
Abstract
Purpose of Review
This review synthesizes recent evidence of secondary sclerosing cholangiopathies, specifically IgG4-sclerosing cholangiopathy, post-transplant cholangiopathies, COVID-19-induced cholangiopathy, and sclerosing cholangiopathies due to critical illness.
Recent Findings
The clinical diagnostic criteria and practice guidelines have been updated for IgG4-sclerosing cholangiopathy. Cholangiopathy associated with livers donated after circulatory death has been further characterized, though incidence is expected to decline significantly as the use of normothermic perfusion technologies expands. COVID-19 sclerosing cholangiopathy, a likely novel entity similar in pathogenesity to sclerosing cholangiopathy of critical illness, has been identified during the SARS-CoV-2 pandemic.
Summary
The evaluation of progressive cholestasis requires consideration of rarer forms of secondary cholangiopathies based on clinical context.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Sclerosing cholangiopathies—progressive inflammatory processes of the bile ducts (BD) that lead to their destruction and consequent liver dysfunction—are categorized as either primary or secondary. Primary sclerosing cholangitis (PSC) is a prototypical, primary, immune-mediated cholangiopathy, often associated with inflammatory bowel disease and with a distinct risk of cholangiocarcinoma, though the precise drivers are not well understood and treatment is usually supportive until the need for liver transplantation [1, 2]. Conversely, secondary sclerosing cholangiopathies have identifiable triggers and varied disease courses and treatments [3]. In this review, we address four distinct secondary sclerosing cholangiopathies: IgG4 sclerosing cholangiopathy, post-transplant ischemic cholangiopathy, sclerosing cholangiopathy of critical illness, and post-COVID-19 cholangiopathy, which represent more epidemiologically important entities.
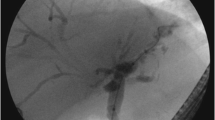
By definition, the hallmark clinical presentation of cholangiopathies is cholestasis, marked by abnormal elevation of serum alkaline phosphatase and gamma glutamyl transferase levels. When cholangiopathies progress to significantly impairing choleresis or liver synthetic function, serum total bilirubin levels may also be elevated. Patients may complain of pruritus and fatigue, and may present with jaundice when total bilirubin is elevated. When disease is prolonged, consequences of cholestasis such as fat-soluble vitamin deficiency and bone mineral loss can be expected. Impaired choleresis due to strictures can lead to acute and chronic cholangitis, and irreversible states are characterized by loss of biliary endothelial integrity causing intrahepatic bilomas and chronic biliary inflammation leading to wasting, liver fibrosis, and eventual liver failure. Depending on the chronicity of inflammation, patients may be at risk of cholangiocarcinoma, but the rapidity of the progression of secondary cholangiopathies appear to suggest this to be of lower likelihood. As biliary injury progresses, sclerosing cholangiopathies are readily diagnosed by magnetic resonance cholangiography (MRC) or endoscopic retrograde cholangiography (ERC) which confirm multifocal biliary strictures and pruning of the peripheral bile ducts [4, 5].
The process of biliary endothelial injury, which ultimately leads to biliary stricturing and the above downstream clinical complications, can be traced to impaired hepatic arterial flow causing ischemic death of cholangiocytes and cholangiocyte regenerative structures like peribiliary glands, immune-mediated attacks on cholangiocytes, and/or bile composition-mediated inflammation of cholangiocytes [6, 7]. The pathophysiology however is often multi-factorial. Nonetheless, this allows for a convenient categorization of secondary sclerosing cholangiopathies and treatments or supportive care that are geared at addressing these specific causes [8, 9]. Besides attempts to address the root cause, supportive therapeutic options for sclerosing cholangitis are limited. Biliary strictures causing obstructive jaundice are best addressed by ERC or percutaneous biliary drainage, in order to restore choleresis and reduce the risk of cholangitis. Advanced cholangiopathies, manifesting in progressive jaundice, wasting, fibrosis, or biloma formation, however, usually represent an irreversible state and are best treated with liver transplantation.
IgG-4 Sclerosing Cholangiopathy
IgG4 sclerosing cholangiopathy (IgG4-SC) is a liver-specific subtype of IgG4-related disease (IgG4-RD), a systemic condition of prolific IgG4-positive plasma deposition and consequent inflammation and fibrosis [10, 11] With IgG4-SC, these plasma cells are deposited transmurally in biliary ductal tissue, leading to an intense lymphoplasmacytic inflammatory response, storiform (cartwheel-like) fibrosis, and obliterative phlebitis [12]. IgG4-related autoimmune pancreatitis (AIP) coexists in 83–92% cases of IgG4-SC [10, 11, 13]. Overall prevalence of IgG4-SC has been difficult to estimate as it is a relatively new disease whose diagnostic criteria were not formally established until the early 2000s. However, based on population studies up until 2020, IgG4-SC has a predilection for older males with an average age at diagnosis of 67 years [14].
The specific triggers of IgG4-SC are not known. Gut microbiome or metabolome dysregulation is believed to have an influence on PSC progression, and small studies have attempted to determine if similar correlations exist with IgG4-SC. For instance, in a study performed by Liu et al., 16 s rRNA amplicon sequencing of fecal samples in patients with PSC, IgG4-SC (n = 34), and healthy controls demonstrated low levels of the Blautia species and elevated levels of succinic acid as a distinct signature of IgG4-SC patients [15]. Whether this correlation is causal or has prognostic implications on the disease is unclear.
The cholangiographic appearance of IgG4-SC allows for identification of four sub-forms of the disease. Type 1 IgG4-SC causes stenosis isolated to the common bile duct (CBD). Type 2a involves CBD stenosis and intrahepatic biliary strictures with associated proximal dilation, and type 2b involves CBD stenosis and intrahepatic biliary strictures without proximal dilation. Type 3 IgG4-SC is characterized by stenosis of the CBD and hilar bile ducts, while type 4 stenoses are isolated to the hilar bile ducts [16, 17•].
Definite diagnosis of Ig4-SC however can be challenging. While serum IgG4 levels are usually elevated in IgG4-SC, this is not sufficient to diagnose the condition [18]. For instance, serum IgG4 and/or tissue IgG4 staining is detectable in up to a quarter of patients with PSC—a condition with markedly different treatment, monitoring, and prognostic implications from IgG4-SC. Attempting to differentiate IgG4-SC and PSC is also challenging as both conditions may have similar cholangiographic appearances. Given the clinical implications, studies have attempted to develop models to differentiate IgG4-SC from PSC. For instance, a model developed by Moon et al. using age, other organ involvement, and beaded cholangiographic appearance were able to discriminate PSC and IgG4-SC with a high degree of certainty, with consequent cutoffs to trigger corticosteroid trials when the score predicts IgG4-SC. However, these usually single-center studies with small sample sizes require further validation [19].
The HISORt (Histology of the bile ducts, Imaging of the bile ducts, Serology (serum Ig4 level), Other organ involvement, Response to corticosteroid Therapy) criteria developed by Mayo Clinic originally for AIP, and the Japanese IgG4-SC Consensus Criteria are the most widely used to aid in the diagnosis of IgG4-SC. Both HISTORt and the Japanese criteria include other organ IgG4 involvement, histology demonstrating lymphoplasmacytic infiltrates with IgG4 staining, serum IgG4 levels, biliary strictures on cholangiography, and response to corticosteroid therapy [13, 17•, 20]. While these remain robust diagnostic criteria for IgG4-SC, both require extensive diagnostic workups and may not be readily generalizable to non-specialist centers.
All forms of IgG4-SC however respond to corticosteroids, and this remains the first-line therapy. This treatment response also distinguishes IgG4-SC from other sclerosing cholangiopathies. While robust clinical trials are lacking, a prednisone dose of 0.6 mg/kg/day for 2–4 weeks followed by a taper over 2–3 months—a regimen extrapolated from the treatment of IgG4-RD—is considered reasonable [21]. Corticosteroid treatment response rate in IgG4-RD exceeds 80% but relapse occurs in upwards of 30% patients [22].
Patients with IgG4-SC being older and more susceptible to corticosteroid-related adverse effects may however require alternative long-term treatment strategies for relapsing disease. Recent data suggest high response rates with the B-cell depleting therapy rituximab in IgG4-RD. A prospective study of 30 patients with IgG4-RD treated with two doses of rituximab demonstrated a favorable response in 77%, with complete remission at 12 months in 40% of patients [23]. In a retrospective analysis, response was seen in 29/33 (93.5%) of IgG4-RD treated with at least one course of rituximab. While relapse occurred in 42% of patients with follow-up, in patients who were placed on maintenance rituximab, relapse free survival was 41 vs 21 months [24]. While there is insufficient data to extrapolate these IgG4-RD treatment responses and relapse rates to IgG4-SC, rituximab may offer a salvage treatment option for IgG4-SC patients with inadequate response or with contraindications to corticosteroids.
Post-liver Transplant Cholangiopathy
In the post-liver transplant setting, biliary strictures can be delineated as anastomotic or non-anastomotic. Anastomotic strictures, owing largely to surgical reconstruction technique of the bile duct, are focal, identified early after transplantation, usually responsive to endoscopic, percutaneous, or surgical interventions to restore patency, and are not discussed in review as they are not causes of secondary cholangiopathies when treated. Non-anastomotic strictures, occurring in the intrahepatic biliary system, however represent a more insidious process as they are significantly more prone to progressing to secondary cholangiopathy and allograft loss.
Non-anastomotic post-transplant cholangiopathies can be broadly classified into two etiologies, those caused by biliary ischemic injury due to hepatic arterial compromise after transplantation, and those caused by biliary ischemic injury during liver procurement, transport, and transplantation.
Post-transplant Cholangiopathy Secondary to Arterial Compromise
The intrahepatic biliary system is exclusively supplied by arterial flow, with no contribution from portal venous blood. In the native state, the biliary system is supplied by the hepatic artery as well as several collateral vessels. The biliary system in a transplanted liver however is entirely reliant on blood flow through the donor hepatic artery [25]. Clinical manifestations of arterial insufficiency can range from cholestasis to jaundice, cholangitis or biliary sepsis, bile leaks, to frank allograft failure, depending on the acuity and degree of insufficiency. Acute processes like early hepatic arterial thrombosis after transplantation manifest in rapid allograft failure rather than cholangiopathy, and are managed with urgent surgical or endovascular revascularization attempts and frequently, re-transplantation [26, 27].
Less severe forms of arterial insufficiency, for instance, from hepatic artery stenosis (HAS), if unaddressed, are prone to ischemic cholangiopathy (IC) and subacute allograft dysfunction and failure. HAS occurs in up to a 3–15% of liver transplants, with the majority occurring at the level of the anastomosis (59%) or in the form of kinks in the donor hepatic artery (41%) [27, 28]. Most patients are asymptomatic but cholestasis is the most common clinical presentation, though HAS can progress to hepatic artery thrombosis and consequent allograft failure [26, 29]. If ischemic injury progresses, multifocal biliary strictures, biliary necrosis and biloma formation, recurrent cholangitis, and consequent allograft failure are the typical clinical presentations.
In several studies, Doppler ultrasonography has demonstrated a sensitivity of 85–100% and specificity of 99% for HAS [27, 28]. Doppler measurement of reduction in the resistive indices, increased velocity, and tardus parvus waveforms in the main, right, and/or left hepatic artery confirm HAS. Angiography of the hepatic artery, either by computed tomography, magnetic resonance imaging, or through endovascular approaches, remain the gold standard diagnostic modalities.
The management of HAS ranges from observation to endovascular therapies (balloon angioplasty, bare metal stents, drug eluting stents, or a combination), and less frequently, surgical revision [30]. While head-to-head trials do not exist, balloon angioplasty alone is thought to not provide as durable a response as angioplasty combined with stent placement [31]. Stent-related complications, including in-stent restenosis from intimal hyperplasia, can hamper revascularization though a clear superiority of drug-eluting stents versus the more restenosis-prone bare metal stents has not been demonstrated [30].
Post-transplant Cholangiopathy in the Absence of Arterial Compromise
IC in the absence of arterial compromise is a progressive, and hence feared complication of liver transplantation. Up to 50% of allografts affected by IC fail despite ERC and other interventions to maintain biliary patency, requiring consideration of re-transplantation [32, 33•].
Rates of IC in livers procured after brain death (DBD) historically ranged 1–—10%, whereas rates in livers procured after circulatory death (DCD) historically ranged 10–30% [34,35,36]. With careful donor and recipient selection and improvements in surgical and preservation techniques, DCD-IC rates have been reduced to 3–12% at experienced centers [33•], but DCD organs remain significantly underutilized in the USA with approximately a 12:1 preference for DBD organs [37]. The degree of this apparent out-of-proportion dissuasion to use DCD organs prompted the United Network for Organ Sharing (UNOS) to allow Model for End-Stage Liver Disease (MELD) exception point allocation for DCD-IC as of June 2022, which facilitates re-transplantation if IC occurs [38•, 39]. The criteria that must be met for MELD exception points to be granted include two of the following within 12 months of transplant: persistent elevation in bilirubin > 2 mg/dl, \(\ge\) 2 episodes of cholangitis associated with bacteremia, and/or non-anastomotic biliary strictures that do not respond to ERC [38•].
The typical onset of clinically significant DBD- and DCD-IC is within 12 months of transplantation, initially manifesting in progressive cholestasis, and then progressing to non-anastomotic biliary stricturing, and the final common pathway of jaundice, cholangitis, and allograft failure [33•, 40]. In DCD allografts, where the majority of the research on this form of cholangiopathy has been done, four distinct biliary stricturing patterns are identified, each with prognostic implications (Table 1) [33•]. Early identification of IC may be possible. Once biliary anastomotic strictures, HAT, HAS, acute cellular rejection, and drug-induced liver injury are excluded, elevated alkaline phosphatase and bilirubin at 1 month and 2 months after transplantation has been shown in a single center study to strongly correlate with IC-related allograft failure. Conversely, in this study, alkaline phosphatase < 100 U/L 2 months after transplantation has a 97% negative predictive value [40].
Key histologic features of early DCD-IC are biliary endothelial necrosis and biliary obstruction which impedes choleresis–findings common to other forms of biliary ischemic injury such as ischemia reperfusion injury, HAT, and HAS. The duration of liver non-perfusion after donor cardiac standstill (warm ischemia) is the most important risk factor for IC that appears to differentiate the rates of IC in DCD versus DBD livers.
Cold ischemia times and preservation methods after organ procurement (i.e., during transport) are common to both DCD and DBD organ procurement, and may explain in part the baseline rate of IC in DBD organs. Prolonged cold ischemia time also has association with early allograft dysfunction and biliary complications. The development of normothermic machine perfusion technologies may circumvent this [41]. The PROTECT trial evaluated the effect of normothermic machine perfusion on early allograft dysfunction and ischemic biliary complications [42]. Across 20 transplant centers in the USA, patients (N = 293) were randomized to normothermic machine perfusion or traditional static cold storage preservation methods. The use of normothermic perfusion led to a lower rate of early allograft dysfunction, an overall increase in the transplant center propensity to use DCD organs, and a decrease in biliary cholangiopathy at 6 and 12 months [42]. Several subsequent studies appear to validate these results, particularly markedly reduced rates of IC to at least levels comparable to DBD organs [43,44,45]. Normothermic perfusion is likely transformative in paving the way for greater acceptance of DCD organs, though the technology is currently limited by cost, perfusionist expertise, and logistics, which may delay its widespread application.
While ischemic injury appears to be the primary precipitant of DCD-IC, it does not explain progressive bile duct destruction after re-establishing arterial perfusion after transplantation. This suggests other ongoing injury processes in the transplanted liver, intervening on which could mitigate the downstream complications of DCD-IC. One pathophysiologic process that may propagate the DCD-IC syndrome is the accumulation of toxic concentrations of bile acids (BA) which have detergent effects on hepatocyte and cholangiocyte lipid membranes and induce apoptosis [36]. Since the antecedent to DCD-IC is impairment of choleresis due to bile duct injury/obstruction triggered by an ischemic insult, it is conceivable that BA stasis and accumulation influence the ongoing progressive injury seen even after re-established physiologic arterial perfusion at transplantation. Recent data suggest that the use of peroxisome proliferator activated receptor alpha (PPAR-alpha) agonists which downregulate bile acid synthesis may significantly impede the progression of IC in DCD transplant recipients, but require further validation [46].
Sclerosing Cholangiopathy in Critical Ill Patients
Critical illness is often associated with abnormal liver enzymes, usually the result of “bystander injury” due to ischemic hepatitis, infection-related pro-inflammatory cytokines, or drug-induced liver injury. The typical manifestation is a mild increase in aminotransferases (< 3 × ULN). This initial insult may evolve into a cholestatic liver injury, with rise in bilirubin and alkaline phosphatase, commonly referred to as cholestasis of sepsis [47]. Rarely, cholestatic liver injury may evolve into a progressive and irreversible sclerosing cholangiopathy and eventually, biliary cirrhosis. Once diagnosed, sclerosing cholangitis in critically ill patients (SC-CIP) has been shown to quickly devolve to biliary cirrhosis as documented by a case series of 16 patients who found that 88% of those with SC-CIP developed findings consistent with cirrhosis in less than 6 months from initial diagnosis [48].
While the pathogenesis is unclear, SC-CIP is documented most often in settings of hemodynamic instability, respiratory failure requiring mechanical ventilation, those requiring cardiothoracic surgery, and in severe trauma or burn victims [49, 50]. Hemodynamic instability instigates cholestatic liver injury via two distinct mechanisms. First, it creates a state of hypoperfusion which directly induces biliary ischemia. Second, the initiation of vasoactive medications (e.g., epinephrine, norepinephrine) reduces perfusion in the splanchnic bed [8, 51, 52]. Separately, high end expiratory pressures and low tidal volumes may reduce splanchnic flow [51, 53].
SC-CIP results in a chronic and progressive cholestatic liver injury that continues long after recovery from the initial critical illness. Imaging with MRC demonstrates multifocal beading and stricturing of the bile ducts, and patients may require ERC to relieve biliary obstructions [4]. Concurrent bacterial cholangitis can be expected, as with any case of biliary obstruction, and this has been borne out in cultures of ERC aspirates [54, 55]. SC-CIP is an ominous development with a median survival of 13 months without liver transplantation [49, 56]. Transplant outcomes however are excellent, and comparable to the 1-year and 5-year survival rates seen in alcohol-related cirrhosis [48, 54, 57].
COVID-19 Sclerosing Cholangiopathy
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the virus responsible for the ongoing coronavirus pandemic (COVID-19), is well-documented to have multi-organ effects besides the respiratory system. Liver enzyme abnormalities is a well-recognized complication of COVID-19, with an incidence rate as high as 67% [58,59,60]. As with critical illness, liver injury is usually mild and limited to aminotransferase elevations < 3 × ULN. A distinct entity of COVID-19 sclerosing cholangiopathy was recognized during the pandemic as a secondary injury that results in cholestatic jaundice and sometimes leading to liver failure. In a small subset of patients, this cholestatic injury can progress into a cholangiopathy leading to fibrosis and liver decompensation. Case series have noted this can occur > 3 months after initial diagnosis of COVID-19 [61•]. Anecdotal experiences with ursodeoxycholic acid has not demonstrated benefit [61•].
In most cases, COVID-19 sclerosing cholangiopathy affects patients with severe COVID-19 disease, which has been most frequently defined as requiring vasopressors for hemodynamic instability, extracorporeal membrane oxygenation and/or mechanical ventilation for respiratory failure. Cholangiopathy has been diagnosed using MRC with findings of intrahepatic biliary duct beading, hyperenhancement of bile duct walls and increased peribiliary diffusion signal [61•]. Liver biopsies have been performed in a number of case series with heterogeneous bile ductule-centered injury patterns [58, 61•, 62]. A unifying feature of most biopsies is evidence of large bile duct obstruction, absence of intravascular thrombi, portal and periportal fibrosis, and hepatic artery endothelial swelling [61•, 62]. The presence of ductopenia was noted in a minority of cases [62]. The angiotensin-converting enzyme 2 (ACE2) with host protein transmembrane serine protease 2 (TMPRSS2)—the main cellular receptors for the SARS-CoV-2 virus—are densely expressed in the gastrointestinal tract, including the vascular and parenchymal cells of within the liver [63,64,65,66]. While this suggests a likelihood of direct liver injury from the virus, whether COVID-19 sclerosing cholangiopathy is a subset of SC-CIP or a distinct entity related to the pathogenesis of the COVID-19 disease, is not clear.
Underlying chronic liver disease appears to be a risk factor for development of COVID-19 sclerosing cholangiopathy [67]. In a retrospective review of 496 patients with COVID-19-related hospitalization, 15.4% of patients with chronic liver disease developed sclerosing cholangiopathy, compared to 4.6% of patients without chronic liver disease, but chronic liver disease had no impact on the rate of severe cholestasis [67]. Metabolic dysfunction associated steatotic liver disease accounted for 60% of underlying liver disease, and this correlation with metabolic disease in patients with sclerosing cholangiopathy is seen in other smaller studies as well [67,68,69,70]. Other correlations of COVID-19 sclerosing cholangiopathy development include functional and medication-induced immunosuppressed states [71]. Finally, while there is an increased comfort level of using SARS-CoV-2-positive livers for transplantation, whether this predisposes to COVID-19 sclerosing cholangiopathy is not known.
Conclusion
Secondary sclerosing cholangiopathies occur due to a variety of liver injury processes ranging from ischemia, autoimmune injury, critical illness, and infection. While cholestasis is the hallmark early marker common to all of these etiologies, late state manifestations—cholestatic jaundice and biliary strictures—are the most clinically apparent. These late-stage manifestations may not have distinct pathognomonic features to differentiate between the forms of secondary sclerosing cholangiopathy, and features may be comparable to primary sclerosing cholangitis. Some forms readily respond to treatment (e.g., corticosteroids in IgG4-sclerosing cholangiopathy), while others rapidly progress (e.g., ischemic cholangiopathy, SC-CIP), and therefore require a judicious clinical-context specific evaluation and management. The rarity of these conditions mandate greater multi-center collaboration to better define etiologic mechanisms, pathogenesis, diagnostic criteria, and management guidelines.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Broomé U, Olsson R, Lööf L, Bodemar G, Hultcrantz R, Danielsson A, et al. Natural history and prognostic factors in 305 Swedish patients with primary sclerosing cholangitis. Gut. 1996;38(4):610–5.
de Valle MB, Björnsson E, Lindkvist B. Mortality and cancer risk related to primary sclerosing cholangitis in a Swedish population-based cohort. Liver Int Off J Int Assoc Study Liver. 2012;32(3):441–8.
Ruemmele P, Hofstaedter F, Gelbmann CM. Secondary sclerosing cholangitis. Nat Rev Gastroenterol Hepatol. 2009;6(5):287–95.
Ludwig DR, Anderson MA, Itani M, Sharbidre KG, Lalwani N, Paspulati RM. Secondary sclerosing cholangitis: mimics of primary sclerosing cholangitis. Abdom Radiol N Y. 2023;48(1):151–65.
Möller K, Braden B, Culver EL, Jenssen C, Zadeh ES, Alhyari A, et al. Secondary sclerosing cholangitis and IgG4-sclerosing cholangitis — a review of cholangiographic and ultrasound imaging. Endosc Ultrasound. 2022;12(2):181–99.
Gudnason HO, Björnsson ES. Secondary sclerosing cholangitis in critically ill patients: current perspectives. Clin Exp Gastroenterol. 2017;23(10):105–11.
Trauner M, Fickert P, Wagner M. MDR3 (ABCB4) defects: a paradigm for the genetics of adult cholestatic syndromes. Semin Liver Dis. 2007;27(1):77–98.
Leonhardt S, Veltzke-Schlieker W, Adler A, Schott E, Hetzer R, Schaffartzik W, et al. Trigger mechanisms of secondary sclerosing cholangitis in critically ill patients. Crit Care. 2015;19(1):131.
Beuers U, Hohenester S, de Buy Wenniger LJM, Kremer AE, Jansen PLM, Elferink RPJO. The biliary HCO(3)(-) umbrella: a unifying hypothesis on pathogenetic and therapeutic aspects of fibrosing cholangiopathies. Hepatol Baltim Md. 2010;52(4):1489–96.
Tanaka A. IgG4-related sclerosing cholangitis and primary sclerosing cholangitis. Gut Liver. 2019;13(3):300–7.
Okazaki K, Uchida K, Ikeura T, Takaoka M. Current concept and diagnosis of IgG4-related disease in the hepato-bilio-pancreatic system. J Gastroenterol. 2013;48(3):303–14.
Brooling J, Leal R. Secondary sclerosing cholangitis: a review of recent literature. Curr Gastroenterol Rep. 2017;19(9):44.
Ghazale A, Chari ST, Zhang L, Smyrk TC, Takahashi N, Levy MJ, et al. Immunoglobulin G4-associated cholangitis: clinical profile and response to therapy. Gastroenterology. 2008;134(3):706–15.
Tanaka A, Mori M, Kubota K, Naitoh I, Nakazawa T, Takikawa H, et al. Epidemiological features of immunoglobulin G4-related sclerosing cholangitis in Japan. J Hepato-Biliary-Pancreat Sci. 2020;27(9):598–603.
Liu Q, Li B, Li Y, Wei Y, Huang B, Liang J, et al. Altered faecal microbiome and metabolome in IgG4-related sclerosing cholangitis and primary sclerosing cholangitis. Gut. 2022;71(5):899–909.
Nakazawa T, Ohara H, Sano H, Ando T, Joh T. Schematic classification of sclerosing cholangitis with autoimmune pancreatitis by cholangiography. Pancreas. 2006;32(2):229.
• Nakazawa T, Kamisawa T, Okazaki K, Kawa S, Tazuma S, Nishino T, et al. Clinical diagnostic criteria for IgG4-related sclerosing cholangitis 2020: (Revision of the clinical diagnostic criteria for IgG4-related sclerosing cholangitis 2012). J Hepato-Biliary-Pancreat Sci. 2021;28(3):235–42 The authors have revised the 2012 clinical diagnostic criteria for IgG4-related disease.
Manganis CD, Chapman RW, Culver EL. Review of primary sclerosing cholangitis with increased IgG4 levels. World J Gastroenterol. 2020;26(23):3126–44.
Moon SH, Kim MH, Lee JK, Baek S, Woo YS, Cho DH, et al. Development of a scoring system for differentiating IgG4-related sclerosing cholangitis from primary sclerosing cholangitis. J Gastroenterol. 2017;52(4):483–93.
Ohara H, Okazaki K, Tsubouchi H, Inui K, Kawa S, Kamisawa T, et al. Clinical diagnostic criteria of IgG4-related sclerosing cholangitis 2012. J Hepato-Biliary-Pancreat Sci. 2012;19(5):536–42.
Kamisawa T, Okazaki K. Diagnosis and treatment of IgG4-related disease. Curr Top Microbiol Immunol. 2017;401:19–33.
Shirakashi M, Yoshifuji H, Kodama Y, Chiba T, Yamamoto M, Takahashi H, et al. Factors in glucocorticoid regimens associated with treatment response and relapses of IgG4-related disease: a multicentre study. Sci Rep. 2018;8(1):10262.
Carruthers MN, Topazian MD, Khosroshahi A, Witzig TE, Wallace ZS, Hart PA, et al. Rituximab for IgG4-related disease: a prospective, open-label trial. Ann Rheum Dis. 2015;74(6):1171–7.
Ebbo M, Grados A, Samson M, Groh M, Loundou A, Rigolet A, et al. Long-term efficacy and safety of rituximab in IgG4-related disease: data from a French nationwide study of thirty-three patients. PLoS ONE. 2017;12(9):e0183844.
Pretter PC, Orons PD, Zajko AB. The bile duct in liver transplantation. Semin Roentgenol. 1997;32(3):202–14.
Mourad MM, Liossis C, Gunson BK, Mergental H, Isaac J, Muiesan P, et al. Etiology and management of hepatic artery thrombosis after adult liver transplantation. Liver Transpl. 2014;20(6):713–23.
Piardi T, Lhuaire M, Bruno O, Memeo R, Pessaux P, Kianmanesh R, et al. Vascular complications following liver transplantation: a literature review of advances in 2015. World J Hepatol. 2016;8(1):36–57.
Abbasoglu O, Levy MF, Vodapally MS, Goldstein RM, Husberg BS, Gonwa TA, et al. Hepatic artery stenosis after liver transplantation–incidence, presentation, treatment, and long term outcome. Transplantation. 1997;63(2):250–5.
Saad WEA, Davies MG, Sahler L, Lee DE, Patel NC, Kitanosono T, et al. Hepatic artery stenosis in liver transplant recipients: primary treatment with percutaneous transluminal angioplasty. J Vasc Interv Radiol. 2005;16(6):795–805.
Naidu S, Alzubaidi S, Knuttinen G, Patel I, Fleck A, Sweeney J, et al. Treatment of hepatic artery stenosis in liver transplant patients using drug-eluting versus bare-metal stents. J Clin Med. 2021;10(3):380.
Hamby BA, Ramirez DE, Loss GE, Bazan HA, Smith TA, Bluth E, et al. Endovascular treatment of hepatic artery stenosis after liver transplantation. J Vasc Surg. 2013;57(4):1067–72.
Croome KP, Taner CB. The changing landscapes in DCD liver transplantation. Curr Transplant Rep. 2020;7(3):194–204.
Croome KP, Mathur AK, Aqel B, Yang L, Taner T, Heimbach JK, et al. Classification of distinct patterns of ischemic cholangiopathy following DCD liver transplantation: distinct clinical courses and long-term outcomes from a multicenter cohort. Transplantation. 2022;106(6):1206–14 These authors performed a multi-center study to classify DCD cholangiopathy into four main subtypes and describe the clinical implications of each subtype.
de Vries Y, von Meijenfeldt FA, Porte RJ. Post-transplant cholangiopathy: classification, pathogenesis, and preventive strategies. Biochim Biophys Acta Mol Basis Dis. 2018;1864(4 Pt B):1507–15.
Mourad MM, Algarni A, Liossis C, Bramhall SR. Aetiology and risk factors of ischaemic cholangiopathy after liver transplantation. World J Gastroenterol WJG. 2014;20(20):6159–69.
Op den Dries S, Sutton ME, Lisman T, Porte RJ. Protection of bile ducts in liver transplantation: looking beyond ischemia. Transplantation. 2011;92(4):373–9.
Kwong AJ, Kim WR, Lake JR, Smith JM, Schladt DP, Skeans MA, et al. OPTN/SRTR 2019 Annual Data Report: Liver. Am J Transplant Off J Am Soc Transplant Am Soc Transpl Surg. 2021;21(Suppl 2):208–315.
Notices of implemented actions - OPTN [Internet]. [cited 2023 Sep 16]. Available from: https://optn.transplant.hrsa.gov/policies-bylaws/notices-of-implemented-actions/ New MELD-exception criteria were enlisted for DCD-cholangiopathy in an effort to encourage use of DCD livers.
Mehta S, Trotter JF. Policy corner: ischemic cholangiopathy associated with donation after cardiac death. Liver Transpl. 2023;29(6):653.
Halldorson JB, Rayhill S, Bakthavatsalam R, Montenovo M, Dick A, Perkins J, et al. Serum alkaline phosphatase and bilirubin are early surrogate markers for ischemic cholangiopathy and graft failure in liver transplantation from donation after circulatory death. Transplant Proc. 2015;47(2):465–8.
Schlegel A, Mergental H, Fondevila C, Porte RJ, Friend PJ, Dutkowski P. Machine perfusion of the liver and bioengineering. J Hepatol. 2023;78(6):1181–98.
Markmann JF, Abouljoud MS, Ghobrial RM, Bhati CS, Pelletier SJ, Lu AD, et al. Impact of portable normothermic blood-based machine perfusion on outcomes of liver transplant: the OCS Liver PROTECT randomized clinical trial. JAMA Surg. 2022;157(3):189–98.
Hessheimer AJ, de la Rosa G, Gastaca M, Ruíz P, Otero A, Gómez M, et al. Abdominal normothermic regional perfusion in controlled donation after circulatory determination of death liver transplantation: outcomes and risk factors for graft loss. Am J Transplant Off J Am Soc Transplant Am Soc Transpl Surg. 2022;22(4):1169–81.
van Rijn R, Schurink IJ, de Vries Y, van den Berg AP, Cortes Cerisuelo M, Darwish Murad S, et al. Hypothermic machine perfusion in liver transplantation — a randomized trial. N Engl J Med. 2021;384(15):1391–401.
Aqel B, Nguyen M, Reddy K, Moss A, Hewitt W, Jadlowiec C, et al. Normothermic mechanical perfusion (NMP) significantly reduces the risk of ischemic cholangiopathy in recipients of donation after cardiac death (DCD) liver transplants [Internet]. San Diego: American Transplant Congress; 2023. p. 2023. https://doi.org/10.1016/j.ajt.2023.05.013.
Jayasekera CR, Barnhill M, Chascsa DM, Aqel B, Carey EJ, Vargas HE. Fenofibrate improves outcomes in ischemic cholangiopathy after liver transplantation. Gastroenterology. 2023;164(7):1321-1323.e2.
Bhogal HK, Sanyal AJ. The molecular pathogenesis of cholestasis in sepsis. Front Biosci Elite Ed. 2013;1(5):87–96.
Leonhardt S, Veltzke-Schlieker W, Adler A, Schott E, Eurich D, Faber W, et al. Secondary sclerosing cholangitis in critically Ill patients: clinical presentation, cholangiographic features, natural history, and outcome: a series of 16 cases. Medicine (Baltimore). 2015;94(49): e2188.
Horvatits T, Drolz A, Trauner M, Fuhrmann V. Liver injury and failure in critical illness. Hepatol Baltim Md. 2019;70(6):2204–15.
Martins P, Verdelho MM. Secondary sclerosing cholangitis in critically Ill patients: an underdiagnosed entity. GE Port J Gastroenterol. 2020;27(2):103–14.
Meier-Hellmann A, Reinhart K, Bredle DL, Specht M, Spies CD, Hannemann L. Epinephrine impairs splanchnic perfusion in septic shock. Crit Care Med. 1997;25(3):399–404.
Lin T, Qu K, Xu X, Tian M, Gao J, Zhang C, et al. Sclerosing cholangitis in critically ill patients: an important and easily ignored problem based on a German experience. Front Med. 2014;8(1):118–26.
Fujita Y. Effects of PEEP on splanchnic hemodynamics and blood volume. Acta Anaesthesiol Scand. 1993;37(4):427–31.
Kirchner GI, Scherer MN, Obed A, Ruemmele P, Wiest R, Froh M, et al. Outcome of patients with ischemic-like cholangiopathy with secondary sclerosing cholangitis after liver transplantation. Scand J Gastroenterol. 2011;46(4):471–8.
Kirchner GI, Rümmele P. Update on sclerosing cholangitis in critically Ill patients. Viszeralmedizin. 2015;31(3):178–84.
Kulaksiz H, Heuberger D, Engler S, Stiehl A. Poor outcome in progressive sclerosing cholangitis after septic shock. Endoscopy. 2008;40(3):214–8.
Kirchner GI, Hartl J, Schnitzbauer A, Scherer MN, Farkas S, Baier L, et al. Ischemic-like cholangiopathy with secondary sclerosing cholangitis: a good indication for liver transplantation?: 1929. Transplantation. 2012;94(10S):419.
Shih AR, Hatipoglu D, Wilechansky R, Goiffon R, Deshpande V, Misdraji J, et al. Persistent cholestatic injury and secondary sclerosing cholangitis in COVID-19 patients. Arch Pathol Lab Med. 2022;146(10):1184–93.
Lagana SM, Kudose S, Iuga AC, Lee MJ, Fazlollahi L, Remotti HE, et al. Hepatic pathology in patients dying of COVID-19: a series of 40 cases including clinical, histologic, and virologic data. Mod Pathol. 2020;33(11):2147–55.
Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet Lond Engl. 2020;395(10223):507–13.
Faruqui S, Okoli FC, Olsen SK, Feldman DM, Kalia HS, Park JS, et al. Cholangiopathy after severe COVID-19: clinical features and prognostic implications. Am J Gastroenterol. 2021;116(7):1414–25 These authors have delineated the clinical features and prognostic implications of COVID-19 cholangiopathy.
Roth NC, Kim A, Vitkovski T, Xia J, Ramirez G, Bernstein D, et al. Post-COVID-19 cholangiopathy: a novel entity. Am J Gastroenterol. 2021;116(5):1077–82.
Chai X, Hu L, Zhang Y, Han W, Lu Z, Ke A, et al. Specific ACE2 expression in cholangiocytes may cause liver damage after 2019-nCoV infection [Internet]. bioRxiv; 2020 [cited 2023 Sep 10]. p. 2020.02.03.931766. Available from:https://www.biorxiv.org/content/10.1101/2020.02.03.931766v1
Nardo AD, Schneeweiss-Gleixner M, Bakail M, Dixon ED, Lax SF, Trauner M. Pathophysiological mechanisms of liver injury in COVID-19. Liver Int. 2021;41(1):20–32.
Wang Y, Liu S, Liu H, Li W, Lin F, Jiang L, et al. SARS-CoV-2 infection of the liver directly contributes to hepatic impairment in patients with COVID-19. J Hepatol. 2020;73(4):807–16.
Sonzogni A, Previtali G, Seghezzi M, Grazia Alessio M, Gianatti A, Licini L, et al. Liver histopathology in severe COVID 19 respiratory failure is suggestive of vascular alterations. Liver Int Off J Int Assoc Study Liver. 2020;40(9):2110–6.
Hartl L, Haslinger K, Angerer M, Semmler G, Schneeweiss-Gleixner M, Jachs M, et al. Progressive cholestasis and associated sclerosing cholangitis are frequent complications of COVID-19 in patients with chronic liver disease. Hepatol Baltim Md. 2022. https://doi.org/10.1002/hep.32582.
Bütikofer S, Lenggenhager D, Wendel Garcia PD, Maggio EM, Haberecker M, Reiner CS, et al. Secondary sclerosing cholangitis as cause of persistent jaundice in patients with severe COVID-19. Liver Int. 2021;41(10):2404–17.
Keta-Cov research group. Electronic address: Vincent.mallet@aphp.fr, Keta-Cov research group. Intravenous ketamine and progressive cholangiopathy in COVID-19 patients. J Hepatol. 2021;74(5):1243–4.
Meersseman P, Blondeel J, De Vlieger G, van der Merwe S, Monbaliu D. Secondary sclerosing cholangitis: an emerging complication in critically ill COVID-19 patients. Intensive Care Med. 2021;47(9):1037–40.
Onuiri J, Fiel MI. COVID Cholangiopathy can occur despite mild COVID Am J Clin Pathol. 2022;158(Supplement_1):S128-9.
Author information
Authors and Affiliations
Contributions
MB and CJ both wrote and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Conflict of Interest
Michele Barnhill, MD, and Channa Jayasekera, MD, MSc, have no conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Barnhill, M.S., Jayasekera, C. Secondary Sclerosing Cholangiopathies. Curr Hepatology Rep 23, 145–152 (2024). https://doi.org/10.1007/s11901-024-00646-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11901-024-00646-7