
Abstract
Heart failure with preserved ejection fraction (HFpEF) is a major cause of HF-related morbidity and mortality, with no medical therapy proven to modify the underlying disease process and result in improvements in survival. With long-standing pulmonary venous congestion, a majority of HFpEF patients develop pulmonary hypertension (PH). Elevated pulmonary pressures have been shown to be a major determinant of mortality in this population. Given the paucity of available disease-modifying therapies for HFpEF, there has been a considerable interest in evaluating new therapeutic options specifically targeting PH in this patient population.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Patients with heart failure with preserved ejection fraction (HFpEF) are being increasingly recognized, with almost 50 % of all patients with HF having a normal left ventricular systolic function [1]. Both morbidity and mortality in patients with HFpEF are similar to those suffering from HF with reduced EF (HFrEF) [2]. Pulmonary hypertension (PH), characterized by elevation of the mean pulmonary artery pressure (PAP) to ≥25 mmHg, can develop in up to 50 % of patients with HFpEF [3, 4]. Importantly, HFpEF patients with PH (HFpEF-PH) have reduced survival and increased hospitalization compared to those without PH [5–7].
HFpEF-PH is highly prevalent and considerably more common than other more investigated forms of PH [5, 7]. Observational studies have shown a 53 % prevalence of PH in a wide cohort of patients with HFpEF, as assessed by right heart catheterization (RHC) [3, 8]. The current management of PH in HFpEF is predominantly based on addressing systemic blood pressure, heart rate, circulating volume, and myocardial ischemia, with no specific focus on pulmonary vasculature remodeling. In addition, the use of drugs with proven efficacy in HFrEF provides little benefit in patients with HFpEF, in general, and little to no response in those with associated PH [9–11]. As a result, clinical outcomes and symptomatic improvement in patients with HFpEF-PH remain poor. The development of therapeutic strategies targeting both the unique pathophysiology of HFpEF and the associated PH offers a unique opportunity to treat these patients and is the focus of this review.
Pathophysiology of HFpEF-PH
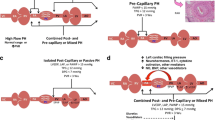
The pathophysiology of HFpEF-PH is complex and probably multifactorial. It is important to recognize that both the incidence of HFpEF and elevations in PAP increase with aging and that both these processes appear to be concordant with age-related elevations in systemic vascular stiffness. Since many patients with HFpEF are elderly, one must consider if the elevations in PAP noted in HFpEF are truly pathological and not merely related to the aging process. However, contrary to this observation is the fact that the degree of PH typically seen in the context of HFpEF clearly exceeds that expected with the normal aging process. Thus, in order to understand the unique contribution of HFpEF to the development of PH, one must also understand the fundamental mechanisms that contribute to the development of HFpEF (Fig. 1).

Multiple factors contribute to the pathophysiology and progression of pulmonary hypertension (PH) in patients with heart failure with preserved ejection fraction (HFpEF). MMP matrix metalloproteinases, TIMP tissue inhibitors of matrix metalloproteinases, IpcPH isolated precapillary pulmonary hypertension, CpcPH combined pre- and postcapillary pulmonary hypertension
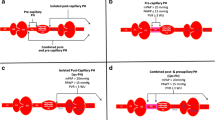
The main drivers of this form of HF are impaired left ventricular (LV) filling (diastolic dysfunction) and ventricular-vascular uncoupling. Diastolic dysfunction related to increased myocardial stiffness occurs both from (a) increased type I collagen synthesis and deposition in the extracellular matrix, as well as from decreased matrix degradation from downregulation of matrix metalloproteinases (MMPs) and upregulation of tissue inhibitors of matrix metalloproteinases (TIMPs) and (b) increased cardiomyocyte stiffness related to myocyte hypertrophy, dysfunction of the cytoskeletal protein, titin hypophosphorylation, and cross-bridge detachment. As these ultrastructual changes progress, elevations in LV end-diastolic and left atrial pressures (LAP) ensue and become the initial triggers for the development of PH. In this early phase, the elevations in PAP in HFpEF are concordant with the LAP and the resultant PH is considered “passive” (isolated postcapillary PH or IpcPH) [12]. However, over time, progressive abnormalities in the pulmonary circulation develop as a result of chronically elevated LAP that mirror the pathologic changes seen in pulmonary arterial hypertension (PAH), including both abnormalities of pulmonary arteries (intimal fibrosis and medial hypertrophy) and pulmonary venules (combined precapillary and postcapillary PH or CpcPH).
Ventricular-vascular uncoupling is characterized by increased pulmonary vascular stiffness, decreased compliance, and abnormal vasorelaxation, both from endothelial dysfunction and vasoconstriction. This results in increased load for the right ventricle (RV) endocardium. Many patients with LV diastolic dysfunction also have septal dysfunction, which can worsen RV function [13]. Even though the LVEF is normal in HFpEF, LV contractility may be impaired [14]. Because a substantial component (∼30 %) of RV contractility is generated by the LV, due to shared short axis fibers and transseptal contributions, impaired LV contractility may impair RV contractility [15]. Though the normal RV can increase contractility to match increases in afterload, in the setting of impaired RV/LV interactions, the RV loses its ability to compensate, causing ventricular-vascular uncoupling [16, 17]. This results in increased ventricular workload per unit of stress and greatly accelerates the onset of RV failure. Adaptive RV remodeling accompanies IpcPH with transition to maladaptation as the condition progresses to CpcPH. The RV is now not only dilated and hypertrophied but also dysfunctional, with increased wall stress and increasingly severe tricuspid valve insufficiency [18, 19]. Progressive fibrosis and pulmonary vascular remodeling ensue, which in association with lung capillary stress failure result in decompensated RV failure, eventually leading to death [20••] (Fig. 2).

Role of right ventricular (RV) failure and progressive remodeling in the morbidity and mortality associated with HFpEF-PH
Thus, from a therapeutic standpoint, drugs or drug combinations that act in concert to improve LV diastology and RV/PA coupling and reduce RV afterload offer the greatest hope of treating the consequences of HFpEF-related PH.
Diagnosis of HFpEF-PH
Although PH in HFpEF is recognized by noninvasive modalities such as echocardiogram, it remains a hemodynamic diagnosis. IpcPH and CpcPH are differentiated hemodynamically by parameters that suggest a component of pulmonary vascular disease (i.e., a precapillary component). These parameters include the transpulmonary gradient (TPG), which is the mean PAP minus the pulmonary artery wedge pressure (PAWP), pulmonary vascular resistance (PVR) [TPG divided by cardiac output], and the diastolic pulmonary gradient (DPG) [diastolic pulmonary artery pressure (dPAP) minus PAWP]. Elevations in TPG and PVR are not specific for pulmonary vascular remodeling or even pulmonary arterial vasoconstriction. TPG is dependent on cardiac output (flow), and both parameters are also influenced by elevations in LAP [21, 22]. On the other hand, the diastolic PAP is not subject to these same effects and therefore the DPG has recently been proposed as a better way to differentiate IpcPH from CpcPH [12]. In one study of patients with left heart failure and an elevated TPG >12 mmHg, a DPG of ≥7 mmHg identified a higher-risk group compared with those with a low DPG [23]. However, even the DPG is influenced by other hemodynamic factors, including heart rate [24], and may be particularly prone to measurement error.
Distinguishing HFpEF-PH from IPAH
Differentiating patients with PH due to HFpEF from those with PAH is challenging and requires careful consideration of clinical, radiologic, and hemodynamic data (Table 1). Demographic and clinical features more predominate in HFpEF-PH compared with PAH include older age, female gender, and features of the metabolic syndrome (hyperlipidemia, obesity, diabetes, and hypertension) [25, 26]. Echocardiographic findings of LA enlargement, increased LA volume index, advanced diastolic dysfunction, and absence of a mid-systolic “notch” in the RV outflow tract Doppler signal are more commonly found in HFpEF-PH [27]. It is important to remember that mild diastolic dysfunction is not uncommon in PAH, so this finding alone is not sufficient to make a diagnosis of HFpEF-PH [28].
In most cases, and in particular if PAH-specific therapies are being considered, invasive hemodynamic measures by RHC must be performed. In the presence of PH, a resting PAWP or left ventricular end-diastolic pressure (LVEDP) >15 mmHg is consistent with a diagnosis of HFpEF. In a retrospective study of 580 patients undergoing simultaneous left and RHC, Halpern and colleagues reported PAWP measurements were approximately 3 mmHg less than LVEDP, and defining PH group by LVEDP (rather than PAWP) led to a reclassification in over half of their cohort [29]. At least some of this discrepancy may be explained by the common but incorrect practice of relying on digitized or computer-generated mean PAWP values, which are averaged over the entire respiratory cycle. Care must be taken to ensure adequate wedging of the balloon-tipped catheter confirmed by typical waveform appearance and by measuring a PAWP saturation whenever possible. In cases where a PAWP cannot be obtained or if there is a concern about its accuracy, direct LVEDP measures should be considered [30].
A resting PAWP ≤15 mmHg does not necessarily rule out HFpEF [31]. Treatment with diuretics and afterload reducers could lower PAWP into the normal range [32], or occult left heart disease may only be realized after volume expansion or additional stressors. Therefore, provocative measures during RHC may be useful in certain situations. In a study of 207 patients who initially met hemodynamic criteria for PAH, Robbins and colleagues reported 22 % had an increase in PAWP >15 mmHg after a 500 cc saline bolus, leading to a reclassification of these patients as occult HFpEF [33]. Importantly, the patient characteristics of those reclassified as occult HFpEF were more similar to those with HFpEF-PH than PAH. Recent recommendations suggest that a 500 cc fluid bolus over 5–10 min is safe and may help distinguish PAH from HFpEF-PH, although there is no consensus cutoff value and results must be interpreted with caution [25, 29]. Exercise has also been used as a provocative measure to help differentiate HFpEF-PH from PAH. Thadani et al. studied 10 sedentary patients without demonstrable cardiovascular disease and showed an average rise in PAWP of 7 mmHg during the supine exercise, though none to an absolute value of greater than 20 mmHg [34]. However, since there are no widely accepted standards regarding the method, level, or position of exercise or well-defined cutoffs for what constitutes an abnormal response, current guidelines do not recommend incorporation of exercise as a provocative measure into routine clinical practice [25].
Management of PH in HFpEF
Current Guidelines
There is limited published data to give guidance on the pharmacologic management of patients with PH from left heart disease, and data for the treatment of HFpEF-PH is even sparser. Studies for the management of HFpEF have previously focused on therapies that have demonstrated benefit in patients with HFrEF. However, results of these trials have been disappointing and have not demonstrated the same morbidity and mortality benefits seen in HFrEF trials [7–9].
For HFpEF-PH management, current guidelines recommend the treatment of symptoms of congestion and volume overload, targeting LV relaxation and comorbidities that contribute to HFpEF [35•, 36, 37]. These include the management of pulmonary congestion, ischemia, sleep apnea, atrial fibrillation, and diabetes. Aggressive management of systemic hypertension should be instituted, especially in patients with LV hypertrophy and diabetes mellitus. Although the evidence of ACE inhibitors, ARBs, beta-blockers, and aldosterone antagonists are lacking in HFpEF, it is appropriate to utilize these agents to treat comorbidities based on current guidelines for these conditions. Nitrates are also oftentimes used to relieve symptoms of congestion and improved blood pressure control. Judicious use of diuretics, along with sodium and fluid restriction, is essential to regulate and maintain a normal LAP in patients with HFpEF. Management of volume overload can be difficult in HFpEF, as these patients are often very sensitive to the rapid changes in preload that occurs with diuresis and inadvertent hypotension and renal insufficiency can commonly occur. However, it is important to remember that the chronic control of elevated LAP is critical to reverse HFpEF-PH and essential if later active pulmonary vasodilator therapies to promote vascular remodeling are pursued. It is unclear whether improvement in associated metabolic conditions, such as obesity, diabetes, sleep apnea, and blood pressure will result in improved pulmonary vascular remodeling, but in the absence of ill effects, we would recommend pursuing these modifications, as they likely have favorable effects on LV diastology.
Both atrial and ventricular tachyarrhythmias contribute to the mortality associated with HF. Treatment of atrial fibrillation improves diastolic filling in patients with HFpEF and hence LAP [38]. Thus, control of dysrhythmias, particularly atrial fibrillation, is an essential part of the early pulmonary vascular remodeling process.
Pharmacological Therapies Directed at Pulmonary Artery Remodeling
Given the lack of therapies for HFpEF-PH, it has been debated whether PAH-targeted therapies can be used to treat this condition. Both endothelin receptor antagonists (ERA) and prostanoids have been successful treatment options for PAH. As patients with long-standing HFpEF-PH may also develop pulmonary vascular remodeling akin to PAH, it makes intuitive sense that selected patients, particularly those with CpcPH, could potentially benefit from these therapies. However, clinical trials utilizing these particular agents in the treatment of PH due to left heart disease have been neutral or even detrimental [39–43]. Selective dilation of the pulmonary vessels in patients with postcapillary PH, without simultaneously ensuring the unloading of the LV, can cause profound pulmonary venous congestion resulting in sudden pulmonary edema, which greatly increases the morbidity in patients with this form of PH [44, 45]. However, some experimental data does exist to suggest that some ERAs may positively affect LV remodeling in HFpEF, thereby offering a means to control both the “trigger” for this form of PH (i.e., impaired LV filling and resultant high LAP) and simultaneously improve PA remodeling. Theoretically, this balance between these two conditions could mitigate the aforementioned rise in pulmonary venous pressures.
Sitaxsentan, a selective ET-A receptor antagonism initially designed to treat PAH, was noted in preclinical trials to improve diastolic dysfunction and LV hypertrophy [46–48]. Its safety and efficacy were evaluated in HFpEF patients with New York Heart Association (NYHA) class II or III symptom in a randomized, double-blind, placebo-controlled trial [49]. Patients treated with sitaxsentan had improved exercise tolerance (p = 0.03) compared to placebo, with no significant difference in NYHA class or echocardiographic measures of diastolic dysfunction. Despite the disappointing results in the ENABLE trials [44], a trial with the nonselective endothelin antagonist, bosentan, was recently completed in Patients with Diastolic Heart Failure and Secondary Pulmonary Hypertension (BADDHY; ClinicalTrials.gov identifier: NCT00820352). The results of this trial are forthcoming. In addition, a trial with macitentan, a tissue-specific, nonselective ERA, is currently underway to study the efficacy and safety of this new ERA in patients with HFpEF-PH (MELODY; ClinicalTrials.gov identifier: NCT02070991).
Currently, the most compelling published data for pharmacological treatment targeting PH in HFpEF involves phosphodiesterase (PDE) inhibitor sildenafil. The PDE-5 isoenzyme is responsible for the degradation of cyclic guanosine monophosphate (cGMP), inhibition of which leads to increased concentrations of cGMP, thereby facilitating smooth muscle relaxation [50]. Guazzi et al. randomized 44 symptomatic HFpEF patients with PASP >40 mmHg to sildenafil vs. placebo for 1 year [51••]. Sildenafil improved the mean PAP (−16.7 ± 3 and −18.2 ± 2.4 mmHg) from baseline and at 6 and 12 months, respectively, compared to no improvement seen in the placebo group (+1.0 ± 0.7 and +2.8 ± 1.1 mmHg). The RELAX trial also studied sildenafil in patients with NYHA class II to IV symptoms in which 216 HFpEF patients were randomized to either sildenafil (n = 113) or placebo (n = 103) [52•]. Overall, there was no significant difference in the primary endpoint of change in peak oxygen consumption at 24 weeks (−0.20 mL/kg/min for sildenafil vs. −0.02 mL/kg/min for placebo, p = 0.90). The seemingly disparate results of these two trials may be explained by differing trial design, patient numbers, and baseline characteristics. For example, there were notable differences in systolic blood pressures (153 vs. 126 mmHg), LV mass (166 vs. 65 g/m2), and PASP (54.5 vs. 41 mmHg) between the study by Guazzi and RELAX, respectively. This later point is particularly relevant for HFpEF-PH, as the patients in the RELAX trial did not have significant PH. In addition, the ability of PDE-5 inhibitors to enhance cGMP in HFpEF might be limited by insufficient endogenous stimulation of soluble guanylate cyclase (sGC), secondary to reduced NO availability in these patients. Additional trials utilizing PDE inhibitors are still underway, including a single-center phase III trial investigating whether sildenafil treatment results in an improvement in exercise capacity and a reduction of PAP without decrease of CO in HFpEF-PH (ClinicalTrials.gov identifier: NCT01726049). Unfortunately, the National Institutes of Health-sponsored trial utilizing the PDE-5 inhibitor, Tadalafil, in PH related to left heart disease (PITCH-HF) was recently terminated because of lack of enrollment (ClinicalTrials.gov identifier: NCT01910389).
Deficient nitric oxide-soluble guanylate cyclase-cGMP (NO-sGC-cGMP) signaling plays a role in endothelial dysfunction and impaired cardiac relaxation in patients with HF. Riociguat is a direct sGC stimulator, thereby increasing the sensitivity of sGC to endogenous NO. In addition, it has anti-fibrotic, anti-proliferative as well as vasodilatory properties, with proven efficacy in patients with PAH and chronic thromboembolic PH. Its hemodynamic effects, safety, and pharmacokinetics in patients with HFpEF-PH were studied in a randomized, double-blind, placebo-controlled phase IIa trial (DILATE-1) [53]. The primary end point was the peak decrease in mPAP from baseline to 6 h after a single dose, which did not change significantly (p = 0.6). However, the hemodynamic and echocardiographic effects of a single dose of riociguat study should not be extrapolated to the potential short- or long-term treatment responses with this agent in HFpEF or HFpEF-PH. A similar agent, Vericiguat (BAY1021189) is currently in a phase II randomized, placebo-controlled clinical trial in patients with HFpEF (SOCRATES-PRESERVED), exploring the pharmacodynamics, safety and tolerability, and pharmacokinetics of the drug. It should be noted though, that patients in this trial, similar to RELAX, are not being selected for the presence or absence or PH. Thus, the results of this trial may only give us insight to the effects of this drug on HFpEF and not HFpEF-related PH.
Other Therapies
The lack of compelling evidence for treating HFpEF patients with standard HF therapies has led to a greater study of agents with direct relaxant effects and anti-inflammatory properties. Ranolazine, a partial fatty acid oxidation inhibitor, which inhibits the late sodium current and reduces sodium-dependent calcium overload, is believed to improve LV relaxation and diastolic function. Interestingly, ranolazine may also have a direct benefit on RV function by improving glucose metabolism in hypertrophied RV. As RVH and RV function have prognostic importance in PH and especially in HFpEF-PH patients, who have compromised cardiac output from not only lower LV stroke volumes but also from marginal RV function, modulators of RV myocardial performance are critically important. In RVH induced by PA banding in an animal model, RV metabolism was shown to switch from glucose oxidation to fatty acid oxidation and glycolysis. Inhibiting fatty acid oxidation improves RV energetics and metabolic efficiency, thus directly enhancing RV function [54]. Ranolazine was shown to improve RV oxygen consumption and RV function as well as promote RVH regression in experimental rats [48]. This preclinical data lends support for future study of ranolazine in HFpEF-PH patients with RV failure.
Additionally, it is well established that patients with HFpEF have limited exercise tolerance due to impaired chronotropic response and vasodilator reserve [55]. In addition, heart rate response is a major determinant of cardiac output in PH patients [56]. Hence, the rationale for beta-blocker use in HFpEF patients to slow heart rates and allow greater diastolic filling time has been challenged. In patients with HFpEF-PH, the reliance on heart rate augmentation may be more important than previously realized and further studies are needed to understand the mechanisms of autonomic dysfunction in these groups. The use of nebivolol in HFpEF-PH is being evaluated in a prospective, open-label clinical study (ClinicalTrials.gov identifier: NCT02053246).
Use of Remote Hemodynamic Monitoring for Management of HFpEF
A rise in PAP has been shown to herald a HF decompensation, often prior to the onset of worsened symptoms. Given that rises in LAP and pulmonary venous congestion are drivers of symptoms in patients with HFpEF-PH and that the current mainstay of management includes volume management, a patient management strategy utilizing remote, direct, or indirect measurements of LV filling pressures is promising. The ability to continually follow hemodynamic measurements and adjust therapy based upon these measurements is aimed at reducing HF exacerbations and hospitalizations.
The COMPASS-HF trial utilized a programmable device with a transvenous sensor lead to measure intracardiac pressure [57]. Patients with implanted device had a nonsignificant 21 % lower rate of all HF-related events compared with the control group (p = 0.33). A subgroup analysis of HFpEF revealed a 20 % reduction in HF-related events (p = 0.66), including a 29 % relative risk reduction in HF hospitalizations (p = 0.43). Given that the primary end point of significant reduction in HF-related events was not achieved, this device has not been approved for use.
The CHAMPION trial included patients with NYHA functional class III HFrEF or HFpEF who had been hospitalized for HF exacerbation within the past 12 months [4]. This trial utilized the CardioMEMS sensor implanted into a distal PA branch, which directly measured PAP. Measurements from the device were obtained on a daily basis and then were transmitted electronically by patients to a centralized database, and treatment decisions were made based upon a predefined protocol. Of the 550 patients analyzed, 314 were identified as WHO group II PH (due to left heart disease), of which 55 (17.5 %) had HFpEF. Irrespective of LVEF, patients without PH were at significantly lower risk for mortality than those with PH (hazard ratio (HR) 0.31, 95 % confidence interval (CI) 0.19 to 0.52, p < 0.0001). In addition, PH patients had higher HF hospitalization rates than nonPH patients (0.77/year vs. 0.37/year; HR 0.49, 95 % CI 0.39 to 0.61, p < 0.001). The primary end point of the rate of HF-related hospitalizations at 6 months was reduced by 28 % in the treatment group (p = 0.0002), including importantly those patients with HFpEF-PH. This suggests that this device may be useful in the management of patients with WHO group II PH, including those with HFpEF-PH. The CardioMEMS device has recently received FDA approval for use.
The LAPTOP device is an indwelling monitor implanted in the left atrium by a transseptal puncture technique, which promises to allow for more accurate direct measurement of LAP. Unfortunately, the phase III clinical trial of the LAPTOP device was recently terminated due to higher incidence of periprocedural complications in the implant group. (ClinicalTrials.Gov identifier: NCT01121107). The device/implant technique may be redesigned in the future, providing another opportunity to evaluate this innovative device in HFpEF-PH.
Future Directions, Personal Observations, and Hypothesis of Interest
As noted in the preceding sections, our understanding of complex underpinnings of HFpEF is still at a nescient level. This lack of clarity has delayed the development and deployment of efficacious molecules to treat this disease. This, coupled with the often-delayed diagnosis, can lead to the development of a progressive pulmonary vasculopathy, which in some patients may eventually evolve into a condition that resembles PAH. Once PH matures, the usual ramifications on RV function ensue, leading to biventricular dysfunction, excess neurohumoral activation, cardiorenal syndrome, and death. Unfortunately, there is no current therapy for treating this form of pulmonary vascular remodeling, especially if attempted in isolation. This later point is the key to developing a graded and combined approach to treating HFpEF-PH, especially if CpcPH is prominent. This approach involves the early identification and tenacious control of LAP, the initial trigger for HFpEF-PH, combined with measures to directly improve diastolic relaxation. If and only if this can occur will the implementation of current PH-specific medications prove to be efficacious in remodeling the pulmonary vasculature without the concomitant increase in morbidity noted in earlier trials of this therapeutics. Future trials in HFpEF-PH should incorporate this philosophy in order to maximize the potential for success. Achievement of this therapeutic goal would be a significant breakthrough for this highly morbid and mortal combined disease state.
Conclusion
HFpEF-PH is a common, poorly understood, and highly morbid disease entity for which there is no current effective therapy. This condition is commonly confused with IPAH in light of their similar hemodynamic profiles and lack of appreciation and assessment of historical risk factors, dynamic LV filling properties, and/or LV/LA morphometrics. Current treatment regimens for HFpEF-PH have either focused either on restoring LV diastolic filling properties or treatment of PH in isolation, resulting in less than satisfactory outcomes. A combined approach, which initially focuses on tenacious control of LAP and improvement of diastolic filling with the later initiation of pulmonary vascular remodeling therapy, could provide a rational and potential efficacious strategy for treating this combined condition. Use of indwelling monitors that allows ongoing assessment of LAP and PA pressures may be useful in this approach.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Hogg K, Swedberg K, McMurray J. Heart failure with preserved left ventricular systolic function; epidemiology, clinical characteristics, and prognosis. J Am Coll Cardiol. 2004;43(3):317–27.
Tribouilloy C, Rusinaru D, Mahjoub H, Souliere V, Levy F, Peltier M, et al. Prognosis of heart failure with preserved ejection fraction: a 5 year prospective population-based study. Eur Heart J. 2008;29(3):339–47.
Guazzi M. Pulmonary hypertension in heart failure preserved ejection fraction: prevalence, pathophysiology, and clinical perspectives. Circ Heart Fail. 2014;7(2):367–77.
Galie N, Hoeper MM, Humbert M, Torbicki A, Vachiery JL, Barbera JA, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J. 2009;30(20):2493–537.
Lam CS, Roger VL, Rodeheffer RJ, Borlaug BA, Enders FT, Redfield MM. Pulmonary hypertension in heart failure with preserved ejection fraction: a community-based study. J Am Coll Cardiol. 2009;53(13):1119–26.
Benza RL, Raina A, Abraham WT, Adamson PB, Lindenfeld J, Miller AB, et al. Pulmonary hypertension related to left heart disease: insight from a wireless implantable hemodynamic monitor. J Heart Lung Transplant. 2014.
Klapholz M, Maurer M, Lowe AM, Messineo F, Meisner JS, Mitchell J, et al. Hospitalization for heart failure in the presence of a normal left ventricular ejection fraction: results of the New York Heart Failure Registry. J Am Coll Cardiol. 2004;43(8):1432–8.
Leung CC, Moondra V, Catherwood E, Andrus BW. Prevalence and risk factors of pulmonary hypertension in patients with elevated pulmonary venous pressure and preserved ejection fraction. Am J Cardiol. 2010;106(2):284–6.
Massie BM, Carson PE, McMurray JJ, Komajda M, McKelvie R, Zile MR, et al. Irbesartan in patients with heart failure and preserved ejection fraction. N Engl J Med. 2008;359(23):2456–67.
Cleland JG, Tendera M, Adamus J, Freemantle N, Polonski L, Taylor J, et al. The perindopril in elderly people with chronic heart failure (PEP-CHF) study. Eur Heart J. 2006;27(19):2338–45.
Yusuf S, Pfeffer MA, Swedberg K, Granger CB, Held P, McMurray JJ, et al. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: the CHARM-Preserved Trial. Lancet. 2003;362(9386):777–81.
Vachiery JL, Adir Y, Barbera JA, Champion H, Coghlan JG, Cottin V, et al. Pulmonary hypertension due to left heart diseases. J Am Coll Cardiol. 2013;62(25 Suppl):D100–8.
Luo C, Ramachandran D, Ware DL, Ma TS, Clark Jr JW. Modeling left ventricular diastolic dysfunction: classification and key indicators. Theor Biol Med Model. 2011;8:14.
Borlaug BA, Lam CS, Roger VL, Rodeheffer RJ, Redfield MM. Contractility and ventricular systolic stiffening in hypertensive heart disease insights into the pathogenesis of heart failure with preserved ejection fraction. J Am Coll Cardiol. 2009;54(5):410–8.
Damiano Jr RJ, La Follette Jr P, Cox JL, Lowe JE, Santamore WP. Significant left ventricular contribution to right ventricular systolic function. Am J Physiol. 1991;261(5 Pt 2):H1514–24.
Kuehne T, Yilmaz S, Steendijk P, Moore P, Groenink M, Saaed M, et al. Magnetic resonance imaging analysis of right ventricular pressure-volume loops: in vivo validation and clinical application in patients with pulmonary hypertension. Circulation. 2004;110(14):2010–6.
Tedford RJ, Mudd JO, Girgis RE, Mathai SC, Zaiman AL, Housten-Harris T, et al. Right ventricular dysfunction in systemic sclerosis-associated pulmonary arterial hypertension. Circ Heart Fail. 2013;6(5):953–63.
Pellegrini P, Rossi A, Pasotti M, Raineri C, Cicoira M, Bonapace S, et al. Prognostic relevance of pulmonary arterial compliance in patients with chronic heart failure. Chest. 2014;145(5):1064–70.
Guazzi M, Gatto P, Giusti G, Pizzamiglio F, Previtali I, Vignati C, et al. Pathophysiology of cardiorenal syndrome in decompensated heart failure: role of lung-right heart-kidney interaction. Int J Cardiol. 2013;169(6):379–84.
Melenovsky V, Hwang SJ, Lin G, Redfield MM, Borlaug BA. Right heart dysfunction in heart failure with preserved ejection fraction. Eur Heart J. 2014. This article underlines the importance and role of right heart function in the pathology and prognosis of patients with HFpEF-PH.
Tedford RJ, Hassoun PM, Mathai SC, Girgis RE, Russell SD, Thiemann DR, et al. Pulmonary capillary wedge pressure augments right ventricular pulsatile loading. Circulation. 2012;125(2):289–97.
Naeije R, Vachiery JL, Yerly P, Vanderpool R. The transpulmonary pressure gradient for the diagnosis of pulmonary vascular disease. Eur Respir J. 2013;41(1):217–23.
Gerges C, Gerges M, Lang MB, Zhang Y, Jakowitsch J, Probst P, et al. Diastolic pulmonary vascular pressure gradient: a predictor of prognosis in “out-of-proportion” pulmonary hypertension. Chest. 2013;143(3):758–66.
Enson Y, Wood JA, Mantaras NB, Harvey RM. The influence of heart rate on pulmonary arterial-left ventricular pressure relationships at end-diastole. Circulation. 1977;56(4 Pt 1):533–9.
Robbins IM, Newman JH, Johnson RF, Hemnes AR, Fremont RD, Piana RN, et al. Association of the metabolic syndrome with pulmonary venous hypertension. Chest. 2009;136(1):31–6.
Thenappan T, Shah SJ, Gomberg-Maitland M, Collander B, Vallakati A, Shroff P, et al. Clinical characteristics of pulmonary hypertension in patients with heart failure and preserved ejection fraction. Circ Heart Fail. 2011;4(3):257–65.
Opotowsky AR, Ojeda J, Rogers F, Prasanna V, Clair M, Moko L, et al. A simple echocardiographic prediction rule for hemodynamics in pulmonary hypertension. Circ Cardiovasc Imaging. 2012;5(6):765–75.
Guazzi M, Borlaug BA. Pulmonary hypertension due to left heart disease. Circulation. 2012;126(8):975–90.
Halpern SD, Taichman DB. Misclassification of pulmonary hypertension due to reliance on pulmonary capillary wedge pressure rather than left ventricular end-diastolic pressure. Chest. 2009;136(1):37–43.
Hoeper MM, Bogaard HJ, Condliffe R, Frantz R, Khanna D, Kurzyna M, et al. Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D42–50.
Prasad A, Hastings JL, Shibata S, Popovic ZB, Arbab-Zadeh A, Bhella PS, et al. Characterization of static and dynamic left ventricular diastolic function in patients with heart failure with a preserved ejection fraction. Circ Heart Fail. 2010;3(5):617–26.
Abraham WT, Adamson PB, Bourge RC, Aaron MF, Costanzo MR, Stevenson LW, et al. Wireless pulmonary artery haemodynamic monitoring in chronic heart failure: a randomised controlled trial. Lancet. 2011;377(9766):658–66.
Robbins IM, Hemnes AR, Pugh ME, Brittain EL, Zhao DX, Piana RN, et al. High prevalence of occult pulmonary venous hypertension revealed by fluid challenge in pulmonary hypertension. Circ Heart Fail. 2014;7(1):116–22.
Thadani U, Parker JO. Hemodynamics at rest and during supine and sitting bicycle exercise in normal subjects. Am J Cardiol. 1978;41(1):52–9.
Yancy CW, Jessup M, Bozkurt B, Butler J, Casey Jr DE, Drazner MH, et al. 2013 ACCF/AHA guideline for the management of heart failure: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation. 2013;128(16):1810–52. These are the most updated guidelines for management of heart failure, including HF-pEF.
Heart Failure Society of A, Lindenfeld J, Albert NM, Boehmer JP, Collins SP, Ezekowitz JA, et al. HFSA 2010 Comprehensive Heart Failure Practice Guideline. J Card Fail. 2010;16(6):e1–194.
McMurray JJ, Adamopoulos S, Anker SD, Auricchio A, Bohm M, Dickstein K, et al. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2012;33(14):1787–847.
January CT, Wann LS, Alpert JS, Calkins H, Cleveland JC, Jr., Cigarroa JE, et al. 2014 AHA/ACC/HRS Guideline for the Management of Patients With Atrial Fibrillation: A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol. 2014.
McMurray JJ, Teerlink JR, Cotter G, Bourge RC, Cleland JG, Jondeau G, et al. Effects of tezosentan on symptoms and clinical outcomes in patients with acute heart failure: the VERITAS randomized controlled trials. JAMA. 2007;298(17):2009–19.
Califf RM, Adams KF, McKenna WJ, Gheorghiade M, Uretsky BF, McNulty SE, et al. A randomized controlled trial of epoprostenol therapy for severe congestive heart failure: The Flolan International Randomized Survival Trial (FIRST). Am Heart J. 1997;134(1):44–54.
Anand I, McMurray J, Cohn JN, Konstam MA, Notter T, Quitzau K, et al. Long-term effects of darusentan on left-ventricular remodelling and clinical outcomes in the EndothelinA Receptor Antagonist Trial in Heart Failure (EARTH): randomised, double-blind, placebo-controlled trial. Lancet. 2004;364(9431):347–54.
Kaluspi E, Kobrin I, Zimlichman R, et al. RITZ-5: randomized intravenous tezosentan (an endothelin-A-B antagonist) for the treatment of pulmonary edema: a prospective, multicenter, double-blind, placebo-controlled study. J Am Coll Cardiol. 2003;41:204–14.
Luscher TF, Enseleit F, Pacher R, Mitrovic V, Schulze MR, Willenbrock R, et al. Hemodynamic and neurohumoral effects of selective endothelin A (ET(A)) receptor blockade in chronic heart failure: the Heart Failure ET(A) Receptor Blockade Trial (HEAT). Circulation. 2002;106(21):2666–72.
Packer M, McMurray J, Massie B, et al. Clinical effects of endothelin receptor antagonism with bosentan in patients with severe chronic heart failure: results of a pilot study. J Card Fail. 2005;11:12–20.
Michelakis E, Tymchak W, Lien D, Webster L, Hashimoto K, Archer S. Oral sildenafil is an effective and specific pulmonary vasodilator in patients with pulmonary arterial hypertension: comparison with inhaled nitric oxide. Circulation. 2002;105(20):2398–403.
Noguchi T, Kihara Y, Begin KJ, Gorga JA, Palmiter KA, LeWinter MM, et al. Altered myocardial thin-filament function in the failing Dahl salt-sensitive rat heart: amelioration by endothelin blockade. Circulation. 2003;107(4):630–5.
Vuurmans TJ, Boer P, Koomans HA. Effects of endothelin-1 and endothelin-1 receptor blockade on cardiac output, aortic pressure, and pulse wave velocity in humans. Hypertension. 2003;41(6):1253–8.
Berger R, Stanek B, Hulsmann M, Frey B, Heher S, Pacher R, et al. Effects of endothelin a receptor blockade on endothelial function in patients with chronic heart failure. Circulation. 2001;103(7):981–6.
Zile MR, Bourge RC, Redfield MM, Zhou D, Baicu CF, Little WC. Randomized, double-blind, placebo-controlled study of sitaxsentan to improve impaired exercise tolerance in patients with heart failure and a preserved ejection fraction. JACC Heart Fail. 2014;2(2):123–30.
Francis SH, Blount MA, Corbin JD. Mammalian cyclic nucleotide phosphodiesterases: molecular mechanisms and physiological functions. Physiol Rev. 2011;91(2):651–90.
Guazzi M, Vicenzi M, Arena R, Guazzi MD. Pulmonary hypertension in heart failure with preserved ejection fraction: a target of phosphodiesterase-5 inhibition in a 1-year study. Circulation. 2011;124(2):164–74. This is one of the few studies showing the positive outcomes in patients with HFpEF-PH using a pulmonary vasodilator agent.
Redfield MM, Chen HH, Borlaug BA, Semigran MJ, Lee KL, Lewis G, et al. Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA. 2013;309(12):1268–77. Large, randomized control trial of use of PDE5i in patients with HFpEF.
Bonderman D, Pretsch I, Steringer-Mascherbauer R, Jansa P, Rosenkranz S, Tufaro C, et al. Acute Hemodynamic Effects of Riociguat in Patients with Pulmonary Hypertension Associated with Diastolic Heart Failure (DILATE-1): A Randomized, Double-Blind, Placebo-Controlled, Single-Dose Study. Chest. 2014.
Fang YH, Piao L, Hong Z, Toth PT, Marsboom G, Bache-Wiig P, et al. Therapeutic inhibition of fatty acid oxidation in right ventricular hypertrophy: exploiting Randle’s cycle. J Mol Med (Berlin). 2012;90(1):31–43.
Borlaug BA, Melenovsky V, Russell SD, Kessler K, Pacak K, Becker LC, et al. Impaired chronotropic and vasodilator reserves limit exercise capacity in patients with heart failure and a preserved ejection fraction. Circulation. 2006;114(20):2138–47.
Deboeck G, Taboada D, Hagan G, Treacy C, Page K, Sheares K, et al. Maximal cardiac output determines 6 minutes walking distance in pulmonary hypertension. PLoS One. 2014;9(3):e92324.
Bourge RC, Abraham WT, Adamson PB, Aaron MF, Aranda Jr JM, Magalski A, et al. Randomized controlled trial of an implantable continuous hemodynamic monitor in patients with advanced heart failure: the COMPASS-HF study. J Am Coll Cardiol. 2008;51(11):1073–9.
Compliance with Ethics Guidelines
Conflict of Interest
Manreet Kanwar, Megan M. Clarke, Claire Walter, and Srinivas Murali have nothing to disclose. Ryan J. Tedford is a consultant for Merck. Richa Agarwal reports grants from Thoratec. George Sokos reports that he is a speaker for Actelion and Bayer. Raymond L. Benza reports grants from Bayer and grants from Actelion.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Pulmonary Hypertension
Rights and permissions
About this article

Cite this article
Kanwar, M., Tedford, R.J., Agarwal, R. et al. Management of Pulmonary Hypertension due to Heart Failure with Preserved Ejection Fraction. Curr Hypertens Rep 16, 501 (2014). https://doi.org/10.1007/s11906-014-0501-5
Published:
DOI: https://doi.org/10.1007/s11906-014-0501-5