggravation of proteinuria, decreased renal function, and ultimately glomerular fibrosis associated with increased expression of TRPC6 in the model of ischemia-reperfusion acute kidney injury [101]. Contribution of TRPC6-mediated calcium pathways and podocyte injury in pre-eclampsia was also proposed [102].
Conclusions
In summary, mutations in TRPC6, as well as upregulation of its expression and activity, are linked to a multitude of renal abnormalities such as albuminuria and podocyte cell loss that all contribute to DKD. Some of these mutations have a strong genetic impetus for the development of FSGS, which has similar renal damage found in DKD. TRPC6 channel activity is highly regulated by Ang II, ATP, PAR agonists, and other endogenous factors in DKD (Fig. 1d), which result in cell hypertrophy, foot processes effacement, podocyte depletion, and proteinuria. Not only is there a connection between TRPC6 and listed above GPCR agonist but TRPC6 activity is also linked to TGF-β, AMPK, and the calcineurin/NFAT pathways, that all play their part in the progression of DKD. Despite this wealth of information, research on the effects of TRPC6, especially in type 2 diabetes, is limited and should be explored further. In short, TRPC6 presents as a strong therapeutic target for the treatment of DKD given its widespread involvement in multiple disease-instigating pathways in the kidney of diabetic patients and experimental models. Importantly, the TRPC6 channel, as are many other channels, is a component of complex pathways. Therefore, in addition to direct targeting of TRPC6, targeting up- or downstream signaling pathways of the TRPC6 channel can be potentially important for drug development in DKD.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
• Dryer SE, Roshanravan H, Kim EY. TRPC channels: Regulation, dysregulation and contributions to chronic kidney diseaseBiochim Biophys Acta Mol basis Dis. 2019. https://doi.org/10.1016/j.bbadis.2019.04.001 Excellent recent review summarizing the role of TRPC6 and other TRPC channels in regulation of renal function. Nice discussion of contradictory topics in the area.
Staruschenko A. Hypertension and diabetes mellitus: the chicken and egg problem. Hypertension. 2017;69(5):787–8. https://doi.org/10.1161/HYPERTENSIONAHA.117.08671.
DeMarco VG, Aroor AR, Sowers JR. The pathophysiology of hypertension in patients with obesity. Nat Rev Endocrinol. 2014;10(6):364–76. https://doi.org/10.1038/nrendo.2014.44.
Rieg T, Vallon V. Development of SGLT1 and SGLT2 inhibitors. Diabetologia. 2018;61(10):2079–86. https://doi.org/10.1007/s00125-018-4654-7.
Vallon V, Thomson SC. Cardiovascular and renal benefits of SGLT2 inhibition: insights from CANVAS. Nat Rev Nephrol. 2017;13:517. https://doi.org/10.1038/nrneph.2017.113.
Giugliano D, De Nicola L, Maiorino MI, Bellastella G, Esposito K. Type 2 diabetes and the kidney: insights from cardiovascular outcome trials. Diabetes Obes Metab. 2019. https://doi.org/10.1111/dom.13743.
Perkovic V, Jardine MJ, Neal B, Bompoint S, Heerspink HJL, Charytan DM, et al. Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. N Engl J Med. 2019. https://doi.org/10.1056/NEJMoa1811744.
Alicic RZ, Rooney MT, Tuttle KR. Diabetic kidney disease: challenges, Progress, and possibilities. Clin J Am Soc Nephrol. 2017;12(12):2032–45. https://doi.org/10.2215/cjn.11491116.
Chaudhari S, Ma R. Store-operated calcium entry and diabetic complications. Exp Biol Med. 2016;241(4):343–52. https://doi.org/10.1177/1535370215609693.
Wang Z, do Carmo JM, Aberdein N, Zhou X, Williams JM, da Silva AA, et al. Synergistic interaction of hypertension and diabetes in promoting kidney injury and the role of endoplasmic reticulum stress. Hypertension. 2017;69(5):879–91. https://doi.org/10.1161/HYPERTENSIONAHA.116.08560.
Trebak M, Putney JW Jr. ORAI calcium channels. Physiology. 2017;32(4):332–42. https://doi.org/10.1152/physiol.00011.2017.
Wang Y, Deng X, Gill DL. Calcium signaling by STIM and Orai: intimate coupling details revealed. Sci Signal. 2010;3(148):pe42. https://doi.org/10.1126/scisignal.3148pe42.
Liao Y, Erxleben C, Yildirim E, Abramowitz J, Armstrong DL, Birnbaumer L. Orai proteins interact with TRPC channels and confer responsiveness to store depletion. Proc Natl Acad Sci U S A. 2007;104(11):4682–7. https://doi.org/10.1073/pnas.0611692104.
Liao Y, Plummer NW, George MD, Abramowitz J, Zhu MX, Birnbaumer L. A role for Orai in TRPC-mediated Ca2+ entry suggests that a TRPC:Orai complex may mediate store and receptor operated Ca2+ entry. Proc Natl Acad Sci U S A. 2009;106(9):3202–6. https://doi.org/10.1073/pnas.0813346106.
Abramowitz J, Birnbaumer L. Physiology and pathophysiology of canonical transient receptor potential channels. FASEB J. 2009;23(2):297–328. https://doi.org/10.1096/fj.08-119495.
Ma R, Chaudhari S, Li W. Canonical transient receptor potential 6 channel: a new target of reactive oxygen species in renal physiology and pathology. Antioxid Redox Signal. 2016;25(13):732–48. https://doi.org/10.1089/ars.2016.6661.
Ilatovskaya DV, Staruschenko A. TRPC6 channel as an emerging determinant of the podocyte injury susceptibility in kidney diseases. Am J Physiol Renal Physiol. 2015;309(5):F393–7. https://doi.org/10.1152/ajprenal.00186.2015.
Dryer SE, Kim EY. Permeation and rectification in canonical transient receptor potential-6 (TRPC6) channels. Front Physiol. 2018;9:1055. https://doi.org/10.3389/fphys.2018.01055.
Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature. 1999;397(6716):259–63. https://doi.org/10.1038/16711.
Venkatachalam K, Zheng F, Gill DL. Regulation of canonical transient receptor potential (TRPC) channel function by diacylglycerol and protein kinase C. J Biol Chem. 2003;278(31):29031–40. https://doi.org/10.1074/jbc.M302751200.
Bousquet SM, Monet M, Boulay G. Protein kinase C-dependent phosphorylation of transient receptor potential canonical 6 (TRPC6) on serine 448 causes channel inhibition. J Biol Chem. 2010;285(52):40534–43. https://doi.org/10.1074/jbc.M110.160051.
Inoue R, Jensen LJ, Jian Z, Shi J, Hai L, Lurie AI, et al. Synergistic activation of vascular TRPC6 channel by receptor and mechanical stimulation via phospholipase C/diacylglycerol and phospholipase A2/omega-hydroxylase/20-HETE pathways. Circ Res. 2009;104(12):1399–409. https://doi.org/10.1161/circresaha.108.193227.
Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science. 2005;308(5729):1801–4.
Reiser J, Polu KR, Moller CC, Kenlan P, Altintas MM, Wei C, et al. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat Genet. 2005;37(7):739–44.
Heeringa SF, Moller CC, Du J, Yue L, Hinkes B, Chernin G, et al. A novel TRPC6 mutation that causes childhood FSGS. PLoS One. 2009;4(11):e7771.
Buscher AK, Kranz B, Buscher R, Hildebrandt F, Dworniczak B, Pennekamp P, et al. Immunosuppression and renal outcome in congenital and pediatric steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol. 2010;5(11):2075–84. https://doi.org/10.2215/cjn.01190210.
Riehle M, Buscher AK, Gohlke BO, Kassmann M, Kolatsi-Joannou M, Brasen JH, et al. TRPC6 G757D loss-of-function mutation associates with FSGS. J Am Soc Nephrol. 2016;27(9):2771–83. https://doi.org/10.1681/ASN.2015030318.
Obeidova L, Reiterova J, Lnenicka P, Stekrova J, Safrankova H, Kohoutova M, et al. TRPC6 gene variants in Czech adult patients with focal segmental glomerulosclerosis and minimal change disease. Folia Biol. 2012;58(4):173–6.
Barua M, Brown EJ, Charoonratana VT, Genovese G, Sun H, Pollak MR. Mutations in the INF2 gene account for a significant proportion of familial but not sporadic focal and segmental glomerulosclerosis. Kidney Int. 2013;83(2):316–22. https://doi.org/10.1038/ki.2012.349.
Santin S, Ars E, Rossetti S, Salido E, Silva I, Garcia-Maset R, et al. TRPC6 mutational analysis in a large cohort of patients with focal segmental glomerulosclerosis. Nephrol Dial Transplant. 2009;24(10):3089–96. https://doi.org/10.1093/ndt/gfp229.
Hofstra JM, Lainez S, van Kuijk WH, Schoots J, Baltissen MP, Hoefsloot LH, et al. New TRPC6 gain-of-function mutation in a non-consanguineous Dutch family with late-onset focal segmental glomerulosclerosis. Nephrol Dial Transplant. 2013;28(7):1830–8. https://doi.org/10.1093/ndt/gfs572.
Buscher AK, Konrad M, Nagel M, Witzke O, Kribben A, Hoyer PF, et al. Mutations in podocyte genes are a rare cause of primary FSGS associated with ESRD in adult patients. Clin Nephrol. 2012;78(1):47–53.
Gigante M, Caridi G, Montemurno E, Soccio M, d'Apolito M, Cerullo G, et al. TRPC6 mutations in children with steroid-resistant nephrotic syndrome and atypical phenotype. Clin J Am Soc Nephrol. 2011;6(7):1626–34. https://doi.org/10.2215/cjn.07830910.
Mir S, Yavascan O, Berdeli A, Sozeri B. TRPC6 gene variants in Turkish children with steroid-resistant nephrotic syndrome. Nephrol Dial Transplant. 2012;27(1):205–9. https://doi.org/10.1093/ndt/gfr202.
Gheissari A, Meamar R, Kheirollahi M, Rouigari M, Dehbashi M, Dehghani L, et al. TRPC6 mutational analysis in Iranian children with focal segmental glomerulosclerosis. Iran J Kidney Dis. 2018;12(6):341–9.
Zhu B, Chen N, Wang ZH, Pan XX, Ren H, Zhang W, et al. Identification and functional analysis of a novel TRPC6 mutation associated with late onset familial focal segmental glomerulosclerosis in Chinese patients. Mutat Res. 2009;664(1–2):84–90. https://doi.org/10.1016/j.mrfmmm.2008.11.021.
Toth-Manikowski S, Atta MG. Diabetic kidney disease: pathophysiology and therapeutic targets. J Diabetes Res. 2015;2015:697010. https://doi.org/10.1155/2015/697010.
Ilatovskaya DV, Palygin O, Chubinskiy-Nadezhdin V, Negulyaev YA, Ma R, Birnbaumer L, et al. Angiotensin II has acute effects on TRPC6 channels in podocytes of freshly isolated glomeruli. Kidney Int. 2014;86(3):506–14. https://doi.org/10.1038/ki.2014.71.
Anderson M, Kim EY, Hagmann H, Benzing T, Dryer SE. Opposing effects of podocin on the gating of podocyte TRPC6 channels evoked by membrane stretch or diacylglycerol. Am J Physiol Cell Physiol. 2013;305(3):C276–89. https://doi.org/10.1152/ajpcell.00095.2013.
Dietrich A, Gudermann T. TRPC6: physiological function and pathophysiological relevance. Handb Exp Pharmacol. 2014;222:157–88. https://doi.org/10.1007/978-3-642-54215-2_7.
Ilatovskaya DV, Palygin O, Levchenko V, Staruschenko A. Pharmacological characterization of the P2 receptors profile in the podocytes of the freshly isolated rat glomeruli. Am J Physiol Cell Physiol. 2013;305(10):C1050–9. https://doi.org/10.1152/ajpcell.00138.2013.
Palygin O, Ilatovskaya DV, Staruschenko A. Protease-activated receptors in kidney disease progression. Am J Physiol Renal Physiol. 2016;311(6):F1140–F4. https://doi.org/10.1152/ajprenal.00460.2016.
Roshanravan H, Dryer SE. ATP acting through P2Y receptors causes activation of podocyte TRPC6 channels: role of podocin and reactive oxygen species. Am J Physiol Renal Physiol. 2014;306(9):F1088–97. https://doi.org/10.1152/ajprenal.00661.2013.
Anderson M, Roshanravan H, Khine J, Dryer SE. Angiotensin II activation of TRPC6 channels in rat podocytes requires generation of reactive oxygen species. J Cell Physiol. 2014;229(4):434–42. https://doi.org/10.1002/jcp.24461.
Wang L, Jirka G, Rosenberg PB, Buckley AF, Gomez JA, Fields TA, et al. Gq signaling causes glomerular injury by activating TRPC6. J Clin Invest. 2015;125(5):1913–26. https://doi.org/10.1172/JCI76767.
Ilatovskaya DV, Palygin O, Levchenko V, Endres BT, Staruschenko A. The role of angiotensin II in glomerular volume dynamics and podocyte calcium handling. Sci Rep. 2017;7(1):299. https://doi.org/10.1038/s41598-017-00406-2.
Ilatovskaya DV, Levchenko V, Lowing A, Shuyskiy LS, Palygin O, Staruschenko A. Podocyte injury in diabetic nephropathy: implications of angiotensin II-dependent activation of TRPC channels. Sci Rep. 2015;5:17637. https://doi.org/10.1038/srep17637.
Eckel J, Lavin PJ, Finch EA, Mukerji N, Burch J, Gbadegesin R, et al. TRPC6 enhances angiotensin II-induced albuminuria. J Am Soc Nephrol. 2011;22(3):526–35.
Tian D, Jacobo SM, Billing D, Rozkalne A, Gage SD, Anagnostou T, et al. Antagonistic regulation of actin dynamics and cell motility by TRPC5 and TRPC6 channels. Sci Signal. 2010;3(145):ra77.
Sonneveld R, van der Vlag J, Baltissen MP, Verkaart SA, Wetzels JF, Berden JH, et al. Glucose specifically regulates TRPC6 expression in the podocyte in an AngII-dependent manner. Am J Pathol. 2014;184(6):1715–26. https://doi.org/10.1016/j.ajpath.2014.02.008.
Nijenhuis T, Sloan AJ, Hoenderop JG, Flesche J, van Goor H, Kistler AD, et al. Angiotensin II contributes to podocyte injury by increasing TRPC6 expression via an NFAT-mediated positive feedback signaling pathway. Am J Pathol. 2011;179(4):1719–32. https://doi.org/10.1016/j.ajpath.2011.06.033.
Zhang H, Ding J, Fan Q, Liu S. TRPC6 up-regulation in Ang II-induced podocyte apoptosis might result from ERK activation and NF-kappaB translocation. Exp Biol Med. 2009;234(9):1029–36.
Hayek SS, Koh KH, Grams ME, Wei C, Ko YA, Li J, et al. A tripartite complex of suPAR, APOL1 risk variants and alphavbeta3 integrin on podocytes mediates chronic kidney disease. Nat Med. 2017;23(8):945–53. https://doi.org/10.1038/nm.4362.
Rinschen MM, Huesgen PF, Koch RE. The podocyte protease web: uncovering the gatekeepers of glomerular disease. Am J Physiol Renal Physiol. 2018;315(6):F1812-F1816.. https://doi.org/10.1152/ajprenal.00380.2018.
Kim EY, Roshanravan H, Dryer SE. Changes in podocyte TRPC channels evoked by plasma and sera from patients with recurrent FSGS and by putative glomerular permeability factors. Biochim Biophys Acta. 2017;1863(9):2342–54. https://doi.org/10.1016/j.bbadis.2017.06.010.
Kim EY, Hassanzadeh Khayyat N, Dryer SE. Mechanisms underlying modulation of podocyte TRPC6 channels by suPAR: role of NADPH oxidases and Src family tyrosine kinases. Biochim Biophys Acta Mol Basis Dis. 2018;1864(10):3527–36. https://doi.org/10.1016/j.bbadis.2018.08.007.
Dande RR, Peev V, Altintas MM, Reiser J. Soluble urokinase receptor and the kidney response in diabetes mellitus. J Diabetes Res. 2017;2017:3232848. https://doi.org/10.1155/2017/3232848.
Hayek SS, Sever S, Ko Y-A, Trachtman H, Awad M, Wadhwani S, et al. Soluble urokinase receptor and chronic kidney disease. N Engl J Med. 2015;373(20):1916–25. https://doi.org/10.1056/NEJMoa1506362.
Guthoff M, Wagner R, Randrianarisoa E, Hatziagelaki E, Peter A, Häring H-U, et al. Soluble urokinase receptor (suPAR) predicts microalbuminuria in patients at risk for type 2 diabetes mellitus. Sci Rep. 2017;7:40627. https://doi.org/10.1038/srep40627.
Guan Y, Nakano D, Zhang Y, Li L, Liu W, Nishida M, et al. A protease-activated receptor-1 antagonist protects against podocyte injury in a mouse model of nephropathy. J Pharmacol Sci. 2017. https://doi.org/10.1016/j.jphs.2017.09.002.
Waasdorp M, Duitman J, Florquin S, Spek CA. Vorapaxar treatment reduces mesangial expansion in streptozotocin-induced diabetic nephropathy in mice. Oncotarget. 2018;9(31):21655–62. https://doi.org/10.18632/oncotarget.25069.
Waasdorp M, Duitman J, Florquin S, Spek CA. Protease-activated receptor-1 deficiency protects against streptozotocin-induced diabetic nephropathy in mice. Sci Rep. 2016;6:33030. https://doi.org/10.1038/srep33030.
Kitamoto Y, Tomita K, Imamura T. Assessment of thrombin in the urine of glomerulonephritic patients by enzyme-linked immunosorbent assay. Ann Clin Biochem. 2004;41(Pt 2):133–7. https://doi.org/10.1258/000456304322880023.
Bauer KA, Weiss LM, Sparrow D, Vokonas PS, Rosenberg RD. Aging-associated changes in indices of thrombin generation and protein C activation in humans. Normative Aging Study. J Clin Invest. 1987;80(6):1527–34. https://doi.org/10.1172/JCI113238.
Konieczynska M, Fil K, Bazanek M, Undas A. Prolonged duration of type 2 diabetes is associated with increased thrombin generation, prothrombotic fibrin clot phenotype and impaired fibrinolysis. Thromb Haemost. 2014;111(4):685–93. https://doi.org/10.1160/th13-07-0566.
Palygin O, Ilatovskaya DV, Levchenko V, Klemens CA, Dissanayake L, Williams AM, et al. Characterization of purinergic receptor expression in ARPKD cystic epithelia. Purinergic Signal. 2018. https://doi.org/10.1007/s11302-018-9632-5.
Marko L, Mannaa M, Haschler TN, Kramer S, Gollasch M. Renoprotection: focus on TRPV1, TRPV4, TRPC6 and TRPM2. Acta Physiol. 2017;219(3):589–612. https://doi.org/10.1111/apha.12828.
•• Ilatovskaya DV, Blass G, Palygin O, Levchenko V, Pavlov TS, Grzybowski MN, et al. A NOX4/TRPC6 pathway in podocyte calcium regulation and renal damage in diabetic kidney disease. J Am Soc Nephrol. 2018;29(7):1917–27. https://doi.org/10.1681/ASN.2018030280 This study demostrate that H 2 O 2 stimulates calcium influx via TRPC6 channels, and podocytes isolated from TRPC6-knockout mice are protected from injury induced by H 2 O 2 . Furthermore, diabetes-induced increase in basal and Ang II–elicited calcium flux in podocytes was blunted in STZ-SS Nox4-/- rats.
Jha JC, Banal C, Okabe J, Gray SP, Hettige T, Chow BSM, et al. NADPH oxidase Nox5 accelerates renal injury in diabetic nephropathy. Diabetes. 2017;66(10):2691–703. https://doi.org/10.2337/db16-1585.
Graham S, Gorin Y, Abboud HE, Ding M, Lee DY, Shi H, et al. Abundance of TRPC6 protein in glomerular mesangial cells is decreased by ROS and PKC in diabetes. Am J Physiol Cell Physiol. 2011;301(2):C304–15. https://doi.org/10.1152/ajpcell.00014.2011.
Chiluiza D, Krishna S, Schumacher VA, Schlondorff J. Gain-of-function mutations in transient receptor potential C6 (TRPC6) activate extracellular signal-regulated kinases 1/2 (ERK1/2). J Biol Chem. 2013;288(25):18407–20. https://doi.org/10.1074/jbc.M113.463059.
Azumaya CM, Sierra-Valdez F, Cordero-Morales JF, Nakagawa T. Cryo-EM structure of the cytoplasmic domain of murine transient receptor potential cation channel subfamily C member 6 (TRPC6). J Biol Chem. 2018;293(26):10381–91. https://doi.org/10.1074/jbc.RA118.003183.
•• Tang Q, Guo W, Zheng L, Wu JX, Liu M, Zhou X, et al. Structure of the receptor-activated human TRPC6 and TRPC3 ion channels. Cell Res. 2018;28(7):746–55. https://doi.org/10.1038/s41422-018-0038-2 This is the first description of the structure of human TRPC6 homotetramer.
Staruschenko A. TRPC6 in diabetic kidney disease: good guy or bad guy? Kidney Int. 2019;95(2):256–8. https://doi.org/10.1016/j.kint.2018.10.027.
Rudemiller NP, Mattson DL. Candidate genes for hypertension: insights from the Dahl S rat. Am J Physiol Renal Physiol. 2015;309(12):F993–5. https://doi.org/10.1152/ajprenal.00092.2015.
Pavlov TS, Levchenko V, O'Connor PM, Ilatovskaya DV, Palygin O, Mori T, et al. Deficiency of renal cortical EGF increases ENaC activity and contributes to salt-sensitive hypertension. J Am Soc Nephrol. 2013;24(7):1053–62. https://doi.org/10.1681/ASN.2012080839.
Feng D, Yang C, Geurts A, Kurth T, Liang M, Lazar J, et al. Increased expression of NAD(P)H oxidase subunit p67 phox in the renal medulla contributes to excess oxidative stress and salt-sensitive hypertension. Cell Metab. 2012;15:201–8.
Slaughter TN, Paige A, Spires D, Kojima N, Kyle PB, Garrett MR, et al. Characterization of the development of renal injury in Type-1 diabetic Dahl salt-sensitive rats. Am J Physiol Regul Integr Comp Physiol. 2013;305(7):R727–34. https://doi.org/10.1152/ajpregu.00382.2012.
Spires D, Ilatovskaya DV, Levchenko V, North PE, Geurts AM, Palygin O, et al. Protective role of Trpc6 knockout in the progression of diabetic kidney disease. Am J Physiol Renal Physiol. 2018;315(4):F1091–F7. https://doi.org/10.1152/ajprenal.00155.2018.
Khayyat NH, Kim EY, Dryer SE. TRPC6 inactivation does not protect against diabetic kidney disease in streptozotocin (STZ)-treated Sprague-Dawley rats. FASEB J. 2019;33(1_supplement):567.3 (Abstract:851.9)). https://doi.org/10.1096/fasebj.2019.33.1_supplement.567.3.
• Kim EY, Yazdizadeh Shotorbani P, Dryer SE. Trpc6 inactivation confers protection in a model of severe nephrosis in rats. J Mol Med. 2018;96(7):631–44. https://doi.org/10.1007/s00109-018-1648-3 This study revealed that genetic inactivation of TRPC6 in Sprague-Dawley rats markedly reduced albuminuria, glomerulosclerosis, and ultrastructural changes in glomeruli that occurred during the chronic phase of PAN nephrosis.
Brosius FC, Alpers CE, Bottinger EP, Breyer MD, Coffman TM, Gurley SB, et al. Mouse models of diabetic nephropathy. J Am Soc Nephrol. 2009;20(12):2503–12. https://doi.org/10.1681/asn.2009070721.
Wang L, Chang JH, Buckley AF, Spurney RF. Knockout of TRPC6 promotes insulin resistance and exacerbates glomerular injury in Akita mice. Kidney Int. 2019;95(2):321–32. https://doi.org/10.1016/j.kint.2018.09.026.
Wang Y, Jarad G, Tripathi P, Pan M, Cunningham J, Martin DR, et al. Activation of NFAT signaling in podocytes causes glomerulosclerosis. J Am Soc Nephrol. 2010;21(10):1657–66. https://doi.org/10.1681/asn.2009121253.
Schlondorff J, del Camino D, Carrasquillo R, Lacey V, Pollak MR. TRPC6 mutations associated with focal segmental glomerulosclerosis cause constitutive activation of NFAT-dependent transcription. Am J Physiol Cell Physiol. 2009;296(3):C558–C69.
Ma R, Xu Y, Zhou H, Zhang D, Yao D, Song L, et al. Participation of the AngII/TRPC6/NFAT axis in the pathogenesis of podocyte injury in rats with type 2 diabetes. Mol Med Rep. 2019;19(3):2421–30. https://doi.org/10.3892/mmr.2019.9871.
Ma R, Liu L, Jiang W, Yu Y, Song H. FK506 ameliorates podocyte injury in type 2 diabetic nephropathy by down-regulating TRPC6 and NFAT expression. Int J Clin Exp Pathol. 2015;8(11):14063–74.
Fu Y, Wang C, Zhang D, Xin Y, Li J, Zhang Y, et al. Increased TRPC6 expression is associated with tubular epithelial cell proliferation and inflammation in diabetic nephropathy. Mol Immunol. 2018;94:75–81. https://doi.org/10.1016/j.molimm.2017.12.014.
Rajani R, Pastor-Soler NM, Hallows KR. Role of AMP-activated protein kinase in kidney tubular transport, metabolism, and disease. Curr Opin Nephrol Hypertens. 2017;26(5):375–83. https://doi.org/10.1097/mnh.0000000000000349.
Rachubik P, Szrejder M, Rogacka D, Audzeyenka I, Rychlowski M, Angielski S, et al. The TRPC6-AMPK pathway is involved in insulin-dependent cytoskeleton reorganization and glucose uptake in cultured rat podocytes. Cell Physiol Biochem. 2018;51(1):393–410. https://doi.org/10.1159/000495236.
Ji T, Zhang C, Ma L, Wang Q, Zou L, Meng K, et al. TRPC6-mediated Ca2+ signaling is required for hypoxia-induced autophagy in human podocytes. Cell Physiol Biochem. 2018;48(4):1782–92. https://doi.org/10.1159/000492351.
Huang H, You Y, Lin X, Tang C, Gu X, Huang M, et al. Inhibition of TRPC6 signal pathway alleviates podocyte injury induced by TGF-beta1. Cell Physiol Biochem. 2017;41(1):163–72. https://doi.org/10.1159/000455985.
Yu L, Lin Q, Liao H, Feng J, Dong X, Ye J. TGF-beta1 induces podocyte injury through Smad3-ERK-NF-kappaB pathway and Fyn-dependent TRPC6 phosphorylation. Cell Physiol Biochem. 2010;26(6):869–78. https://doi.org/10.1159/000323996.
Liu B, He X, Li S, Xu B, Birnbaumer L, Liao Y. Deletion of diacylglycerol-responsive TRPC genes attenuates diabetic nephropathy by inhibiting activation of the TGFbeta1 signaling pathway. Am J Transl Res. 2017;9(12):5619–30.
• Lin BL, Matera D, Doerner JF, Zheng N, Del Camino D, Mishra S, et al. In vivo selective inhibition of TRPC6 by antagonist BI 749327 ameliorates fibrosis and dysfunction in cardiac and renal disease. Proc Natl Acad Sci U S A. 2019. https://doi.org/10.1073/pnas.1815354116 In this study, newly developed orally bioavailable TRPC6-specific inhibitor, BI 749327, revealed its capacity to improve cardiac function in the context of abnormal hemodynamic stress. Similarly, BI 749327 suppressed myofibroblast activation and fibrosis in a renal disease model.
Kim EY, Shotorbani PY, Dryer SE. TRPC6 inactivation does not affect loss of renal function in nephrotoxic serum glomerulonephritis in rats, but reduces severity of glomerular lesions. Biochem Biophys Rep. 2019;17:139–50. https://doi.org/10.1016/j.bbrep.2018.12.006.
Li W, Ding Y, Smedley C, Wang Y, Chaudhari S, Birnbaumer L, et al. Increased glomerular filtration rate and impaired contractile function of mesangial cells in TRPC6 knockout mice. Sci Rep. 2017;7(1):4145. https://doi.org/10.1038/s41598-017-04067-z.
Wu YL, Xie J, An SW, Oliver N, Barrezueta NX, Lin MH, et al. Inhibition of TRPC6 channels ameliorates renal fibrosis and contributes to renal protection by soluble klotho. Kidney Int. 2017;91(4):830–41. https://doi.org/10.1016/j.kint.2016.09.039.
Hofstra JM, Coenen MJ, Schijvenaars MM, Berden JH, van der Vlag J, Hoefsloot LH, et al. TRPC6 single nucleotide polymorphisms and progression of idiopathic membranous nephropathy. PLoS One. 2014;9(7):e102065. https://doi.org/10.1371/journal.pone.0102065.
Moller CC, Wei C, Altintas MM, Li J, Greka A, Ohse T, et al. Induction of TRPC6 channel in acquired forms of proteinuric kidney disease. J Am Soc Nephrol. 2007;18(1):29–36.
Chen Y, Lin L, Tao X, Song Y, Cui J, Wan J. The role of podocyte damage in the etiology of ischemia-reperfusion acute kidney injury and post-injury fibrosis. BMC Nephrol. 2019;20(1):106. https://doi.org/10.1186/s12882-019-1298-x.
Yu Y, Zhang L, Xu G, Wu Z, Li Q, Gu Y, et al. Angiotensin II type I receptor agonistic autoantibody induces podocyte injury via activation of the TRPC6- calcium/calcineurin pathway in pre-eclampsia. Kidney Blood Press Res. 2018;43(5):1666–76. https://doi.org/10.1159/000494744.
Funding
Research in the authors’ laboratories is supported by the American Heart Association 16EIA26720006, 17SDG33660149, and 18PRE34030127, the National Heart, Lung, and Blood Institute R35 HL135749, and the Department of Veteran Affairs I01 BX004024.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no conflicts of interest relevant to this manuscript.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Mechanisms of Hypertension and Target-Organ Damage
Rights and permissions
About this article

Cite this article
Staruschenko, A., Spires, D. & Palygin, O. Role of TRPC6 in Progression of Diabetic Kidney Disease. Curr Hypertens Rep 21, 48 (2019). https://doi.org/10.1007/s11906-019-0960-9
Published:
DOI: https://doi.org/10.1007/s11906-019-0960-9