
Abstract
Diabetic nephropathy (DN) is the leading cause of kidney failure in the world and has a natural history of progressive albuminuria. Furthermore, diabetic patients with albuminuria have a greatly increased chance of dying due to cardiovascular events such as stroke and heart attacks. It is now clear that the podocyte is a key cell in the prevention of albuminuria and an important early target cell in DN. Therefore, understanding the critical factors and signalling pathways that control this cell in the setting of diabetes are highly desirable. In this chapter we review recent work which has increased the understanding of the role of alterations in the unique biology of the podocyte in the pathogenesis of DN.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Podocyte
- Albuminuria
- Hyperglycaemia
- Insulin resistance
- Hypertrophy
- Epithelial-mesenchymal transition (EMT)
- Endoplasmic reticulum stress
- Autophagy
- Epigenetics
Introduction
Diabetic nephropathy (DN) is the leading cause of kidney failure, accounting for approximately a third of patients in the UK and half of patients in the USA entering end-stage renal failure and requiring dialysis or kidney transplantation. It is increasing rapidly due to the global epidemic of type 2 diabetes. In most cases DN is characterized by progressive albuminuria due to damage to the glomerular filtration barrier of the kidney. Initially this begins with the loss of small amounts of urinary albumin in the range of 30–300 mg/L (microalbuminuria), but as the kidney damage progresses, this develops into macroalbuminuria (>300 mg/L). Diabetic patients with albuminuria have a greatly increased chance of dying due to cardiovascular events such as stroke and heart attacks [1]. It is now clear that the glomerular podocyte is a key target cell in the prevention of albuminuria. Landmark genetic and biological studies over the last decade point compellingly to the podocyte as a critical cell in maintaining glomerular filtration barrier function and an important early target cell in DN [1, 2]. There are now over 50 human genetic mutations associated with albuminuria , all of which code for proteins found in the podocyte [3]. Podocyte cell injury plays a pivotal role in the pathogenesis of diabetic nephropathy. The characteristic podocyte response to injury or cell stress is actin cytoskeleton reorganization, which typically leads to foot process effacement, resulting in proteinuria. This is followed by irreversible podocyte depletion which coincides with the progression of glomerular disease, as these cells do not have the ability to proliferate and regenerate. Numerous human and experimental studies have demonstrated that the podocyte number or density was diminished during DN. Reduced podocyte number is also a predictor of DN progression [4,5,6,7].
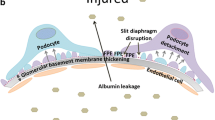
The glomerulus is the filtration unit of the kidney and is composed of a bundle of capillaries, which are highly permeable to water, and yet can selectively allow passage of solutes while retaining larger macromolecules. This selectivity is achieved through the action of the glomerular filtration barrier . The glomerular filtration barrier consists of the glomerular basement membrane, glomerular endothelial cells and glomerular epithelial cells or podocytes. Podocytes are terminally differentiated epithelial cells that are critical in preventing protein passage across the filtration barrier. Podocytes have branching and interdigitating processes, and filtration takes place through slits between these processes. The slit diaphragm , a critical component of the filtration barrier, is an ultra-thin zipper-like structure that bridges the gap between interdigitating podocyte foot processes. The slit diaphragm is a cell junction and signalling complex essential for regulating podocyte cytoskeletal dynamics. Podocytes have a remarkably elaborate and highly specialized cell biology and morphology which are essential for maintaining glomerular function and integrity in healthy kidneys [2]. The podocyte is critically important in preventing albuminuria. Damage to or loss of podocytes is linked to the development of albuminuria and occurs early in the progression of diabetic nephropathy [4, 6, 8, 9]. Although good progress has been made in recent years, the mechanisms underlying podocyte injury have not yet been fully identified. Therefore, understanding the critical factors and signalling pathways that control this cell in the setting of diabetes is highly desirable.
Damage/changes to the specialized cell biology of the podocyte have been reported to be due to podocyte hypertrophy, endoplasmic reticulum stress, autophagy, epithelial-to-mesenchymal transition (EMT) , detachment and apoptosis. Historically this was thought to be due to the diabetic milieu especially high glucose. However, we have shown that diminished insulin signalling to the podocyte is also detrimental to glomerular function [10,11,12]. We have recently described the role of the podocyte in the pathogenesis of DN [13]. In this chapter, we will discuss new insights into the mechanisms underlying podocyte injury in the progression of DN (displayed in Fig. 11.1) which may point to novel therapeutic targets to develop important renoprotective treatments for DN.

Schematic overview of pathophysiological mechanisms of podocyte damage in the pathogenesis of diabetic nephropathy. VEGF vascular endothelial growth factor, ER endoplasmic reticulum, HDAC histone deacetylase, SHP-1 protein tyrosine phosphatase type 1, mIR micro-RNA
Diabetic Environment and Insulin Sensitivity
Hyperglycaemia has been demonstrated to be a key factor underlying podocyte injury [14], and podocytes have been shown to be the direct target of circulating hormones, lipids and adipokines whose levels are altered in diabetes [13]. For example, free fatty acids (FFA) are elevated in insulin-resistant states, are involved in the pathogenesis of diabetic nephropathy and induce insulin resistance in human podocytes [15]. Interestingly insulin resistance has emerged as a key driver of impaired glomerular function and the development of renal complications. Insulin resistance plays a major role in the pathogenesis of both type 1 and type 2 diabetes [16, 17] being associated with albuminuria and nephropathy [18, 19]. Insulin resistance is also associated with the development of albuminuria in nondiabetic individuals [20]. Furthermore, renal disease is also common among people with severe forms of genetic insulin resistance [21]. We have shown that the podocyte is a direct target for insulin action and that loss of this signalling leads to a diabetic nephropathy like phenotype importantly in the absence of hyperglycaemia [10]. Recently we have shown that degradation of the insulin receptor, caused by high levels of insulin, drives early podocyte insulin resistance and that both the IR and nephrin are required for full insulin sensitivity of this cell [22]. This could be highly relevant for the development of nephropathy in type 2 diabetic patients who are commonly hyperinsulinaemic in the early phases of their disease. In both type 1 and type 2 diabetes, glomerular insulin signalling is lost early in the development of kidney disease [23] suggesting that targeting, and enhancing, this pathway, in these settings, could be beneficial.
Podocyte EMT
A number of phenotypic and morphological changes are seen in the injured podocyte which are described as an epithelial-to-mesenchymal transition . These result from hyperglycaemia induced changes to several podocyte signalling pathways such as upregulation of the TGFβ/Smad, Wnt/β catenin/ and jagged/Notch pathways which have been described in detail in a recent review article [24]. Interestingly it has been demonstrated that high glucose induces glomerular endothelial cells to secrete exosomes that are internalized by podocytes causing podocyte EMT possibly via TGF-β1 in the exosomes and activation of podocyte Wnt/β-catenin signalling [25].
Podocyte Endoplasmic Reticular (ER) Stress and Autophagy
In a diabetic environment cellular metabolic overload results in increased cellular oxidative stress and ER-stress which leads to the activation of unfolded protein response (UPR) [26]. UPR is a positive cellular response that in its early phase either refolds accumulated unfolded proteins or degrades unfolded protein by the ubiquitin-proteasome pathway. This is probably extremely important for the podocyte as it a terminally differentiated cell with minimal capacity to replicate, so maintaining its cellular function under stress is crucial. Misfolded proteins are detected as a result of ER membrane stress which, in turn, activates several signalling events and triggers a compensatory response to prevent further accumulation of misfolded protein. However, when the unfolded protein and cellular damage exceed a threshold, chronic and unresolved stress results in a change from an adaptive to pro-apoptotic responses [26].
There is now evidence that glucose/oxidative stress-mediated ER stress plays a role in chronic vascular complications in DN [27]. Hyperglycaemia or increased glycation of proteins has been shown to mediate apoptosis partly through increases in ER stress in cultured murine podocytes [28, 29]. Activation of the UPR has also been observed in mouse glomerular mesangial cells exposed to glucose and glucosamine [30], and in kidneys from diabetic rats administered streptozotocin for 16 weeks [31]. Furthermore, microarray analysis of human biopsies from patients with established DN showed that UPR genes were upregulated proportionally to the severity of diabetic renal lesions [32].
Recently a link between podocyte insulin sensitivity and ER stress has been demonstrated. Madhusudhan et al. have elegantly shown that under diabetic conditions ER adaptive mechanisms are impaired in the podocyte and that this is exacerbated when the cell is rendered more insulin resistant by partially knocking down its insulin receptor in a podocyte -specific manner. Studying human and murine DN, they have shown that nephropathy was associated with alterations in the UPR with impairment of the nuclear translocation of XBP-1. Genetic ablation of the transcription factor XBP-1 or activation of ATF6 (downstream of XBP-1) in the podocyte of diabetic mice aggravates DN. Of interest, mice with genetically impaired podocyte insulin signalling exhibited impaired UPR (XBP-1 activation) that was associated with more severe diabetic kidney disease when compared with diabetic controls [33].
Autophagy, regulated by the mammalian target of rapamycin complex 1 (mTORC1) is, with the UPR, also essential to maintain cellular homeostasis and in the context of ER stress contributes towards the elimination of toxic and damaged cellular components [34]. Genetic loss of mTORC1 in podocytes or administration of rapamycin (a mTORC1 inhibitor) resulting in activation of autophagy [35] has been shown to prevent progressive DN [36, 37]. In contrast, mTORC1 activation in podocytes, resulting in inhibition of autophagy, leads to accelerated DN [38]. Loss of insulin sensitivity in cultured podocytes results in suppression of autophagy and addition of rapamycin in these cells attenuates insulin resistance [39]. In the future, understanding how to manipulate podocyte ER stress and autophagic pathways may prove fruitful in developing novel therapies for DN.
Podocyte Vascular Endothelial Growth Factors
In the past decade, it has become clear that several vascular endothelial growth factors are produced by the podocyte and are altered in diabetes. A key factor produced by the podocyte , which signals to the endothelium, is vascular endothelial growth factor A (VEGFA). Podocyte produced VEGFA is crucial for glomerular function both during development [40] and also in maturity [41]. It is also clear that its production needs to be tightly regulated as either too much or too little is detrimental [40].
Recent studies have shown a connection between insulin resistance and the subsequent production of VEGFA in podocytes [42]. This finding is likely to be important in the setting of DN with many elegant studies using transgenic mice highlighting the importance of podocyte VEGFA levels in the progression of this condition [43]. A new aspect of VEGFA signalling in the glomerulus is potential crosstalk between VEGFA secreted from podocytes and the GECs glycocalyx in the setting of diabetes. There is clear evidence that the GECs glycocalyx is lost both systemically and within the diabetic glomerulus and that this contributes to both cardiovascular and renal complications [44]. Mechanistically there are a number of pathways which led to loss of the glomerular glycocalyx including hyperglycaemia [45] and reactive oxygen species (ROS) [46].
During the early phases of diabetes, an increase in VEGFA causes glycocalyx shedding from the GECs. Furthermore, the inhibitory isoform of VEGFA, called VEGF-A165b, also plays a role in maintaining the GECs glycocalyx in diabetes. Oltean et al. [47] have shown that in diabetic patients with progressive nephropathy, the renal expression of VEGF-A165b is lost. They went on to develop several murine models of DN and have shown that genetic overexpression or pharmacological administration of VEGF-A165b to the mouse, acting through VEGF receptor 2 in the GECs, restores damaged glomerular endothelial glycocalyx and improves renal function. VEGF-A165b also improved the permeability of isolated human diabetic glomeruli suggesting the response is conserved across murine and human species [47].
Very recently another member of the VEGF family, VEGF-B, has been implicated in the development of diabetic nephropathy through causing increased podocyte lipid uptake and subsequently inducing insulin resistance in this cell type. This is believed to be through VEGF-B engaging with the VEGFR1 (Flt1) and Neuropilin-1 (NRP1) in glomerular endothelial cells and upregulation of fatty acid transporter protein 3 (FATP3) and FATP4 which facilitates the passage of free fatty acids through the filtration barrier and into the podocyte . Elegant studies by Falkevall et al. [48] in which VEGF-B was systemically genetically or pharmacologically inhibited in diabetic and high-fat fed mice have revealed that podocyte insulin sensitivity is increased and that mice are subsequently protected from DN. Furthermore, they show that in patients with diabetic nephropathy that their glomerular VEGF-B levels are increased, and this closely correlates with their degree of albuminuria. Genetic overexpression of VEGF-B in the podocyte mirrors these findings when the mice are fed a high-fat diet. Going forward it will be interesting to understand the precise mechanism through which lipids are taken up by podocytes when VEGF-B is increased in the glomerulus, the precise cellular origin of VEGF-B and if there are any VEGF-B receptors in the podocyte that facilitate lipid uptake. These could reveal novel therapeutic targets to prevent the progression of DN in the future.
Epigenetics
There is increasing evidence that epigenetic modifications , resulting from prolonged exposure to hyperglycaemia, play an important role in podocyte injury and diabetic nephropathy. These modifications include methylation of cytosine residues of the DNA and acetylation and methylation of lysine residues of the histone proteins which are the principal component of chromatin. These changes have been shown to continue even after the normalization of glucose levels explaining in part why diabetic complications persist in patients even after hyperglycaemia is controlled [49]. One of the first reported demonstrations of these epigenetic modifications in podocytes involved the adapter protein P66Shc which mediates receptor tyrosine kinase signalling and oxidative stress-induced apoptosis [50]. This protein has been strongly implicated in the pathogenesis of diabetic nephropathy as p66Shc-deficient mice are protected against this condition [51]. High glucose was shown in podocytes to induce hypomethylation and hyperacetylation of the p66Shc promoter resulting in high levels of p66Shc expression leading to mitochondrial p66Shc translocation, ROS generation and oxidative stress [50]. Subsequently, hyperglycaemia has also been shown to result in increased histone H3 lysine acetylation and histone H3 lysine methylation of the promotor of the tyrosine phosphatase SHP1 resulting in high expression of this protein and inhibition of insulin signalling pathways even after glycaemic control had been achieved [52, 53]. Furthermore, elevated circulating lipids, which are known to cause podocyte insulin resistance, have been shown to alter histone modifications of the FOXO1 promotor in podocytes, an effect again which is sustained after lipid levels have returned to normal [15, 54]. FOXO1 is a key regulator of gluconeogenic genes, and overexpression has been reported to ameliorate podocyte injury in diabetic animal models and high glucose-treated podocytes by decreasing apoptosis and promoting mitophagy, via regulation of the PTEN-induced PINK1/Parkin-dependent signalling and inhibiting epithelial-mesenchymal transition [55,56,57,58].
Histone acetylation , which is regulated by histone acetyltransferases (HATs) and histone deacetylases (HDAC), plays a crucial role in the regulation of gene expression [49]. The histone deacetylases are a family of enzymes (HDACS 1–11 and Sirts 1–7), several of which have been implicated in podocyte injury and the pathogenesis of diabetic nephropathy [59, 60]. Sirt 1 and 6 levels and activity are downregulated in high-glucose and AGE-BSA-treated podocytes and in diabetic animal models [59, 61,62,63]. Sirt1 deficiency has been shown to exacerbate podocyte injury in both diabetic and glomerular disease models [59, 60]. Sirt1, via crosstalk with AMPK, plays a key role in the regulation of insulin action in podocyte [61]. Furthermore, downregulation of Sirt1 in podocytes has been shown to result in epigenetic modulation and subsequent upregulation of the tight junction protein Claudin 1 . Upregulation of Claudin 1 activates the β-catenin/Snail pathway leading to downregulation of the slit diaphragm protein podocin and the actin-binding protein Synaptopodin resulting in podocyte foot process effacement [59]. Interestingly the effects of hyperglycaemia on the downregulation of both Sirt1 and AMPK could be ameliorated by treatment of the podocytes with metformin, a commonly used diabetes treatment, suggesting a potential mechanism by which this drug improves the insulin sensitivity of podocytes and prevents diabetes-related complications [64]. Sirt6 has been shown to protect against podocyte injury by reducing inflammation, blocking apoptosis, maintaining the actin cytoskeleton and promoting autophagy through epigenetic regulation of Notch signalling [63]. Therefore, both Sirt1 and 6 are required for the maintenance of a healthy podocyte phenotype and downregulation of these proteins as seen in diabetes leads to podocyte damage.
HDAC proteins have been shown to also have non-epigenetic roles in regulating the progression of podocyte injury in diabetes by deacetylating several nonhistone proteins. Sirt1 deacetylates the transcription factors FOXO4, NFκB and STAT 3, all of which play a role in diabetic nephropathy through increased expression of their target genes, and increased acetylation of these proteins has been observed in diabetic animal models and human diabetic kidneys [65]. Sirt1 also has a role in the maintenance of the podocyte actin cytoskeleton integrity by regulating the acetylation of the actin-binding and polymerizing protein cortactin [60]. HDAC4 expression is upregulated in streptozotocin-induced diabetic rats, kidneys from diabetic patients and in podocytes treated with high glucose, advanced glycation end products or TGF-β. HDAC4 regulates podocyte autophagy via deacetylation of the transcription factor STAT1 [66]. HDAC4 has also been shown to deacetylate nephrin, and high glucose leads to decreased nephrin acetylation leading to increased nephrin loss and podocyte apoptosis which is ameliorated by HDAC4 knockdown in podocytes [67]. This control of nephrin acetylation and loss is part of a complex pathway involving the reciprocal regulation of HDAC4 and the micro RNA(mIR)-29a [67]. In contrast to HDAC4, the levels of mIR-29a are reduced in high glucose-treated podocytes and in diabetic animals. HDAC4 reduces the expression of mIR-29a via decreased acetylation of its proximal promoter. In high glucose-treated podocytes, knockdown of HDAC4 leads to increased acetylation of the mIR-29a promoter, upregulated levels of mIR-29, increased nephrin acetylation and reduced podocyte apoptosis. Importantly HDAC4 is a target for mIR-29a and thus mIR-29a can reciprocally regulate the expression of this protein. In diabetic animals overexpressing mIR-29a nephrin levels are restored, HDAC signalling is reduced and podocyte viability and renal function are improved. Knockdown of mIR-29a leads to increased HDAC4 activity, podocyte apoptosis and renal damage. Therefore, a tight choreography of this reciprocal pathway is important for the health of the podocyte , and the above results suggest that this is deleteriously altered in diabetic nephropathy [67].
Interestingly, several other podocyte mIRs have been implicated recently in the pathogenesis of diabetic nephropathy. For example, the expression of mIR-27a is stimulated by high glucose in cultured podocytes and is upregulated in renal biopsy samples from patients with diabetic nephropathy. Increased mIR-27a expression in podocytes leads to decreased expression of PPARγ and subsequent activation of β-catenin signalling resulting in increased podocyte EMT and apoptosis [68]. Increases in the levels of miR-217 are also seen in high glucose-treated podocytes, and this has been linked to podocyte injury and insulin resistance via regulation of PTEN-mediated autophagy signalling [69]. Conversely loss of mIR-146a in podocytes, which is seen in both glomeruli from diabetic patients and animal models, leads to increased susceptibility to diabetes induced damage via upregulation of ErbB4 and Notch1 [70]. Levels of mIR-93 are also downregulated in the kidneys of experimental animal models of diabetes. mIR-93 through modulation of its target Msk2, a histone kinase and its target H3S10 plays a critical role in chromatin reorganization in podocytes. Importantly inducible expression of mIR-93 specifically in podocytes led to major improvements in key features of diabetic nephropathy in diabetic db/db mice including much reduced mesangial matrix expansion, and increased synaptopodin, and nephrin levels [71].
Finally, long noncoding RNAs have also been shown to play a role in the podocyte in the development of diabetic nephropathy. Long noncoding RNA (lncRNA) taurine-upregulated 1 (Tug1) was shown to regulate mitochondrial bioenergetics in podocytes by epigenetic targeting of expression of the transcription factor PPARγ coactivator 1α (PGC-1α). Tug1 expression is reduced in db/db mouse model of diabetes and overexpression of TUG1 in these animals led to key improvements in biochemical and histological features associated with diabetic nephropathy including rescued expression of PGC-1α and its downstream targets and improvements in podocyte mitochondrial bioenergetics [72].
Podocyte Targeted Treatment of Diabetic Nephropathy
There is evidence that strategies that enhance cellular insulin-sensitivity, including metformin and peroxisome proliferator-activated receptor gamma (PPARγ) agonists, such as rosiglitazone, are beneficial in preventing kidney damage in models of diabetic nephropathy in both type 1 and type 2 diseases, as well as other nondiabetic chronic kidney diseases [64, 73]. It is possible that these drugs are exerting part, or all, of their beneficial effects by directly enhancing insulin sensitivity of the podocyte . For example, we have shown that rosiglitazone directly augments insulin signalling in human immortalized podocytes in vitro [74]. Resveratrol, a non-flavonoid polyphenol, has been shown to have beneficial effects in the treatment of diabetic kidney disease and has recently been shown in a diabetic mouse model to protect podocytes against apoptosis by stimulating autophagy [75]. Furthermore, astragaloside IV, a traditional Chinese herbal remedy, has been reported to prevent the progression of diabetic nephropathy in streptozotocin-induced diabetic mice by attenuating ER stress and promoting autophagy in podocytes [76]. Therefore, understanding the mechanisms behind podocyte damage during diabetes is an important step in treating this condition and directly targeting the podocyte may be beneficial in kidney disease states especially diabetic nephropathy.
References
Brosius FC, Coward RJ. Podocytes, signaling pathways, and vascular factors in diabetic kidney disease. Adv Chronic Kidney Dis. 2014;21(3):304–10.
Welsh GI, Saleem MA. The podocyte cytoskeleton--key to a functioning glomerulus in health and disease. Nat Rev Nephrol. 2012;8(1):14–21.
Bierzynska A, Soderquest K, Koziell A. Genes and podocytes – new insights into mechanisms of podocytopathy. Front Endocrinol (Lausanne). 2014;5:226.
Pagtalunan ME, et al. Podocyte loss and progressive glomerular injury in type II diabetes. J Clin Invest. 1997;99(2):342–8.
Steffes MW, et al. Glomerular cell number in normal subjects and in type 1 diabetic patients. Kidney Int. 2001;59(6):2104–13.
Toyoda M, et al. Podocyte detachment and reduced glomerular capillary endothelial fenestration in human type 1 diabetic nephropathy. Diabetes. 2007;56(8):2155–60.
Meyer TW, Bennett PH, Nelson RG. Podocyte number predicts long-term urinary albumin excretion in pima Indians with type II diabetes and microalbuminuria. Diabetologia. 1999;42(11):1341–4.
Wolf G, Chen S, Ziyadeh FN. From the periphery of the glomerular capillary wall toward the center of disease: podocyte injury comes of age in diabetic nephropathy. Diabetes. 2005;54(6):1626–34.
Reddy GR, et al. The podocyte and diabetes mellitus: is the podocyte the key to the origins of diabetic nephropathy? Curr Opin Nephrol Hypertens. 2008;17(1):32–6.
Welsh GI, et al. Insulin signaling to the glomerular podocyte is critical for normal kidney function. Cell Metab. 2010;12(4):329–40.
Lay A, Coward RJ. Recent advances in our understanding of insulin signalling to the podocyte. Nephrol Dial Transplant. 2014;29(6):1127–33.
Coward RJ, et al. The human glomerular podocyte is a novel target for insulin action. Diabetes. 2005;54(11):3095–102.
Gnudi L, Coward RJM, Long DA. Diabetic nephropathy: perspective on novel molecular mechanisms. Trends Endocrinol Metab. 2016;27(11):820–30.
Welsh GI, Coward RJ. Podocytes, glucose and insulin. Curr Opin Nephrol Hypertens. 2010;19(4):379–84.
Lennon R, et al. Saturated fatty acids induce insulin resistance in human podocytes: implications for diabetic nephropathy. Nephrol Dial Transplant. 2009;24(11):3288–96.
Greenbaum CJ. Insulin resistance in type 1 diabetes. Diabetes Metab Res Rev. 2002;18(3):192–200.
Reaven GM, Banting lecture. Role of insulin resistance in human disease. Diabetes. 1988;37(12):1595–607.
Orchard TJ, et al. Nephropathy in type 1 diabetes: a manifestation of insulin resistance and multiple genetic susceptibilities? Further evidence from the Pittsburgh epidemiology of diabetes complication study. Kidney Int. 2002;62(3):963–70.
Bjornstad P, et al. Early diabetic nephropathy: a complication of reduced insulin sensitivity in type 1 diabetes. Diabetes Care. 2013;36(11):3678–83.
Pilz S, et al. Insulin sensitivity and albuminuria: the RISC study. Diabetes Care. 2014;37(6):1597–603.
Musso C, et al. Spectrum of renal diseases associated with extreme forms of insulin resistance. Clin J Am Soc Nephrol. 2006;1(4):616–22.
Lay AC, et al. Prolonged exposure of mouse and human podocytes to insulin induces insulin resistance through lysosomal and proteasomal degradation of the insulin receptor. Diabetologia. 2017;60:2299.
Mima A, et al. Glomerular-specific protein kinase C-beta-induced insulin receptor substrate-1 dysfunction and insulin resistance in rat models of diabetes and obesity. Kidney Int. 2011;79(8):883–96.
Ying Q, Wu G. Molecular mechanisms involved in podocyte EMT and concomitant diabetic kidney diseases: an update. Ren Fail. 2017;39(1):474–83.
Wu X, et al. Exosomes from high glucose-treated glomerular endothelial cells trigger the epithelial-mesenchymal transition and dysfunction of podocytes. Sci Rep. 2017;7(1):9371.
Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13(2):89–102.
Zhuang A, Forbes JM. Stress in the kidney is the road to pERdition: is endoplasmic reticulum stress a pathogenic mediator of diabetic nephropathy? J Endocrinol. 2014;222(3):R97–111.
Cao Y, et al. Role of endoplasmic reticulum stress in apoptosis of differentiated mouse podocytes induced by high glucose. Int J Mol Med. 2014;33(4):809–16.
Chen Y, et al. Effect of taurine-conjugated ursodeoxycholic acid on endoplasmic reticulum stress and apoptosis induced by advanced glycation end products in cultured mouse podocytes. Am J Nephrol. 2008;28(6):1014–22.
Cheng DW, et al. An analysis of high glucose and glucosamine-induced gene expression and oxidative stress in renal mesangial cells. Arch Physiol Biochem. 2006;112(4–5):189–218.
Liu G, et al. Apoptosis induced by endoplasmic reticulum stress involved in diabetic kidney disease. Biochem Biophys Res Commun. 2008;370(4):651–6.
Lindenmeyer MT, et al. Proteinuria and hyperglycemia induce endoplasmic reticulum stress. J Am Soc Nephrol. 2008;19(11):2225–36.
Madhusudhan T, et al. Defective podocyte insulin signalling through p85-XBP1 promotes ATF6-dependent maladaptive ER-stress response in diabetic nephropathy. Nat Commun. 2015;6:6496.
Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40(2):280–93.
Kim J, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–41.
Godel M, et al. Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J Clin Invest. 2011;121(6):2197–209.
Xiao T, et al. Rapamycin promotes podocyte autophagy and ameliorates renal injury in diabetic mice. Mol Cell Biochem. 2014;394(1–2):145–54.
Inoki K, et al. mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J Clin Invest. 2011;121(6):2181–96.
Xu Y, et al. Autophagy downregulation contributes to insulin resistance mediated injury in insulin receptor knockout podocytes in vitro. PeerJ. 2016;4:e1888.
Eremina V, et al. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Invest. 2003;111(5):707–16.
Eremina V, et al. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med. 2008;358(11):1129–36.
Hale LJ, et al. Insulin directly stimulates VEGF-A production in the glomerular podocyte. Am J Physiol Renal Physiol. 2013;305(2):F182–8.
Gnudi L, et al. Vascular growth factors play critical roles in kidney glomeruli. Clin Sci (Lond). 2015;129(12):1225–36.
Salmon AH, et al. Loss of the endothelial glycocalyx links albuminuria and vascular dysfunction. J Am Soc Nephrol. 2012;23(8):1339–50.
Singh A, et al. High glucose causes dysfunction of the human glomerular endothelial glycocalyx. Am J Physiol Renal Physiol. 2011;300(1):F40–8.
Singh A, et al. Reactive oxygen species modulate the barrier function of the human glomerular endothelial glycocalyx. PLoS One. 2013;8(2):e55852.
Oltean S, et al. Vascular endothelial growth factor-A165b is protective and restores endothelial glycocalyx in diabetic nephropathy. J Am Soc Nephrol. 2015;26:1889–904.
Falkevall A, et al. Reducing VEGF-B signaling ameliorates renal lipotoxicity and protects against diabetic kidney disease. Cell Metab. 2017;25(3):713–26.
Majumder S, Advani A. The epigenetic regulation of podocyte function in diabetes. J Diabetes Complicat. 2015;29(8):1337–44.
Bock F, et al. Activated protein C ameliorates diabetic nephropathy by epigenetically inhibiting the redox enzyme p66Shc. Proc Natl Acad Sci U S A. 2013;110(2):648–53.
Menini S, et al. Deletion of p66Shc longevity gene protects against experimental diabetic glomerulopathy by preventing diabetes-induced oxidative stress. Diabetes. 2006;55(6):1642–50.
Drapeau N, et al. Expression of SHP-1 induced by hyperglycemia prevents insulin actions in podocytes. Am J Physiol Endocrinol Metab. 2013;304(11):E1188–98.
Lizotte F, et al. Persistent insulin resistance in podocytes caused by epigenetic changes of SHP-1 in diabetes. Diabetes. 2016;65(12):3705–17.
Kumar S, Pamulapati H, Tikoo K. Fatty acid induced metabolic memory involves alterations in renal histone H3K36me2 and H3K27me3. Mol Cell Endocrinol. 2016;422:233–42.
Li W, et al. FoxO1 promotes Mitophagy in the podocytes of diabetic male mice via the PINK1/Parkin pathway. Endocrinology. 2017;158(7):2155–67.
Du M, et al. Overexpression of FOXO1 ameliorates the podocyte epithelial-mesenchymal transition induced by high glucose in vitro and in vivo. Biochem Biophys Res Commun. 2016;471(4):416–22.
Li W, et al. Effects of overexpressing FoxO1 on apoptosis in glomeruli of diabetic mice and in podocytes cultured in high glucose medium. Biochem Biophys Res Commun. 2016;478(2):612–7.
Guo F, et al. Effects of FoxO1 on podocyte injury in diabetic rats. Biochem Biophys Res Commun. 2015;466(2):260–6.
Hasegawa K, et al. Renal tubular Sirt1 attenuates diabetic albuminuria by epigenetically suppressing Claudin-1 overexpression in podocytes. Nat Med. 2013;19(11):1496–504.
Motonishi S, et al. Sirtuin1 maintains actin cytoskeleton by deacetylation of Cortactin in injured podocytes. J Am Soc Nephrol. 2015;26(8):1939–59.
Rogacka D, et al. SIRT1-AMPK crosstalk is involved in high glucose-dependent impairment of insulin responsiveness in primary rat podocytes. Exp Cell Res. 2016;349(2):328–38.
Chuang PY, et al. Alteration of forkhead box O (foxo4) acetylation mediates apoptosis of podocytes in diabetes mellitus. PLoS One. 2011;6(8):e23566.
Liu M, et al. Sirt6 deficiency exacerbates podocyte injury and proteinuria through targeting notch signaling. Nat Commun. 2017;8(1):413.
Rogacka D, et al. Metformin overcomes high glucose-induced insulin resistance of podocytes by pleiotropic effects on SIRT1 and AMPK. Biochim Biophys Acta. 2017;1864(1):115–25.
Liu R, et al. Role of transcription factor acetylation in diabetic kidney disease. Diabetes. 2014;63(7):2440–53.
Wang X, et al. Histone deacetylase 4 selectively contributes to podocyte injury in diabetic nephropathy. Kidney Int. 2014;86(4):712–25.
Lin CL, et al. MicroRNA-29a promotion of nephrin acetylation ameliorates hyperglycemia-induced podocyte dysfunction. J Am Soc Nephrol. 2014;25(8):1698–709.
Zhou Z, et al. MicroRNA-27a promotes podocyte injury via PPARgamma-mediated beta-catenin activation in diabetic nephropathy. Cell Death Dis. 2017;8(3):e2658.
Sun J, et al. Repression of miR-217 protects against high glucose-induced podocyte injury and insulin resistance by restoring PTEN-mediated autophagy pathway. Biochem Biophys Res Commun. 2017;483(1):318–24.
Lee HW, et al. Absence of miR-146a in podocytes increases risk of diabetic Glomerulopathy via up-regulation of ErbB4 and Notch-1. J Biol Chem. 2017;292(2):732–47.
Badal SS, et al. miR-93 regulates Msk2-mediated chromatin remodelling in diabetic nephropathy. Nat Commun. 2016;7:12076.
Long J, et al. Long noncoding RNA Tug1 regulates mitochondrial bioenergetics in diabetic nephropathy. J Clin Invest. 2016;126(11):4205–18.
Platt C, Coward RJ. Peroxisome proliferator activating receptor-gamma and the podocyte. Nephrol Dial Transplant. 2017;32(3):423–33.
Lennon R, et al. Rosiglitazone enhances glucose uptake in glomerular podocytes using the glucose transporter GLUT1. Diabetologia. 2009;52(9):1944–52.
Huang SS, et al. Resveratrol protects podocytes against apoptosis via stimulation of autophagy in a mouse model of diabetic nephropathy. Sci Rep. 2017;7:45692.
Guo H, et al. Astragaloside IV protects against podocyte injury via SERCA2-dependent ER stress reduction and AMPKalpha-regulated autophagy induction in streptozotocin-induced diabetic nephropathy. Sci Rep. 2017;7(1):6852.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Welsh, G.I., Coward, R.J. (2019). The Podocyte in Diabetic Nephropathy: Recent Advances. In: Roelofs, J., Vogt, L. (eds) Diabetic Nephropathy. Springer, Cham. https://doi.org/10.1007/978-3-319-93521-8_11
Download citation
DOI: https://doi.org/10.1007/978-3-319-93521-8_11
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-93520-1
Online ISBN: 978-3-319-93521-8
eBook Packages: MedicineMedicine (R0)