
Abstract
Purpose of Review
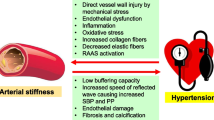
Increased arterial stiffness, an abnormal structural and functional change in the vascular wall, is a precursor for hypertension, coronary heart disease, stroke, and associated cardiovascular disease (CVD). The aim of this paper is to review the etiology of arterial stiffening and potential therapeutic approaches to modulate arterial fibrosis and stiffness.
Recent Findings
The Framingham Heart Study demonstrated that arterial stiffness is an independent predictor of CVD and related morbidity and mortality. Dysfunction of endothelial cells, vascular smooth muscle cells, extracellular matrix, and other functional elements of the vessel wall contribute to underlying pathophysiology of increased arterial stiffness. An activated renin-angiotensin-aldosterone system, oxidative stress, abnormal peri-vascular adipose tissue, inflammation, and increased sympathetic nervous system activity are associated with the development and progression of arterial fibrosis, stiffening, and associated CVD.
Summary
In this review, we will discuss the structural and function changes and mechanisms of the vessel wall in arterial stiffness and provide potential therapeutic strategies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Physiological arterial elasticity is an important vascular property for maintaining normal blood pressure. In individuals with increased arterial stiffness due to obesity, diabetes, aging, and atherosclerosis, this elasticity is compromised. With increased pulse wave velocity (PWV), reflected waves return faster and merge with the forward wave in systole, resulting in augmentation of systolic blood pressure and pulse pressure [1]. The excessive arterial stiffening ascertained by an increased PWV is a consequence of structural and functional changes in the vascular wall [1], and diverse variables such as genetic determinants, obesity, insulin resistance, diabetes, and aging are important risk factors in the pathogenesis of excessive arterial stiffening [1]. Therefore, due to the importance of arterial stiffness in cardiovascular disease (CVD) and its association with significant risk factors, in 2015 the American Heart Association (AHA) Council for High Blood Pressure Research recommended carotid–femoral PWV (cfPWC) as the appropriate method to measure arterial stiffness [2•]. Here, we will focus on recent studies investigating the pathophysiological processes and mechanisms promoting arterial stiffening as well as the contemporary understanding of potential therapeutic strategies.
Arterial Stiffness and Hypertension
Excessive arterial stiffness is associated with damage to target organs such as the arteries, heart, and kidney [3]. The Framingham Heart Study found that increased arterial stiffening is an independent predictor of CVD in the general population, the elderly, and hypertensive patients [4]. A 1 m/s increase in PWV increased the occurrence of CVD events by 14%, CVD mortality by 15%, and all-cause mortality by 15% [5]. Importantly, there is an important interaction between arterial stiffness and hypertension. In this regard, arterial stiffness has been associated with brachial blood pressure in pregnant women [6]. There are increases in forearm vascular resistance in young men with first-degree relatives suffering from essential hypertension [7]. Hypertension is associated with arterial dysfunction characterized by changes in cytoskeletal organization, cell calcification, inflammation, collagens, and arterial fibrosis [8]. These pathophysiological abnormalities induce arterial remodeling and reduce nitric oxide (NO)-mediated vasodilator capacity [7]. Increased arterial stiffness may exist prior to the development of hypertension. Recent research has shown that diet-induced obesity is associated with increased aortic stiffness prior to development of hypertension [9, 10].
Dysregulation of Vascular Cells and Extracellular Matrix in Arterial Stiffness
The arterial endothelial cells (ECs) provide a barrier between the elements of blood and the vessel wall and play an important role in maintaining arterial homeostasis and normal physiological function partly through actions of EC-derived vasodilatory or vasoconstrictory substances including NO, prostacyclin, and endothelin 1. Recent research has underscored the role of activated EC Na channels (EnNaC) in promoting a stiff endothelium and associated impaired endothelial NO synthase (eNOS) activation in aortic and mesenteric arteries [11•, 12]. RAAS-mediated activation of EnNaC induces serum and glucocorticoid-regulated kinase 1 (SGK1) activation which impairs ENaC ubiquitination/degradation, leading to its accumulation in the plasma membrane, and a net increase in Na+ channel activity [11•]. Increased EnNaC expression and membrane abundance in ECs leads to enhanced Na+ influx, polymerization of G-actin to F-actin, reduced EC eNOS activity and NO production, and the development of arterial stiffening [13•, 14•] (Fig. 1). Consistent with this notion, our recent research in obese mice indicated that inhibition of ENaC with very low doses of amiloride, an EnNaC inhibitor, decreases oxidative stress, endothelium permeability, inflammation, arterial fibrosis, and aortic stiffness, as well as cardiac diastolic dysfunction without affecting blood pressure or Na+ retention [11•, 12].

Schematic diagram illustrating EC and VSMC dysfunction in arterial stiffness. Risk factors such as RAAS activation induce activation of SGK1 that increases EnNaC expression and membrane abundance in ECs, leading to enhanced Na+ influx, polymerization of G-actin to F-actin, reduced eNOS activity, NO production, and the development of arterial stiffness
Vascular smooth muscle cells (VSMCs), which are the major cellular component of the arterial wall, are also involved in the genesis of arterial fibrosis and stiffness. Vascular flow-mediated NO diffuses into neighboring VSMCs and activates guanylyl cyclase/cyclic guanosine monophosphate signal pathways, resulting in vascular relaxation (Fig. 1). This process is compromised in conditions of obesity, aging, and insulin resistance. For example, VSMCs in Zucker obese insulin-resistant rats manifest greater concentrations of reactive oxygen species (ROS), impaired activation of the NO/cyclic guanosine monophosphate/protein kinase G pathway, and increased cell stiffness [15]. VSMCs are capable of osteoblast trans-differentiation by promoting alkaline phosphatase activity, the formation of mineralized nodules, and osteocalcin expression in VSMCs [16]. Thus, VSMC calcification is another important contributor in the development of excessive arterial stiffness.
Changes in extracellular matrix (ECM), composition, and arterial structure play an important role in reduction of arterial compliance and increased arterial stiffness. Transforming growth factor beta 1 (TGF-β1)/Smad signaling can stimulate synthesis of ECM proteins including collagens and fibronectin [17]. Increased TGF-β1 increases synthesis and accumulation of ECM proteins partly by associated increases in matrix metalloproteinases (MMPs) [18], which degrade elastin [18]. Excessive arterial stiffening is a complex property that is also mediated by abnormal ECM and matrix-cell interactions [19]. For instance, VSMC dysfunction changes the adhesive interactions with the ECM during active relaxation and contraction. Moreover, modulation of the elasticity of the cortical cytoskeleton occurs in parallel with changes in VSMC adhesion properties [20, 21]. Further, modulation of the elasticity of the cortical cytoskeleton occurs in parallel with changes in VSMC adhesion properties [20, 21]. Angiotensin II (Ang II) induces VSMCs to synthesize collagen, fibronectin, and ECM proteins, as well as activation of the ECM-modifying MMPs [16]. These data underscore the importance of interactions between cellular components and the ECM in the pathogenesis of arterial stiffening.
Pathological Mechanisms of Excessive Arterial Stiffness
Activated RAAS
Enhanced systemic and tissue RAAS activation induce arterial stiffening. Cell-specific RAAS signaling in ECs, VSMCs, and macrophages are involved [22]. Evaluation of the role of vascular RAAS signaling has been facilitated recently by the availability of vascular cell-specific knock out (KO) rodent models [23,24,25]. Indeed, both Ang II and aldosterone directly induce abnormal arterial stiffening by activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) and inhibition of NO bioavailability [26]. Importantly, regulation of RAAS in the vasculature is increased by consumption of western diets, which contribute to arterial fibrosis and increased arterial stiffness [1]. Recent research indicates that aldosterone and diet-induced obesity increase EnNaC expression and activation, leading to reduced NO production that is associated with increases in EC cortical stiffness [11•, 12]. There is also an interaction between specific interactive components of the RAAS. For instance, improvement of Ang II-induced arterial stiffness by mineralocorticoid receptor (MR) antagonist and suppression of aldosterone-induced arterial dysfunctional by inhibition of Ang II receptor 1 (AT-1R) are consistent with the notion of arterial cross-talk between the Ang II and aldosterone signaling [27, 28]. Recent research suggests that the RAAS is involved in regulating parathyroid hormone, which increases the concentration of calcium in the blood and promotes arterial stiffening [29]. Ang II is likely an acute modulator of parathyroid hormone and directly induces release of parathyroid hormone via the AT-1R, whereas aldosterone is thought to be a chronic modulator of parathyroid hormone via indirect and direct mechanisms [29].
Oxidative Stress
Elevated oxidative stress promotes arterial stiffening and CVD. For example, in 386 elderly patients with essential hypertension, superoxide dismutase and antioxidant status are significantly reduced with an increased branchial-ankle artery PWV [30]. Chronic supplementation with a mitochondrial antioxidant (MitoQ) represses oxidative stress and improves vascular function in healthy aging individuals [31]. NOX is one of the important enzymes and plays a key role in the generation of ROS in arterial tissue. For instance, aldosterone induces expression of p47phox through both MR-dependent and AT-1R-dependent mechanisms, and expression of the p22phox subunit and the NOX2 isoform is MR-dependent in vascular tissues [32]. The signaling pathways of the mitochondrial monoamine oxidase, cyclooxygenase 2, and p66Shc signaling are also involved in Ang II-induced activation of NOX induction and oxidative stress in arterial tissues [33]. Some other sources of ROS include peroxisomal β-oxidation of fatty acids, arachidonic acid metabolism, xanthine oxidase, microsomal P-450 enzymes, and pro-oxidant heme molecule that have been recognized to contribute to arterial stiffening and hypertension [18]. Therefore, increased ROS and oxidative stress impair deoxyribonucleic acid, lipid, protein, as well as mitochondrial function. Recent data further support that increased ROS decreases bioavailable NO and thus impairs arterial relaxation [34]. Recent research has also shown that enhanced EC-specific MR activation promotes oxidative stress, vascular fibrosis, increased arterial stiffness, and impairment of flow-mediated mesenteric artery relaxation [25].
Peri-vascular Adipose Tissue and Inflammation
Peri-vascular adipose tissue (PVAT) is a special local deposit of adipose tissue and has functions in mechanical protection and controlling vessel tone [35]. Data from the Framingham Offspring and Third Generation cohorts found that elevated PVAT volume is associated with increased thoracic and abdominal aortic dimensions and abnormal increased arterial stiffness even after adjusting for age, sex, and body mass index [36]. Indeed, adipose tissue is an endocrine organ and releases various biological activities including adipokines [37]. PVAT produces some protective substances such as adiponectin, which helps maintain normal physiological arterial function [38, 39]. However, in individuals with obesity and insulin resistance, there is increased release of pro-inflammatory adipokines including interleukin-6, interleukin-8, and tumor necrosis factor alpha, as well as toll-like receptor-4 [18]. Activated nuclear factor kappa B (NF-κB) is regarded as an important mechanism on increased arterial stiffening. While p50 and p65 subunits of NF-κB are maintained in the cytoplasm, phosphorylation of NF-κB promotes translocation of the heterodimer to the nucleus and release of pro-inflammatory cytokines [18]. Macrophages are an important driver of vascular inflammation and associated increased arterial stiffness. Typically, macrophage polarization is traditionally dichotomized into M1 phenotypes (F4/80+ CD11c+) and M2 phenotypes (F4/80+ CD11c− CD301+ Arg1+ CD206+) [18]. Pro-inflammatory M1 phenotypes are associated with an increase of vascular inflammatory responses, while macrophage M2 phenotypes are involved in anti-inflammatory responses and tissue repair [40]. Interestingly, cell-specific macrophage MR KO displays an increase in M2 polarization and CV protective effects [41]. Our recent data show that diet-induced obesity causes an increase in macrophage infiltration, M1 phenotype polarization, and associated increased aortic stiffness. Importantly, cell-specific ECMR KO prevents these pathophysiological changes [25].
Sympathetic Activity
The role of increased sympathetic nervous system activity in arterial stiffness and hypertension is increasingly recognized. Elevated sympathetic outflow is associated with increases in circulating catecholamines, urinary norepinephrine, and muscle sympathetic nerve activity in obese non-hypertensive individuals [42]. Autonomic ganglionic blockade decreases aortic augmented pressure and PWV in women [43]. Sympathetic activation can be mediated by reflex mechanisms including arterial baroreceptor impairment, oxidative stress, inflammation, and psychological stress, as well as obstructive sleep apnea [18]. There is an interaction between activated RAAS and sympathetic nervous system activation. For example, elevated Ang II induces permeability of the blood-brain barrier and sympathetic activation that increases renin secretion and Na+ retention [44]. Aldosterone infusion increases muscle sympathetic activation and impairs baroreflex responses [45], whereas inhibition of MR with spironolactone prevents chlorthalidone-induced sympathetic activation in individuals with hypertension [46].
Assessment Methods of Arterial Stiffness
There are three noninvasive measurements for measuring arterial stiffness including assessment of pulse transit time, analysis of wave contour in the arterial pulse, and, direct detection of arterial geometry and pressure. Aortic PWV is widely regarded as the gold standard in detecting arterial stiffness. cfPWV is recommended as an appropriate method to measure arterial stiffness as established in 2015 by the American Heart Association Council for High Blood Pressure [2•]. New technologies such as atomic force microscopy also provide a powerful investigative tool in detecting cell’s and tissue’s stiffness at the nano-scale as shown in our recent studies [11•, 25]. The European Society of Hypertension (ESH)/European Society of Cardiology (ESC) further suggest that a threshold value (12 m/s) in PVW is recommended as an indicator of increased arterial stiffness [47].
Therapeutic Strategies in Excessive Arterial Stiffness
Life style modifications comprising exercise, consumption of low sodium, and better dietary habits are effective methods for the prevention and management of increased arterial stiffness and hypertension but patient compliance is one of the concerns in long-term management [48, 49]. Although several studies have shown suppression of arterial stiffness with antihypertensive medication, the magnitude of lowering arterial stiffness is variable among antihypertensive drugs [50,51,52]. In this regard, inhibition of RAAS using angiotensin converting enzyme (ACE), AT-1R, and MR antagonists appears to be superior compared to other antihypertensive medications [53,54,55,56]. Targeting the MR is emerging as a useful approach in combination therapy that reduces arterial stiffness [57,58,59]. This is also supported by studies showing reversal of the increased arterial stiffness in chronic kidney disease patients after undergoing renal transplantation [60] and in subjects with aldosterone producing adenomas after adrenectomy [61]. These beneficial effects on arterial stiffness are independent of blood pressure-lowering effects [52]. These findings suggest that reversibility of arterial stiffness by blood pressure independent effects may also be attributable to local remodeling effects of these drugs on large artery stiffening [62]. A combination of ACE inhibitors and AT-1R antagonists caused significant decrease in PWV in chronic kidney patients [63]. Calcium channel blockers, diuretics, and beta blockers are less effective in reducing arterial stiffness compared to ACE inhibitors and AT-1R antagonists perhaps because of less impact on fibrosis and vascular remodeling [64,65,66,67,68]. In this regard, targeting EnNaC is an attractive strategy. Indeed, very low doses of amiloride, an ENaC inhibitor, can substantially decrease dietary obesity-related vascular and cardiac fibrosis [11•, 12]. In addition, deletion of ECMR decreases arterial stiffness in an amiloride-sensitive manner [25]. Preliminary studies also support a role for Ang II in development of arterial stiffness in mice through stimulation of EnNaC. We have also observed that xanthine oxidase inhibition reduces diet-induced increased aortic fibrosis and stiffness [69]. The clinical relevance of these finding is supported by studies demonstrating greater effectiveness of a combination of low-dose amiloride and spironolactone but not by the use of individual drugs [70]. One of the limiting factors of amiloride is an increase in potassium levels. The development of amiloride analogues [71, 72] with effectiveness at lower concentration with more specificity towards ENaC in preventing arterial stiffness is being pursued.
Conclusions
Increased arterial stiffness is an important precursor and risk factor for hypertension and CVD. Dysregulation of structure and function of ECs, VSMCs, and ECM contribute to the pathogenesis of arterial stiffening and fibrosis. Activation of RAAS and sympathetic nervous activity, increases in oxidative stress, abnormal PVAT, tissue inflammation, and sympathetic outflow are all associated with the development and progression of arterial fibrosis, stiffening, and CVD. A better understanding of the underlying mechanisms increasing arterial stiffness should provide new insights for future therapeutic strategies for CVD.
Sources of Funding
Dr. Sowers receives funding from the Veterans Affairs Merit System (2 I01 BX001981-05A1) and the NIH (R01 HL73101-01A and R01 HL107910-01). Dr. Jia receives funding from the American Diabetes Association (Innovative Basic Science Award #1-17-IBS-201). Dr. Martinez-Lemus receives funding from the NIH (R01-HL-088105).
Abbreviations
- ACE:
-
Angiotensin converting enzyme
- Ang II:
-
Angiotensin II
- AT-1R:
-
Ang II receptor 1
- cfPWC:
-
Carotid–femoral PWV
- CVD:
-
Cardiovascular disease
- EC:
-
Endothelial cells
- ECM:
-
Extracellular matrix
- ENaC:
-
Epithelial Na+ channel
- EnNaC:
-
Endothelial ENaC
- eNOS:
-
Endothelial NO synthase
- MMPs:
-
Matrix metalloproteinases
- MR:
-
Mineralocorticoid receptor
- Na+ :
-
Sodium
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- NF-κB:
-
Nuclear factor kappa B
- NO:
-
Nitric oxide
- NOX:
-
NADPH oxidase
- PVAT:
-
Peri-vascular adipose tissue
- PWV:
-
Pulse wave velocity
- RAAS:
-
Activated renin-angiotensin-aldosterone system
- ROS:
-
Reactive oxygen specie
- SGK1:
-
Serum and glucocorticoid-regulated kinase 1
- TGF-β1:
-
Transforming growth factor beta 1
- VSMC:
-
Vascular smooth muscle cell
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Jia G, Aroor AR, DeMarco VG, Martinez-Lemus LA, Meininger GA, Sowers JR. Vascular stiffness in insulin resistance and obesity. Front Physiol. 2015;6:231.
• Townsend RR, Wilkinson IB, Schiffrin EL, Avolio AP, Chirinos JA, Cockcroft JR, et al. Recommendations for improving and standardizing vascular research on arterial stiffness: a scientific statement from the American Heart Association. Hypertension. 2015;66:698–722. This study recommended that carotid–femoral PWV is an appropriate method to measure arterial stiffness.
Schiffrin EL. Vascular stiffening and arterial compliance. Implications for systolic blood pressure. Am J Hypertens. 2004;17:39S–48S.
Mitchell GF, Hwang SJ, Vasan RS, Larson MG, Pencina MJ, Hamburg NM, et al. Arterial stiffness and cardiovascular events: the Framingham Heart Study. Circulation. 2010;121:505–11.
Vlachopoulos C, Aznaouridis K, Stefanadis C. Prediction of cardiovascular events and all-cause mortality with arterial stiffness: a systematic review and meta-analysis. J Am Coll Cardiol. 2010;55:1318–27.
Tomimatsu T, Fujime M, Kanayama T, Mimura K, Koyama S, Kanagawa T, et al. Abnormal pressure-wave reflection in pregnant women with chronic hypertension: association with maternal and fetal outcomes. Hypertens Res. 2014;37:989–92.
Takeshita A, Imaizumi T, Ashihara T, Yamamoto K, Hoka S, Nakamura M. Limited maximal vasodilator capacity of forearm resistance vessels in normotensive young men with a familial predisposition to hypertension. Circ Res. 1982;50:671–7.
Touyz RM, Alves-Lopes R, Rios FJ, Camargo LL, Anagnostopoulou A, Arner A, et al. Vascular smooth muscle contraction in hypertension. Cardiovasc Res. 2018;114:529–39.
Weisbrod RM, Shiang T, Al Sayah L, Fry JL, Bajpai S, Reinhart-King CA, et al. Arterial stiffening precedes systolic hypertension in diet-induced obesity. Hypertension. 2013;62:1105–10.
DeMarco VG, Habibi J, Jia G, Aroor AR, Ramirez-Perez FI, Martinez-Lemus LA, et al. Low-dose mineralocorticoid receptor blockade prevents western diet-induced arterial stiffening in female mice. Hypertension. 2015;66:99–107.
• Martinez-Lemus LA, Aroor AR, Ramirez-Perez FI, Jia G, Habibi J, DeMarco VG, et al. Amiloride improves endothelial function and reduces vascular stiffness in female mice fed a western diet. Front Physiol. 2017;8:456. This study indicated EnNaC mediates western diet-induced endothelial dysfunction and aretrial stiffness in female mice.
Jia G, Habibi J, Aroor AR, Hill MA, DeMarco VG, Lee LE, et al. Enhanced endothelium epithelial sodium channel signaling prompts left ventricular diastolic dysfunction in obese female mice. Metabolism. 2018;78:69–79.
• Kusche-Vihrog K, Jeggle P, Oberleithner H. The role of ENaC in vascular endothelium. Pflugers Arch. 2014;466:851–9. This study emphasized that EnNaC is an aldosterone-regulated plasma membrane protein of the vascular endothelium that contributes to endothelium stiffness and hypertension.
• Zeng Y, Waters M, Andrews A, Honarmandi P, Ebong EE, Rizzo V, et al. Fluid shear stress induces the clustering of heparan sulfate via mobility of glypican-1 in lipid rafts. Am J Physiol Heart Circ Physiol. 2013;305:H811–20. This study showed the role of changes in glycocalyx organization that underlie mechanisms of mechanotransduction.
Merdzo I, Rutkai I, Tokes T, Sure VN, Katakam PV, Busija DW. The mitochondrial function of the cerebral vasculature in insulin-resistant Zucker obese rats. Am J Physiol Heart Circ Physiol. 2016;310:H830–8.
Lacolley P, Regnault V, Segers P, Laurent S. Vascular smooth muscle cells and arterial stiffening: relevance in development, aging, and disease. Physiol Rev. 2017;97:1555–617.
Jia G, Habibi J, Bostick BP, Ma L, DeMarco VG, Aroor AR, et al. Uric acid promotes left ventricular diastolic dysfunction in mice fed a western diet. Hypertension. 2015;65:531–9.
Jia G, Hill MA, Sowers JR. Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ Res. 2018;122:624–38.
Hill MA, Meininger GA. Small artery mechanobiology: roles of cellular and non-cellular elements. Microcirculation. 2016;23:611–3.
Hong Z, Sun Z, Li M, Li Z, Bunyak F, Ersoy I, et al. Vasoactive agonists exert dynamic and coordinated effects on vascular smooth muscle cell elasticity, cytoskeletal remodelling and adhesion. J Physiol. 2014;592:1249–66.
Hong Z, Sun Z, Li Z, Mesquitta WT, Trzeciakowski JP, Meininger GA. Coordination of fibronectin adhesion with contraction and relaxation in microvascular smooth muscle. Cardiovasc Res. 2012;96:73–80.
Lother A, Hein L. Vascular mineralocorticoid receptors: linking risk factors, hypertension, and heart disease. Hypertension. 2016;68:6–10.
Kim SK, McCurley AT, DuPont JJ, Aronovitz M, Moss ME, Stillman IE, et al. Smooth muscle cell-mineralocorticoid receptor as a mediator of cardiovascular stiffness with aging. Hypertension. 2018;71:609–21.
Tarjus A, Maase M, Jeggle P, Martinez-Martinez E, Fassot C, Loufrani L, et al. The endothelial alphaENaC contributes to vascular endothelial function in vivo. PLoS One. 2017;12:e0185319.
Jia G, Habibi J, Aroor AR, Martinez-Lemus LA, DeMarco VG, Ramirez-Perez FI, et al. Endothelial mineralocorticoid receptor mediates diet-induced aortic stiffness in females. Circ Res. 2016;118:935–43.
Manrique C, DeMarco VG, Aroor AR, Mugerfeld I, Garro M, Habibi J, et al. Obesity and insulin resistance induce early development of diastolic dysfunction in young female mice fed a western diet. Endocrinology. 2013;154:3632–42.
Aroor AR, Demarco VG, Jia G, Sun Z, Nistala R, Meininger GA, et al. The role of tissue renin-angiotensin-aldosterone system in the development of endothelial dysfunction and arterial stiffness. Front Endocrinol (Lausanne). 2013;4:161.
Briet M, Schiffrin EL. Vascular actions of aldosterone. J Vasc Res. 2013;50:89–99.
Brown JM, Williams JS, Luther JM, Garg R, Garza AE, Pojoga LH, et al. Human interventions to characterize novel relationships between the renin-angiotensin-aldosterone system and parathyroid hormone. Hypertension. 2014;63:273–80.
Liu Q, Han L, Du Q, Zhang M, Zhou S, Shen X. The association between oxidative stress, activator protein-1, inflammatory, total antioxidant status and artery stiffness and the efficacy of olmesartan in elderly patients with mild-to-moderate essential hypertension. Clin Exp Hypertens. 2016;38:365–9.
Rossman MJ, Santos-Parker JR, Steward CAC, Bispham NZ, Cuevas LM, Rosenberg HL, et al. Chronic supplementation with a mitochondrial antioxidant (MitoQ) improves vascular function in healthy older adults. Hypertension. 2018;71:1056–63.
Hirono Y, Yoshimoto T, Suzuki N, Sugiyama T, Sakurada M, Takai S, et al. Angiotensin II receptor type 1-mediated vascular oxidative stress and proinflammatory gene expression in aldosterone-induced hypertension: the possible role of local renin-angiotensin system. Endocrinology. 2007;148:1688–96.
Spescha RD, Glanzmann M, Simic B, Witassek F, Keller S, Akhmedov A, et al. Adaptor protein p66(Shc) mediates hypertension-associated, cyclic stretch-dependent, endothelial damage. Hypertension. 2014;64:347–53.
Ren X, Ren L, Wei Q, Shao H, Chen L, Liu N. Advanced glycation end-products decreases expression of endothelial nitric oxide synthase through oxidative stress in human coronary artery endothelial cells. Cardiovasc Diabetol. 2017;16:52.
Xia N, Horke S, Habermeier A, Closs EI, Reifenberg G, Gericke A, et al. Uncoupling of endothelial nitric oxide synthase in perivascular adipose tissue of diet-induced obese mice. Arterioscler Thromb Vasc Biol. 2016;36:78–85.
Thanassoulis G, Massaro JM, Corsini E, Rogers I, Schlett CL, Meigs JB, et al. Periaortic adipose tissue and aortic dimensions in the Framingham Heart Study. J Am Heart Assoc. 2012;1:e000885.
Fasshauer M, Bluher M. Adipokines in health and disease. Trends Pharmacol Sci. 2015;36:461–70.
Omar A, Chatterjee TK, Tang Y, Hui DY, Weintraub NL. Proinflammatory phenotype of perivascular adipocytes. Arterioscler Thromb Vasc Biol. 2014;34:1631–6.
Gil-Ortega M, Somoza B, Huang Y, Gollasch M, Fernandez-Alfonso MS. Regional differences in perivascular adipose tissue impacting vascular homeostasis. Trends Endocrinol Metab. 2015;26:367–75.
Jia G, Aroor AR, Martinez-Lemus LA, Sowers JR. Overnutrition, mTOR signaling, and cardiovascular diseases. Am J Physiol Regul Integr Comp Physiol. 2014;307:R1198–206.
Usher MG, Duan SZ, Ivaschenko CY, Frieler RA, Berger S, Schutz G, et al. Myeloid mineralocorticoid receptor controls macrophage polarization and cardiovascular hypertrophy and remodeling in mice. J Clin Invest. 2010;120:3350–64.
Huang CJ, Webb HE, Zourdos MC, Acevedo EO. Cardiovascular reactivity, stress, and physical activity. Front Physiol. 2013;4:314.
Harvey RE, Barnes JN, Hart EC, Nicholson WT, Joyner MJ, Casey DP. Influence of sympathetic nerve activity on aortic hemodynamics and pulse wave velocity in women. Am J Physiol Heart Circ Physiol. 2017;312:H340–6.
Biancardi VC, Stern JE. Compromised blood-brain barrier permeability: novel mechanism by which circulating angiotensin II signals to sympathoexcitatory centres during hypertension. J Physiol. 2016;594:1591–600.
Monahan KD, Leuenberger UA, Ray CA. Aldosterone impairs baroreflex sensitivity in healthy adults. Am J Physiol Heart Circ Physiol. 2007;292:H190–7.
Raheja P, Price A, Wang Z, Arbique D, Adams-Huet B, Auchus RJ, et al. Spironolactone prevents chlorthalidone-induced sympathetic activation and insulin resistance in hypertensive patients. Hypertension. 2012;60:319–25.
Mancia G, De Backer G, Dominiczak A, Cifkova R, Fagard R, Germano G, et al. The task force for the management of arterial hypertension of the European Society of H and the task force for the management of arterial hypertension of the European Society of C. 2007 guidelines for the management of arterial hypertension: the task force for the management of arterial hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). Eur Heart J. 2007;28:1462–536.
Madden KM, Lockhart C, Cuff D, Potter TF, Meneilly GS. Short-term aerobic exercise reduces arterial stiffness in older adults with type 2 diabetes, hypertension, and hypercholesterolemia. Diabetes Care. 2009;32:1531–5.
Niiranen TJ, Lyass A, Larson MG, Hamburg NM, Benjamin EJ, Mitchell GF, et al. Prevalence, correlates, and prognosis of healthy vascular aging in a western community-dwelling cohort: the Framingham Heart Study. Hypertension. 2017;70:267–74.
Safar ME. Arterial stiffness as a risk factor for clinical hypertension. Nat Rev Cardiol. 2018;15:97–105.
Duprez DA. Is vascular stiffness a target for therapy? Cardiovasc Drugs Ther. 2010;24:305–10.
Janic M, Lunder M, Sabovic M. Arterial stiffness and cardiovascular therapy. Biomed Res Int. 2014;2014:621437.
London GM, Asmar RG, O’Rourke MF, Safar ME, Investigators RP. Mechanism(s) of selective systolic blood pressure reduction after a low-dose combination of perindopril/indapamide in hypertensive subjects: comparison with atenolol. J Am Coll Cardiol. 2004;43:92–9.
Mitchell GF, Dunlap ME, Warnica W, Ducharme A, Arnold JM, Tardif JC, et al. Prevention of events with angiotensin-converting enzyme inhibition I. Long-term trandolapril treatment is associated with reduced aortic stiffness: the prevention of events with angiotensin-converting enzyme inhibition hemodynamic substudy. Hypertension. 2007;49:1271–7.
Dahlof B, Devereux RB, Kjeldsen SE, Julius S, Beevers G, de Faire U, et al. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet. 2002;359:995–1003.
Julius S, Kjeldsen SE, Weber M, Brunner HR, Ekman S, Hansson L, et al. Outcomes in hypertensive patients at high cardiovascular risk treated with regimens based on valsartan or amlodipine: the VALUE randomised trial. Lancet. 2004;363:2022–31.
Kalizki T, Schmidt BMW, Raff U, Reinold A, Schwarz TK, Schneider MP, et al. Low dose-eplerenone treatment decreases aortic stiffness in patients with resistant hypertension. J Clin Hypertens (Greenwich). 2017;19:669–76.
Cameron AC, Lang NN, Touyz RM. Drug treatment of hypertension: focus on vascular health. Drugs. 2016;76:1529–50.
Shibata T, Tsutsumi J, Hasegawa J, Sato N, Murashima E, Mori C, et al. Effects of add-on therapy consisting of a selective mineralocorticoid receptor blocker on arterial stiffness in patients with uncontrolled hypertension. Intern Med. 2015;54:1583–9.
Karras A, Boutouyrie P, Briet M, Bozec E, Haymann JP, Legendre C, et al. Reversal of arterial stiffness and maladaptative arterial remodeling after kidney transplantation. J Am Heart Assoc. 2017;6:e006078.
Liao CW, Lin LY, Hung CS, Lin YT, Chang YY, Wang SM, et al. Time course and factors predicting arterial stiffness reversal in patients with aldosterone-producing adenoma after adrenalectomy: prospective study of 102 patients. Sci Rep. 2016;6:20862.
Chen Y, Shen F, Liu J, Yang GY. Arterial stiffness and stroke: de-stiffening strategy, a therapeutic target for stroke. Stroke Vasc Neurol. 2017;2:65–72.
Frimodt-Moller M, Kamper AL, Strandgaard S, Kreiner S, Nielsen AH. Beneficial effects on arterial stiffness and pulse-wave reflection of combined enalapril and candesartan in chronic kidney disease—a randomized trial. PLoS One. 2012;7:e41757.
Doi M, Miyoshi T, Hirohata S, Kamikawa S, Usui S, Kaji Y, et al. Combination therapy of calcium channel blocker and angiotensin II receptor blocker reduces augmentation index in hypertensive patients. Am J Med Sci. 2010;339:433–9.
Mahmud A, Feely J. Effect of angiotensin ii receptor blockade on arterial stiffness: beyond blood pressure reduction. Am J Hypertens. 2002;15:1092–5.
Ferguson JM, Minas J, Siapantas S, Komesaroff PA, Sudhir K. Effects of a fixed-dose ACE inhibitor-diuretic combination on ambulatory blood pressure and arterial properties in isolated systolic hypertension. J Cardiovasc Pharmacol. 2008;51:590–5.
Manisty CH, Hughes AD. Meta-analysis of the comparative effects of different classes of antihypertensive agents on brachial and central systolic blood pressure, and augmentation index. Br J Clin Pharmacol. 2013;75:79–92.
Lindholm LH, Carlberg B, Samuelsson O. Should beta blockers remain first choice in the treatment of primary hypertension? A meta-analysis. Lancet. 2005;366:1545–53.
Aroor AR, Jia G, Habibi J, Sun Z, Ramirez-Perez FI, Brady B, et al. Uric acid promotes vascular stiffness, maladaptive inflammatory responses and proteinuria in western diet fed mice. Metabolism. 2017;74:32–40.
Pratt JH, Eckert GJ, Newman S, Ambrosius WT. Blood pressure responses to small doses of amiloride and spironolactone in normotensive subjects. Hypertension. 2001;38:1124–9.
Hirsh AJ, Molino BF, Zhang J, Astakhova N, Geiss WB, Sargent BJ, et al. Design, synthesis, and structure-activity relationships of novel 2-substituted pyrazinoylguanidine epithelial sodium channel blockers: drugs for cystic fibrosis and chronic bronchitis. J Med Chem. 2006;49:4098–115.
Hirsh AJ, Zhang J, Zamurs A, Fleegle J, Thelin WR, Caldwell RA, et al. Pharmacological properties of N-(3,5-diamino-6-chloropyrazine-2-carbonyl)-N′-4-[4-(2,3-dihydroxypropoxy)phenyl] butyl-guanidine methanesulfonate (552-02), a novel epithelial sodium channel blocker with potential clinical efficacy for cystic fibrosis lung disease. J Pharmacol Exp Ther. 2008;325:77–88.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare no conflicts of interest relevant to this manuscript.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Hypertension and the Kidney
Rights and permissions
About this article

Cite this article
Jia, G., Aroor, A.R., Martinez-Lemus, L.A. et al. Potential Role of Antihypertensive Medications in Preventing Excessive Arterial Stiffening. Curr Hypertens Rep 20, 76 (2018). https://doi.org/10.1007/s11906-018-0876-9
Published:
DOI: https://doi.org/10.1007/s11906-018-0876-9