
Abstract
Purpose of Review
Nonalcoholic fatty liver disease (NAFLD) and its more aggressive form nonalcoholic steatohepatitis (NASH) are a leading cause of chronic liver disease worldwide. Thus far, there are no FDA-approved therapeutic options for NASH. This review discusses relevant and recent findings in the development of pharmacotherapy that targets the metabolic processes implicated in NASH.
Recent Findings
Several key drugs have been identified across various drug classes. Among inhibitors of de novo lipogenesis, the SCD-1 inhibitor aramchol and the ACC inhibitor firsocostat are the most advanced. Within nuclear hormone receptor agonists, PPARα/δ agonist elafibranor and PPARα/γ agonist saroglitazar show promise with respect to improvement in NASH histology and hepatic steatosis. Additionally, THR-β agonist resmetirom showed significant reduction in hepatic steatosis and NASH resolution. Larger studies with longer treatment duration are needed to establish safety and efficacy of these metabolic drugs.
Summary
Significant progress has been made over the past decade in testing drugs that modulate the metabolic targets responsible for NASH progression.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Nonalcoholic fatty liver disease (NAFLD) has become one of the most common causes for chronic liver disease worldwide. Likely related to the rise in obesity and metabolic syndrome, NAFLD is now the second most common cause of liver transplant and hepatocellular carcinoma in the United States [1, 2]. In a large meta-analysis by Younossi et al., the global prevalence of NAFLD was estimated to be approximately 25%—the vast majority of whom have metabolic syndrome. In the United States, NAFLD is seen in up to 75 million people [3].
NAFLD is a spectrum that ranges from (1) fatty liver disease, characterized by progressive fatty infiltration of the liver parenchyma; (2) nonalcoholic steatohepatitis (NASH), characterized by the development of inflammation and hepatocyte injury in the form of ballooning; (3) progressive liver fibrosis; and (4) eventual cirrhosis with associated complications [4]. The histologic severity of NAFLD is determined by the NAFLD activity score (NAS) which is a semiquantitative grading system for the main histologic features of steatosis, lobular inflammation, and ballooning [5].
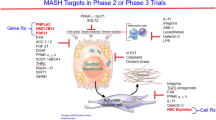
Currently, there are no pharmacotherapeutic options approved by the Food and Drug Administration (FDA) for the treatment of the aggressive form of NASH [6]. Our increased grasp of mechanisms that fuel NASH and fibrosis progression has led to the development of a rich pipeline of medications that can be divided into four major categories: (a) metabolic targets that improve insulin sensitivity, inhibit different enzymes involved in de novo lipogenesis, or improve mitochondrial utilization of fatty acids; (b) targets of cellular injury that inhibit the recruitment of inflammatory cells or inhibit hepatocyte apoptosis; (c) liver-gut axis targets that modulate bile acid enterohepatic circulation and signaling, such as farnesoid X receptor agonists; and (d) anti-fibrotic targets that directly target hepatic stellate cells to decrease collagen deposition in the liver. This review aims to discuss the key metabolic targets in NASH and highlight the future of pharmacotherapy for this pervasive disease.
Pathophysiology of NAFLD
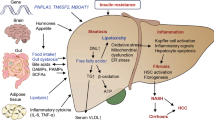
NAFLD pathophysiology is explained by multiple parallel hits that lead to lipid loading into the hepatocytes followed by the formation of reactive oxygen species and activation of inflammatory cascades leading to fibrinogenesis [7].
Estimated to affect 70–80% of patients with diabetes and obesity, NAFLD is strongly associated with insulin resistance [8]. Obesity and the accumulation of central adiposity lead to the dysregulation of the several inflammatory adipokines such as tumor necrosis factor (TNFα) and interleukins (IL-1 and IL-6) [7]. Additionally, adiponectin is a key cytokine that is reduced in obesity. Traditionally responsible for improving insulin sensitivity, a deficiency of adiponectin leads to increased insulin resistance and metabolic disease. Conversely, leptin, a hormone which downregulates appetite and upregulates energy expenditure, is increased in obese patients due to the presence of leptin resistance. Leptin has been found to increase the expression and circulation of pro-inflammatory cytokines and contribute to hepatic fibrinogenesis by directly acting on hepatic stellate cells [9].
Triglyceride accumulation in the cytoplasm of hepatocytes is the key mechanism in the development of hepatic steatosis. This excess of fat is made possible by an imbalance between the hepatocytes’ increased uptake of free fatty acids and their inability to expel fat (via fatty acid oxidation and very low-density lipoprotein (VLDL) secretion) [10]. Circulating free fatty acids are released from adipocytes via lipolysis and are taken at higher rates by hepatocytes in the setting of insulin resistance. Concomitantly, hepatocytes in response to increased levels of circulating insulin will increase de novo lipogenesis. These two mechanisms lead to a profound increase in intracellular triglycerides [11].
This lipotoxicity creates a pro-inflammatory state in the liver, secondary to oxidative stress from reactive oxygen species, mitochondrial dysfunction, and endoplasmic reticulum dysfunction [7]. In response to these free radicals, the smooth endoplasmic reticulum is stressed in an effort to prevent apoptosis and preserve cellular function. This derangement in cellular homeostasis leads to mitochondrial instability and ultimately hepatocyte apoptosis, which can activate the hepatic stellate cells to transform into fibrogenic myofibroblasts [12].
In summary, the complex pathophysiology of NAFLD reveals that hepatocyte lipid loading (due to increased availability of free fatty acids from lipolysis and de novo lipogenesis in a state of systemic insulin resistance) is an early step in disease development with downstream effects that lead to lipotoxicity and disease progression. Therefore, metabolic drugs (Table 1) to reverse this process early on represent an attractive therapeutic option for NASH at the metabolic root.
Metabolic Drugs that Inhibits De Novo Lipogenesis
Acetyl Coenzyme A Carboxylase (ACC) Inhibitors
ACC is a key regulatory enzyme in the de novo lipogenesis pathway (Fig. 1). Through its ACC1 isoform, this enzyme catalyzes the conversion of acetyl-CoA with carbonate to form malonyl-CoA and in the triglyceride synthesis pathway. ACC2 is a second isoenzyme that inhibits the transfer of fatty acids into the mitochondria for β-oxidation [4]. While ACC2 resides primarily in cardiac and skeletal muscles, ACC1 is the primary isoenzyme responsible for hepatic metabolism. Given it dual actions on fatty acid metabolism, the inhibition of ACC has become a leading therapeutic target for NASH [13].

The hepatocyte is the setting of de novo lipogenesis—a biochemical pathway in which glucose enters the cells and is converted to triglycerides. This process is the key in the development of hepatic steatosis and several drugs target enzymes along this pathway
Soraphen A was one of the first discovered ACC inhibitors—studied mostly in murine models. Inhibiting both fatty acid oxidation and de novo lipogenesis, soraphen A showed promise in vitro but was of limited effect in vivo [14]. Studied in vivo rodent models, the nonselective ACC inhibitor CP-640186 reduced hepatic triglyceride content and promoted fatty acid oxidation in liver and skeletal muscles [15].
Among the first studies conducted in humans, Griffith et al. showed significant reduction in de novo lipogenesis and whole body fatty acid oxidation with a nonselective ACC inhibitor Compound 9 (PF-05175157) [16]. In a more recent study, Kim et al. showed reduction in hepatic triglycerides and associated hepatic steatosis in humans with administration of a liver-specific ACC inhibitor MK-4074. However, in the rodent model, there was a twofold increase in serum triglyceride levels [17]. In a similar study in rodents, Goedeke et al. showed a 20% decrease in hepatic de novo lipogenesis and insulin resistance, though an increase in serum triglycerides by 30–130% [18]. While demonstrating promise in alteration of hepatic metabolism, these studies shed light on a potentially impactful consequence of ACC inhibitors—reduction of hepatic lipogenesis and lipolysis may lead to worsening serum triglyceridemia.
Further clinical trials in humans have shown similar results among ACC inhibitors. In a study of 96 healthy subjects, Bergman et al. set out to characterize the pharmacokinetics, pharmacodynamics, safety, and tolerability of a liver-targeted ACC inhibitor PF-05221304. The drug was associated with 70% reduction in de novo hepatic lipogenesis but achieved up to 98% reduction in some patients. Notably, the authors found a significant dose-dependent increase in serum triglycerides and decrease in platelets in patients who achieved > 90% reduction in lipogenesis. While this study showed the potential efficacy in reduction of hepatic steatosis, the authors’ inclusion of only healthy subjects leaves ambiguity with respect to NAFLD patients [19].
The most advanced ACC inhibitor in terms of drug development is firsocostat (GS-0976). In an open-label proof-of-concept trial of 10 patients, Lawitz et al. aimed to evaluate the response of NAFLD patients to firsocostat following 12 weeks of therapy. The researchers saw a significant reduction in de novo lipogenesis, hepatic steatosis (as measured by magnetic resonance proton density fat fraction (PDFF)), and liver stiffness [20] . In a larger phase II trial of 126 patients with NASH or fibrosis, Loomba et al. demonstrated up to 30% reduction in PDFF in nearly half of all patients. However, as with preceding studies, the researchers saw dose-independent hypertriglyceridemia in up to 18% of patients [21]. Though the cardiovascular consequences have not yet been studied, the results of these studies raise important concerns about the long-term safety of this class of medications [22].
Fatty Acid Synthase (FAS) Inhibitors
Fatty acid synthase (FAS) is the second key step in the de novo lipogenesis pathway in the liver—catalyzing the conversion of malonyl-CoA and acetyl-CoA to form the fatty acid palmitate [23]. Present in high concentrations in cancer cells, FAS inhibitors have been a target for oncologic therapy for several years [24]. More recently, FAS inhibition to treat NASH has gained attention.
In a recent phase I proof-of-concept clinical trial of FAS inhibitor TVB-2640, Syed-Abdul et al. showed a significant, dose-dependent reduction in de novo lipogenesis in a cohort of 12 obese males with risk factors for NAFLD. In addition, the authors demonstrated a reduction in serum alanine aminotransferase (ALT) levels. Notably, the authors found no significant aberration in serum triglyceride levels, and the drug had a reasonable safety profile. Due to the study’s small sample size, short duration (10 days), and inclusion of only men, further investigations are required to establish safety and efficacy [25]. While more clinical trials to study FAS inhibitors are needed, this class of medications is relevant in the future of drug development [26].
Stearoyl-Coenzyme A Desaturase-1 (SCD-1) Inhibitors
SCD-1 is an enzyme in the de novo lipogenesis pathway that aids in the formation of monounsaturated fatty acids. Induced by insulin and suppressed by leptin, SCD-1 has become an attractive target for NASH medications given its importance in the formation of triglycerides [27]. In a proof-of-concept, randomized, controlled trial in 60 patients with biopsy-proven NAFLD, Safadi et al. aimed to measure the effects of SCD-1 inhibitor aramchol 100 mg and 300 mg versus placebo. The authors found a dose-dependent reduction in liver fat as measured by MRI-PDFF. However, this study was limited by its smaller sample size and shorter treatment duration (3 months) [28].
In the ARREST trial (NCT02279524), in a phase IIb randomized, controlled study, the authors stratified 247 patients with biopsy-proven NASH into three groups: placebo, aramchol 400 mg, and 600 mg. The researchers found a dose-dependent reduction in absolute liver fat as measured by magnetic resonance spectroscopy after 1 year of treatment. Furthermore, aramchol was associated with decreased liver enzymes (AST, ALT) and improvement in hemoglobin A1c (HbA1c) with a trend toward improvement in NASH resolution [29]. Currently, a phase 3/4 study ARMOR (NCT04104321) is recruiting 2000 patients to receive 600-mg aramchol vs placebo. The researchers plan to assess the resolution of NASH without worsening of fibrosis as a primary endpoint [26, 30].
Monoacylglycerol Acyltransferase (MGAT) Inhibitors
MGAT is a family of three isoenzymes that are responsible for the conversion of monoacylglycerol to diacylglycerol. The MGAT enzymes vary primarily based on location. While all forms of MGAT can be found in the small intestine, only MGAT2 and MGAT3 have been found in the hepatocyte [31].
While several MGAT2 inhibitors have been synthesized, very few have advanced to testing in animal models. Despite the development of several potential molecular agents as reviewed in detail by Devasthale et al., no MGAT1 inhibitor has entered phase I clinical trials [32]. In a murine model, a novel MGAT inhibitor JTP-103237 demonstrated efficacy in reducing triglyceride synthesis, de novo lipogenesis, plasma glucose concentration, and total cholesterol [33]. Thus, MGAT inhibitors offer another potential drug target in the treatment of NASH.
Diacylglycerol Acyltransferase (DGAT) Inhibitors
DGAT is a crucial catalyst in the final step of triglyceride synthesis. Responsible for converting esterified diacylglycerol to triacyclglycerol, two DGAT enzymes are responsible for the formation of triglycerides. While DGAT1 primarily regulates fat absorption in the small bowel, DGAT2 exists primarily in the liver and adipose tissue. However, these are not mutually exclusive as DGAT1 also plays a key role in the generation of triglycerides within the hepatocyte [34]. Gluchowski et al. conducted a recently published study in mice which aimed to characterize which DGAT enzyme is more significant to the development of hepatic steatosis. The authors found a significant reduction in de novo lipogenesis and hepatic steatosis in DGAT2-defecient mice without any derangements in glucose or insulin metabolism [35].
Given the novelty of this class of medication, limited data are available in human subjects. In a murine model, Amin et al. demonstrated a reasonable safety profile of PF-06427878—a liver-specific DGAT2 inhibitor. The authors showed significant reductions in de novo lipogenesis as well as reductions in hepatic and serum triglycerides. Furthermore, the authors demonstrated improvement in NAFLD activity score with key reductions in inflammation and ballooning—which are characteristic of NASH. Currently, PF-06427878 is in two phase I clinical trials [NCT02855177 and NCT02391623] but the results have not been published [36] .
These studies contribute to a significant opportunity to drug development targeted at the DGAT enzymes. A recently published study by Hong et al. synthesized and validated a novel thienopyrimidine derivative DGAT1 inhibitor with improved pharmacokinetics in rodent and canine models [37]. Two recent studies by Okour et al. were recently published testing the safety and pharmacologic profiles of a novel DGAT1 inhibitor GSK3008356 which returned with promising results [38, 39]. Furthermore, several DGAT2 inhibitors are undergoing clinical trials, including IONIS-DGAT2Rx (NCT03334214), SNP-610 (NCT03468556), SNP-612 (NCT03868566), and PF-06865571(NCT04091061) [26].
Metabolic Drugs that Target Nuclear Receptors
Peroxisome Proliferator-Activated Receptor (PPAR) Agonists
The PPAR family includes three isoenzymes: PPARα, PPARβ/δ, and PPARγ. PPARα is a nuclear receptor that is key to glucose and lipid metabolism but is also involved in key inflammatory pathways in the liver. Activated during catabolic states, PPARα is responsible for decreasing low-density lipoprotein (LDL), upregulating high-density lipoprotein (HDL), and decreasing serum triglycerides by downregulation of lipoprotein lipase—the key regulatory enzyme in the hydrolysis of triglycerides [40]. Uniquely, PPARγ is involved in insulin sensitization, and its activation has already been capitalized on by medications intended to treat type 2 diabetes. Additionally, PPARγ has a key role as an anti-inflammatory mediator due to its indirect effects on nuclear factor-κB [41]. PPARβ/δ is somewhat less studied and more controversial mechanistically than either PPARα and PPARγ. Though there has been some conflicting evidence, PPARβ/δ most likely works in the fasting state to suppress lipogenesis and in Kupffer cells in the liver to reduce key inflammatory cytokines [42]. Though seemingly distinct, PPAR receptors show significant overlap in their functions. As the PPAR family controls key regulatory mechanisms toward reduction of hepatic steatosis and inflammation, PPAR agonists have been identified as a relevant drug target in the treatment of NAFLD.
In a study of obese, insulin-resistant mice, Larter et al. identified that activation of PPARα with WY-14643 could reverse insulin resistance, improve hyperglycemia, and reduce hepatic steatosis and ballooning, but not inflammation [43]. Less potent than the agonist WY-14643, fibrates have shown efficacy as PPARα agonists in murine and rodent models as well. In several studies, mice and rats with diet induced NAFLD showed improvement in hepatic steatosis, inflammation, and macrophage accumulation [44, 45]. Given their efficacy in animal models, several studies were conducted to evaluate the efficacy of fibrates in humans.
In the first study of its kind, Laurin et al. aimed to evaluate the efficacy of clofibrate in the treatment of NASH. The authors administered daily clofibrate to 16 patients with biopsy-proven NASH for 1 year. The results showed no significant reduction in liver enzymes or hepatic steatosis [46]. Conversely, in a study of 16 patients treated with 48 weeks of fenofibrate, the authors reported that fenofibrate administration was associated with a reduction in aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels, prevalence of metabolic syndrome, and hepatocellular ballooning but not steatosis or inflammation [47]. Given the limited efficacy of fibrates on hepatic histology and weak modification of PPARα by fibrates, selective PPARα modulators (SPPARMα) such as pemafibrate have demonstrated an association with improvement in liver metabolism in animal models [48, 49]. Thus far, the data surrounding exclusive PPARα agonists have not been extremely promising; however, there may still be a role for activation of the PPARα receptors in combination with other members of the PPAR family.
Isolated agonism of the PPARβ/δ receptor has produced controversial results. In mice with diet-induced NAFLD, Lee et al. demonstrated improvement in hepatic steatosis and reduction in pro-inflammatory cytokines such as caspase-1 and interleukin-1β with treatment with PPARβ/δ agonist GW501516 [50]. However, another study in mice showed increased profibrotic and pro-inflammatory cytokine production in response to PPARβ/δ agonist [51]. Another PPAR β/δ agonist GW0742 demonstrated promise in reduction of serum ALT levels, in addition to suppression of inflammatory cytokines including tumor necrosis factor-α (TNFα)—despite an upregulation of hepatic stellate cell proliferation [52] . However, animal trials on both GW501516 and GW0742 were discontinued after the discovery that these agents were potentially inducing solid organ malignancies [53]. Seladelpar, a selective PPARβ/δ agonist, was tested in NASH patients (NCT03551522). In November 2019, the sponsor terminated the phase II study in NASH based on initial histological findings that showed the development of interface hepatitis in some patients [54].
Thiazolidinediones (TZDs) are the only class of medications acting on the PPAR family that are currently approved by the Food and Drug Administration (FDA), though only for the treatment of type 2 diabetes. TZDs are PPARγ agonists improve insulin resistance by directly acting on adipocytes to modulate adipokines, decrease fatty acid oxidation, and decrease circulating free fatty acids [55]. Given its FDA approval and overall safety and tolerability, several human studies have been conducted on TZDs and NASH—some of which will be discussed below.
The PIVENS trial was a randomized, placebo-controlled trial that compared vitamin E with pioglitazone in the treatment of NASH. Conducted in patients without diabetes, this study demonstrated histologic improvement in steatosis and inflammation without improvement in fibrosis. Though the results trended toward significance, the authors were not able to meet their primary outcome of NASH improvement after 96 weeks of treatment with pioglitazone [56]. More recently, Bril et al. conducted a study meant to compare efficacy of pioglitazone in diabetics with prediabetics. The authors found that a roughly half of all patients were able to meet the primary endpoint of improvement in NAFLD activity score; however, there was more significant association between NASH resolution, reduction in liver fibrosis, and insulin sensitivity in diabetics compared with prediabetics [57]. In another study of 90 patients, Yaghoubi et al. found an improvement in AST, ALT, and blood pressure after treatment with pioglitazone. However, this study is limited in that the patients did not have biopsy-proven disease, nor did the authors assess a histologic primary or secondary endpoint [58]. In a recent randomized, placebo-controlled trial of 101 patients with diabetes or prediabetes and biopsy-proven NASH, 58% of patients met the primary endpoint of improvement in NAFLD activity score, while 51% of patients actually achieved resolution of NASH. Notably, these patients were treated with pioglitazone for 18 months and saw a mean weight gain of 2.5 kg—the most commonly reported deleterious class effect among TZDs [59]. In a recent meta-analysis that reviewed 8 studies on TZDs, administration of TZD was associated with improvement in fibrosis score and resolution of NASH in both diabetics and nondiabetics [60]. These studies suggest that pioglitazone may have a role in the treatment of NASH, independent of a patient’s glycemic status.
Though individual PPAR agonists have been controversial historically, a new class of medications have taken the forefront. Acting on multiple different substrates, a combination of PPAR agonists is showing more promise than their predecessors.
The PPARα/δ dual agonist, elafibranor (GFT505), demonstrated efficacy in the modulation of key metabolic markers, including reducing LDL, increasing HDL, reducing triglycerides, reducing fasting glucose, and improving insulin sensitivity [61,62,63]. In murine and rodent models, treatment with elafibranor was associated with reduction in hepatic steatosis, fat fraction, inflammation, weight, and severity of liver fibrosis [64,65,66,67,68]. In the largest human study to date, the GOLDEN-505 trial randomized 274 patients into three groups: placebo, 80 mg elafibranor, and 120 mg elafibranor. Ratziu et al. showed that patients treated with either dose of elafibranor had improvement in liver enzymes (ALT and AST), lipid parameters (cholesterol, triglycerides, LDL, and HDL), and key inflammatory markers (C-reactive protein, fibrinogen, and haptoglobin). Furthermore, the authors found that treatment with higher dose elafibranor met its protocol-defined primary outcome—resolution of NASH as defined by a score of 0 in any subgroup of the NAFLD activity score (steatosis, inflammation, or ballooning) without worsening of fibrosis. Notably, this study failed to meet its modified primary outcome as defined by complete resolution of ballooning with minimal fibrosis. The lower dose of elafibranor failed to meet either primary outcome [69]. Although the authors failed to meet the modified primary outcome, the inclusion of patients with mild disease and minimal fibrosis may have limited their results as often these patients do not need pharmacotherapy. RESOLVE-IT (NCT02704403) is a phase III, randomized control trial which aims to compare elafibranor to placebo in the resolution of biopsy proven NASH. The recently published interim results showed no significant reduction in the primary endpoint of NASH resolution [70].
The second generation PPARα/γ agonist saroglitazar has been identified as a potential drug target in the treatment of NASH. In a murine model, administration of saroglitazar was associated with significant improvement in NAFLD activity score and reduction in liver transaminases when compared with pioglitazone and fenofibrate [71]. The phase II EVIDENCES IV randomized, controlled trial evaluated the safety and efficacy of different doses of saroglitazar in 106 patients with NASH. The high dose saroglitazar of 4 mg daily significantly reduced ALT and liver fat at the end of the trial with 41% of patients achieving more than 30% relative fat fraction reduction on MRI-PDFF. There was no significant weight gain or edema with saroglitazar [72]. Several trials are ongoing with saroglitazar (NCT03639623, NCT03617263, NCT03061721, NCT03863574, NCT02265276, NCT04193982) [26]. In fact, saroglitazar was approved in India in 2013 for the treatment of diabetic dyslipidemia, and more recently, it received approval by the Drug Controller General of India (DCGI) for the treatment of NASH making it the first drug to receive regulatory approval for this indication [73].
Finally, a pan-PPAR agonist (α/δ/γ) called lanifibranor (IVA337) received fast track designation for treatment of NASH. It is being evaluated in the phase IIb NATIVE study (NCT03008070), and the results are anticipated to be released in the near future [74] .
The family of PPAR agonists shows significant promise in the treatment of NASH, especially combination receptors like elafibranor and saroglitazar. Acting on multiple substrates, this class of medications is uniquely able to modulate insulin sensitivity while improving histopathologic steatohepatitis.
Thyroid Hormone Receptor-β (THRβ) Agonists
Thyroid hormones T3 and T4 have a significant effect on the body’s metabolism. While THRα is the major isoform in cardiac and skeletal muscle, THRβ is predominantly found in the liver [75]. In the liver, THRβ is a key regulator of hepatocellular metabolism by upregulating gene transcription for crucial downstream enzymes involved in de novo lipogenesis including fatty acid synthase and acetyl-CoA carboxylase. In addition, thyroid hormones upregulate the expression of 3-hydroxy-3-methylglutaryl Coenzyme-A (HMG CoA) reductase which acts to decrease serum LDL and increase HDL [76]. Several compounds have been identified as THRβ agonists including: GC-1 (sobetirome), KB-2115 (eprotirome), MB07811/VK2809 (prodrug MB07344), and MGL-3196 (resmetirom). Early trials for sobetirome and eprotirome showed promise; however, clinical trials for both medications were halted after eprotirome was associated with significant elevations in liver function tests and cartilage damage [77]. More recently, hepatocyte-specific VK2809 and resmetirom have come to the forefront in clinical research.
By regulating key metabolic pathways in the liver, modulation of THRβ demonstrated efficacy in the reduction of hepatic steatosis and serum triglycerides in animal models [78,79,80,81]. In a phase IIA, randomized, placebo-controlled trial of 45 patients, Loomba et al. aimed to evaluate the safety, tolerability, and reduction of hepatic steatosis with VK2809 at different dose regimens. The authors found that treatment with VK2809 was associated with roughly a 10% absolute (60% relative) reduction in PDFF. The authors found no significant difference between different doses in efficacy, safety, or tolerability [82]. Given that this study did not evaluate histopathologic change, the clinical significance of sole reduction in hepatic steatosis is somewhat limited and further studies are needed to evaluate its efficacy in reduction of hepatocellular ballooning and fibrosis. Currently, a phase IIB clinical trial (NCT04173065) named VOYAGE is recruiting and aims to characterize safety and efficacy of VK2809 over a 52-week time period [26].
Perhaps the most promising among this class of medication is MGL-3196 (resmetirom). In a recently published phase II clinical trial, resmetirom demonstrated efficacy in reduction of hepatic steatosis. In a study of 125 patients with biopsy-proven NASH, the authors found a dose-dependent association between administration of resmetirom and an absolute reduction in median PDFF of 7.0% (33% relative reduction) at 12 weeks and 8% (37% relative reduction) at 36 weeks. Patients taking higher dose resmetirom showed absolute and relative reductions in PDFF of 10% and 50%, respectively. The authors also found dose-dependent reductions in LDL, triglycerides, ALT, AST, and key inflammatory biomarkers [83]. The phase III MAESTRO-NASH trial (NCT03900429) plans to enroll 2000 participants with non-cirrhotic NASH and stage 2 or 3 fibrosis to evaluate the effect of resmetirom at 80 mg or 100 mg compared with placebo on achieving NASH resolution on liver histology obtained at 52 weeks [26].
Drugs that Target Metabolic Hormones
Fibroblast Growth Factor (FGF) 21 Analogs
FGF21 is a metabolic hormone produced in the liver and upregulated in times of stress. Elevated FGF21 levels have been identified in patients with diabetes mellitus and NASH indicating a state of FGF21 resistance in these conditions. In addition, increased levels of FGF21 are associated with improved insulin sensitivity, lowering serum triglycerides, and mitigating hepatic lipotoxicity [84]. Several studies have drawn significant association between FGF21 and NAFLD, which are reviewed extensively by Tucker et al. [85]. Most notably, FGF21 is an independent predictor of hepatic steatosis, though studies have been unsuccessful in using FGF21 as a prognosticator for NASH [86, 87]. These studies have illuminated the path for drug identification targeting the FGF21.
In animal models, FGF21 analogs have demonstrated efficacy in improving hepatic steatosis, fibrosis, and inflammation [88,89,90]. In a study of 120 obese patients with presumed NAFLD, the authors aimed to characterize the effects of the pegylated FGF21 analog pegbelfermin (BMS-986036) on insulin sensitivity and surrogate markers of liver injury. The authors found that administration of pegbelfermin was associated with improvement in metabolic parameters (HDL, triglycerides) and markers of liver fibrosis (adiponectin and type III collagen propeptide (PRO-C3)) [91]. In a study of 75 patients with biopsy-proven NASH, Sanyal et al. found that treatment with pegbelfermin for 16 weeks was associated with a 6.8% reduction in hepatic steatosis as measured by PDFF. In addition, treatment with pegbelfermin was associated with reduction in ALT and AST and similar improvements in metabolic and fibrosis markers (viz., adiponectin). Consistent with previous studies, pegbelfermin was generally well tolerated but was associated with gastrointestinal side effects [92]. Small sample size and short study duration limit these studies’ generalizability to a larger population of NASH patients; further study is needed.
In the phase IIA BALANCED study on FGF21 analog AKR-001, the researchers compared placebo with three dose groups: 28 mg, 50 mg, and 70 mg. The investigators found that all three dosages met the primary endpoint of absolute reduction in liver fat as measured by MRI-PDFF at week 12. Additionally, the higher dosage arms achieved a relative reduction in liver fat of > 70% [93].
Anti-Diabetic Agents
Glucagon-Like Peptide 1 (GLP1) Agonists
Glucagon-like peptide-1 (GLP-1) is an intestinal incretin hormone that is produced by intestinal enteroendocrine L-cells that acts to stimulate insulin secretion and decrease glucose levels. In patients with type 2 diabetes, agents such as liraglutide and semaglutide have been utilized due to their beneficial role in treating insulin-resistant metabolic dysfunction and their favorable side effects of weight loss and lower risk of hypoglycemia. The mechanism by which GLP-1 acts on the liver to improve steatosis, inflammation, and injury remains unclear; however several studies have shown promising results. In mice models, both liraglutide and semaglutide have been shown to reduce cardiovascular risk in type 2 diabetics and affect atherosclerosis through an anti-inflammatory mechanism [94].
The liraglutide efficiency and action in NASH (LEAN) trial evaluated the use of liraglutide (1.8 mg daily) in participants who were overweight with clinical evidence of NASH for a total of 48 weeks. When compared with placebo, participants treated with liraglutide showed resolution of NASH and improvement in ballooning [95]. Semaglutide, which requires only weekly dosing, is now currently being studied in an interventional clinical trial evaluating three dose levels (0.1 mg, 0.2 mg and 0.4 mg) in subjects with NASH. The primary outcomes include NASH resolution without worsening of fibrosis (NCT02970942) [26].
Sodium Glucose Cotransporter 2 (SGLT2) Inhibitors
Sodium glucose cotransporter 2 (SGLT2) inhibitors target the proximal tubule of the kidney which is responsible for approximately 90% of reabsorption of glucose. These agents are preferred due to their lower risk of inducing hypoglycemia and are shown to have a role in reducing risk factors for cardiovascular disease, improving glycemic control through decreasing insulin resistance, reduction in inflammation along with aiding in weight loss, thus making them an attractive option for potential NASH pharmacotherapy [96].
In a proof-of-concept study in 10 patients with diabetes and biopsy-proven NASH, administration of canagliflozin was associated with reduction in liver markers (ALT, AST) and improvement in metabolic parameters (HbA1c, weight) [97]. In the E-LIFT trial, 50 patients with diabetes and NAFLD (MRI-PDFF >6%) were randomized to receive empagliflozin for 20 weeks. The treatment group showed a statistically significant reduction in liver fat [98]. In a study of 9 patients with diabetes and biopsy-proven NASH, Lai et al. sought to characterize the histopathologic effects of empagliflozin on NASH. In this pilot study, treatment was associated with improvements in metabolic profile (blood pressure, body mass index (BMI), total cholesterol) and histology (steatosis, ballooning and fibrosis) [99]. In a non-randomized study of 16 patients with diabetes and biopsy-proven NASH, treatment with dapagliflozin was associated with improvement in metabolic parameters (BMI, waist circumference) and liver markers (ALT, AST, ferritin) [100]. Notably, this study did not assess liver histology or measure liver fat. These studies add to a growing body of evidence that supports treatment of diabetes, insulin resistance, and metabolic syndrome which are essential in the treatment NASH.
Conclusion
Several drugs that target the metabolic pathways that lead to hepatocellular lipotoxicity have been developed and are now in clinical trials. Targeting these upstream pathways that lead to disease development and progression is an important step in mitigating the downstream effects of liver inflammation, cellular injury, and eventually fibrosis and its consequences.
Over the next few years, more than one metabolic drug will likely become approved for the treatment of NASH, which raises the question of how clinicians will choose an initial agent. Adverse effects and the presence of comorbidities will play a key role in the decision-making process. For example, clinicians may be weary using firsocostat in patients with hypertriglyceridemia and extensive coronary disease, although fenofibrates or DGAT inhibitors may mitigate these effects. In diabetic patients with NASH, GLP-1 agonists and SGLT-2 inhibitors may be preferred due to their unique cardioprotection. Whether these metabolic drugs are used as mono- or combination-therapy will also depend on several factors including efficacy, severity of the baseline disease, and establishing synergy with other classes of medications such as anti-fibrotic agents. Undoubtedly, there is a bright future in the treatment of NASH with medications that attack the disease at the metabolic root.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Noureddin M, Vipani A, Bresee C, Todo T, Kim IK, Alkhouri N, et al. NASH leading cause of liver transplant in women: Updated analysis of indications for liver transplant and ethnic and gender variances. Am J Gastroenterol. 2018;113:1649–59. https://doi.org/10.1038/s41395-018-0088-6.
Fazel Y, Koenig AB, Sayiner M, Goodman ZD, Younossi ZM. Epidemiology and natural history of non-alcoholic fatty liver disease. Metabolism. 2016;65:1017–25. https://doi.org/10.1016/j.metabol.2016.01.012.
Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64:73–84. https://doi.org/10.1002/hep.28431.
Esler WP, Bence KK. Metabolic targets in nonalcoholic fatty liver disease. Cell Mol Gastroenterol Hepatol. 2019;8:247–67.
Brunt EM, Kleiner DE, Wilson LA, Belt P, Neuschwander-Tetri BA. NASH Clinical Research Network (CRN). Nonalcoholic fatty liver disease (NAFLD) activity score and the histopathologic diagnosis in NAFLD: Distinct clinicopathologic meanings. Hepatology. 2011;53:810–20. https://doi.org/10.1002/hep.24127.
Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, et al. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the american association for the study of liver diseases. Hepatology. 2018;67:328–57. https://doi.org/10.1002/hep.29367.
Buzzetti E, Pinzani M, Tsochatzis EA. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism. 2016;65:1038–48. https://doi.org/10.1016/j.metabol.2015.12.012.
Kitade H, Chen G, Ni Y, Ota T. Nonalcoholic fatty liver disease and insulin resistance: new insights and potential new treatments. Nutrients. 2017;9. https://doi.org/10.3390/nu9040387.
Jung UJ, Choi MS. Obesity and its metabolic complications: The role of adipokines and the relationship between obesity, inflammation, insulin resistance, dyslipidemia and nonalcoholic fatty liver disease. Int J Mol Sci. 2014;15:6184–223. https://doi.org/10.3390/ijms15046184.
Manne V, Handa P, Kowdley KV. Pathophysiology of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Clin Liver Dis. 2018;22:23–37.
Alves-Bezerra M, Cohen DE. Triglyceride metabolism in the liver. Compr Physiol. 2017;8:1–8. https://doi.org/10.1002/cphy.c170012.
Caligiuri A, Gentilini A, Marra F. Molecular pathogenesis of NASH. Int J Mol Sci. 2016;17. https://doi.org/10.3390/ijms17091575.
Kreuz S, Schoelch C, Thomas L, Rist W, Rippmann JF, Neubauer H. Acetyl-CoA carboxylases 1 and 2 show distinct expression patterns in rats and humans and alterations in obesity and diabetes. Diabetes Metab Res Rev. 2009;25:577–86. https://doi.org/10.1002/dmrr.997.
Chen L, Duan Y, Wei H, Ning H, Bi C, Zhao Y, et al. Acetyl-CoA carboxylase (ACC) as a therapeutic target for metabolic syndrome and recent developments in ACC1/2 inhibitors. Expert Opin Investig Drugs. 2019;28:917–30. https://doi.org/10.1080/13543784.2019.1657825.
Harwood HJ, Petras SF, Shelly LD, Zaccaro LM, Perry DA, Makowski MR, et al. Isozyme-nonselective N-substituted bipiperidylcarboxamide acetyl-CoA carboxylase inhibitors reduce tissue malonyl-CoA concentrations, inhibit fatty acid synthesis, and increase fatty acid oxidation in cultured cells and in experimental animals. J Biol Chem. 2003;278:37099–111. https://doi.org/10.1074/jbc.M304481200.
Griffith DA, Kung DW, Esler WP, Amor PA, Bagley SW, Beysen C, et al. Decreasing the rate of metabolic ketone reduction in the discovery of a clinical acetyl-CoA carboxylase inhibitor for the treatment of diabetes. J Med Chem. 2014;57:10512–26. https://doi.org/10.1021/jm5016022.
Kim CW, Addy C, Kusunoki J, Anderson NN, Deja S, Fu X, et al. Acetyl CoA carboxylase inhibition reduces hepatic steatosis but elevates plasma triglycerides in mice and humans: A bedside to bench investigation. Cell Metab. 2017;26:394–406.e6.
Goedeke L, Bates J, Vatner DF, Perry RJ, Wang T, Ramirez R, et al. Acetyl-CoA carboxylase inhibition reverses NAFLD and hepatic insulin resistance but promotes hypertriglyceridemia in rodents. Hepatology. 2018;68:2197–211. https://doi.org/10.1002/hep.30097.
Bergman A, Carvajal-Gonzalez S, Tarabar S, Saxena AR, Esler WP, Amin NB. Safety, tolerability, pharmacokinetics, and pharmacodynamics of a liver-targeting acetyl-CoA carboxylase inhibitor (PF-05221304): A three-part randomized phase 1 study. Clin Pharmacol Drug Dev. 2020. https://doi.org/10.1002/cpdd.782.
Lawitz EJ, Coste A, Poordad F, Alkhouri N, Loo N, McColgan BJ, et al. Acetyl-CoA carboxylase inhibitor GS-0976 for 12 weeks reduces hepatic de novo lipogenesis and steatosis in patients with nonalcoholic steatohepatitis. Clin Gastroenterol Hepatol. 2018;16:1983–1991.e3.
• Loomba R, Kayali Z, Noureddin M, Ruane P, Lawitz EJ, Bennett M, et al. GS-0976 reduces hepatic steatosis and fibrosis markers in patients with nonalcoholic fatty liver disease. Gastroenterology. 2018;155:1463–73. https://doi.org/10.1053/j.gastro.2018.07.027Largest study to date demonstrating efficacy of firsocostat in humans with NASH.
Alkhouri N, Lawitz E, Noureddin M, DeFronzo R, Shulman GI. GS-0976 (firsocostat): An investigational liver-directed acetyl-CoA carboxylase (ACC) inhibitor for the treatment of non-alcoholic steatohepatitis (NASH). Expert Opin Investig Drugs. 2020;29:135–41. https://doi.org/10.1080/13543784.2020.1668374.
Jones SF, Infante JR. Molecular pathways: Fatty acid synthase. Clin Cancer Res. 2015;21:5434–8. https://doi.org/10.1158/1078-0432.CCR-15-0126.
Huang C, Freter C. Lipid metabolism, apoptosis and cancer therapy. Int J Mol Sci. 2015;16:924–49. https://doi.org/10.3390/ijms16010924.
Syed-Abdul MM, Parks EJ, Gaballah AH, Bingham K, Hammoud GM, Kemble G, et al. First-in-class fatty acid synthase inhibitor TVB-2640 reduces hepatic de novo lipogenesis in males with metabolic abnormalities. Hepatology. 2019. https://doi.org/10.1002/hep.31000.
Clinical trials. clinicaltrials.gov. Accessed March 26, 2020.
Koeberle A, Loser K, Thurmer M. Stearoyl-CoA desaturase-1 and adaptive stress signaling. Biochim Biophys Acta. 1861;2016:1719–26.
Safadi R, Konikoff FM, Mahamid M, Zelber-Sagi S, Halpern M, Gilat T, et al. The fatty acid-bile acid conjugate aramchol reduces liver fat content in patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2014;12:2085–91.e1. https://doi.org/10.1016/j.cgh.2014.04.038.
•• Ratziu V, de Guevara L, Safadi R, Poordad F, Fuster F, Flores-Figueroa J, et al. One-year results of the global phase 2b randomized placebo- controlled ARREST trial of aramchol, a stearoyl CoA desaturase modulator in NASH patients. Hepatology. 2018; This ARREST trial demonstrates the efficacy of aramchol in treating NASH as treatment was associated with resolution of NASH and improvement in fibrosis.
Nehemya G. Galmed pharmaceuticals initiated ARMOR, a phase 3/4 registrational study of aramchol in subjects with NASH and fibrosis. http://galmedpharma.investorroom.com/2019-09-26-Galmed-Pharmaceuticals-Initiated-ARMOR-a-Phase-3-4-Registrational-Study-of-Aramchol-in-Subjects-With-NASH-and-Fibrosis. Updated 2019.
Yang M, Nickels JT. MOGAT2: A new therapeutic target for metabolic syndrome. Diseases. 2015;3:176–92. https://doi.org/10.3390/diseases3030176.
Devasthale P, Cheng D. Monoacylglycerol acyltransferase 2 (MGAT2) inhibitors for the treatment of metabolic diseases and nonalcoholic steatohepatitis (NASH). J Med Chem. 2018;61:9879–88. https://doi.org/10.1021/acs.jmedchem.8b00864.
Okuma C, Ohta T, Tadaki H, Ishigure T, Sakata S, Taniuchi H, et al. JTP-103237, a monoacylglycerol acyltransferase inhibitor, prevents fatty liver and suppresses both triglyceride synthesis and de novo lipogenesis. J Pharmacol Sci. 2015;128:150–7. https://doi.org/10.1016/j.jphs.2015.06.007.
Bhatt-Wessel B, Jordan TW, Miller JH, Peng L. Role of DGAT enzymes in triacylglycerol metabolism. Arch Biochem Biophys. 2018;655:1–11.
Gluchowski NL, Gabriel KR, Chitraju C, Bronson RT, Mejhert N, Boland S, et al. Hepatocyte deletion of triglyceride-synthesis enzyme acyl CoA: Diacylglycerol acyltransferase 2 reduces steatosis without increasing inflammation or fibrosis in mice. Hepatology. 2019;70:1972–85. https://doi.org/10.1002/hep.30765.
Amin NB, Carvajal-Gonzalez S, Purkal J, Zhu T, Crowley C, Perez S, et al. Targeting diacylglycerol acyltransferase 2 for the treatment of nonalcoholic steatohepatitis. Sci Transl Med. 2019;11:eaav9701. https://doi.org/10.1126/scitranslmed.aav9701.
Hong DJ, Jung SH, Kim J, Jung D, Ahn YG, Suh KH, et al. Synthesis and biological evaluation of novel thienopyrimidine derivatives as diacylglycerol acyltransferase 1 (DGAT-1) inhibitors. J Enzyme Inhib Med Chem. 2020;35:227–34. https://doi.org/10.1080/14756366.2019.1693555.
Okour M, Gress A, Zhu X, Rieman D, Lickliter JD, Brigandi RA. First-in-human pharmacokinetics and safety study of GSK3008356, a selective DGAT1 inhibitor, in healthy volunteers. Clin Pharmacol Drug Dev. 2019;8:1088–99. https://doi.org/10.1002/cpdd.691.
Okour M, Brigandi RA, Tenero D. A population analysis of the DGAT1 inhibitor GSK3008356 and its effect on endogenous and meal-induced triglyceride turnover in healthy subjects. Fundam Clin Pharmacol. 2019;33:567–80. https://doi.org/10.1111/fcp.12455.
Bougarne N, Weyers B, Desmet SJ, Deckers J, Ray DW, Staels B, et al. Molecular actions of PPARalpha in lipid metabolism and inflammation. Endocr Rev. 2018;39:760–802. https://doi.org/10.1210/er.2018-00064.
Marion-Letellier R, Savoye G, Ghosh S. Fatty acids, eicosanoids and PPAR gamma. Eur J Pharmacol. 2016;785:44–9.
Chen J, Montagner A, Tan NS, Wahli W. Insights into the role of PPARbeta/delta in NAFLD. Int J Mol Sci. 2018;19. https://doi.org/10.3390/ijms19071893.
Larter CZ, Yeh MM, Van Rooyen DM, Brooling J, Ghatora K, Farrell GC. Peroxisome proliferator-activated receptor-alpha agonist, wy 14,643, improves metabolic indices, steatosis and ballooning in diabetic mice with non-alcoholic steatohepatitis. J Gastroenterol Hepatol. 2012;27:341–50. https://doi.org/10.1111/j.1440-1746.2011.06939.x.
Zhang N, Lu Y, Shen X, Bao Y, Cheng J, Chen L, et al. Fenofibrate treatment attenuated chronic endoplasmic reticulum stress in the liver of nonalcoholic fatty liver disease mice. Pharmacology. 2015;95:173–80. https://doi.org/10.1159/000380952.
Shiri-Sverdlov R, Wouters K, van Gorp PJ, Gijbels MJ, Noel B, Buffat L, et al. Early diet-induced non-alcoholic steatohepatitis in APOE2 knock-in mice and its prevention by fibrates. J Hepatol. 2006;44:732–41.
Laurin J, Lindor KD, Crippin JS, Gossard A, Gores GJ, Ludwig J, et al. Ursodeoxycholic acid or clofibrate in the treatment of non-alcohol-induced steatohepatitis: a pilot study. Hepatology. 1996;23:1464–7.
Fernandez-Miranda C, Perez-Carreras M, Colina F, Lopez-Alonso G, Vargas C, Solis-Herruzo JA. A pilot trial of fenofibrate for the treatment of non-alcoholic fatty liver disease. Dig Liver Dis. 2008;40:200–5. https://doi.org/10.1016/j.dld.2007.10.002.
Takei K, Han SI, Murayama Y, Satoh A, Oikawa F, Ohno H, et al. Selective peroxisome proliferator-activated receptor-alpha modulator K-877 efficiently activates the peroxisome proliferator-activated receptor-alpha pathway and improves lipid metabolism in mice. J Diabetes Investig. 2017;8:446–52. https://doi.org/10.1111/jdi.12621.
Honda Y, Kessoku T, Ogawa Y, Tomeno W, Imajo K, Fujita K, et al. Pemafibrate, a novel selective peroxisome proliferator-activated receptor alpha modulator, improves the pathogenesis in a rodent model of nonalcoholic steatohepatitis. Sci Rep. 2017;7:42477. https://doi.org/10.1038/srep42477.
Lee HJ, Yeon JE, Ko EJ, Yoon EL, Suh SJ, Kang K, et al. Peroxisome proliferator-activated receptor-delta agonist ameliorated inflammasome activation in nonalcoholic fatty liver disease. World J Gastroenterol. 2015;21:12787–99. https://doi.org/10.3748/wjg.v21.i45.12787.
Kostadinova R, Montagner A, Gouranton E, Fleury S, Guillou H, Dombrowicz D, et al. GW501516-activated PPARbeta/delta promotes liver fibrosis via p38-JNK MAPK-induced hepatic stellate cell proliferation. Cell Biosci. 2012;2:34. https://doi.org/10.1186/2045-3701-2-34.
Shan W, Palkar PS, Murray IA, McDevitt EI, Kennett MJ, Kang BH, et al. Ligand activation of peroxisome proliferator-activated receptor beta/delta (PPARbeta/delta) attenuates carbon tetrachloride hepatotoxicity by downregulating proinflammatory gene expression. Toxicol Sci. 2008;105:418–28. https://doi.org/10.1093/toxsci/kfn142.
Silva AKS, Peixoto CA. Role of peroxisome proliferator-activated receptors in non-alcoholic fatty liver disease inflammation. Cell Mol Life Sci. 2018;75:2951–61. https://doi.org/10.1007/s00018-018-2838-4.
Hans Vitzthum. CymaBay therapeutics halts clinical development of seladelpar. https://ir.cymabay.com/press-releases/detail/476/cymabay-therapeutics-halts-clinical-development-of-seladelpar. Updated 2019.
Liss KH, Finck BN. PPARs and nonalcoholic fatty liver disease. Biochimie. 2017;136:65–74.
Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. 2010;362:1675–85. https://doi.org/10.1056/NEJMoa0907929.
Bril F, Kalavalapalli S, Clark VC, Lomonaco R, Soldevila-Pico C, Liu IC, et al. Response to pioglitazone in patients with nonalcoholic steatohepatitis with vs without type 2 diabetes. Clin Gastroenterol Hepatol. 2018;16:558–566.e2. https://doi.org/10.1016/j.cgh.2017.12.001.
Yaghoubi M, Jafari S, Sajedi B, Gohari S, Akbarieh S, Heydari AH, et al. Comparison of fenofibrate and pioglitazone effects on patients with nonalcoholic fatty liver disease. Eur J Gastroenterol Hepatol. 2017;29:1385–8. https://doi.org/10.1097/MEG.0000000000000981.
Cusi K, Orsak B, Bril F, Lomonaco R, Hecht J, Ortiz-Lopez C, et al. Long-term pioglitazone treatment for patients with nonalcoholic steatohepatitis and prediabetes or type 2 diabetes mellitus: A randomized trial. Ann Intern Med. 2016;165:305–15. https://doi.org/10.7326/M15-1774.
Musso G, Cassader M, Paschetta E, Gambino R. Thiazolidinediones and advanced liver fibrosis in nonalcoholic steatohepatitis: A meta-analysis. JAMA Intern Med. 2017;177:633–40. https://doi.org/10.1001/jamainternmed.2016.9607.
Cariou B, Hanf R, Lambert-Porcheron S, Zair Y, Sauvinet V, Noel B, et al. Dual peroxisome proliferator-activated receptor alpha/delta agonist GFT505 improves hepatic and peripheral insulin sensitivity in abdominally obese subjects. Diabetes Care. 2013;36:2923–30. https://doi.org/10.2337/dc12-2012.
Cariou B, Zair Y, Staels B, Bruckert E. Effects of the new dual PPAR alpha/delta agonist GFT505 on lipid and glucose homeostasis in abdominally obese patients with combined dyslipidemia or impaired glucose metabolism. Diabetes Care. 2011;34:2008–14. https://doi.org/10.2337/dc11-0093.
Colin S, Briand O, Touche V, Wouters K, Baron M, Pattou F, et al. Activation of intestinal peroxisome proliferator-activated receptor-alpha increases high-density lipoprotein production. Eur Heart J. 2013;34:2566–74. https://doi.org/10.1093/eurheartj/ehs227.
Tolbol KS, Stierstorfer B, Rippmann JF, Veidal SS, Rigbolt KTG, Schonberger T, et al. Disease progression and pharmacological intervention in a nutrient-deficient rat model of nonalcoholic steatohepatitis. Dig Dis Sci. 2019;64:1238–56. https://doi.org/10.1007/s10620-018-5395-7.
Briand F, Heymes C, Bonada L, Angles T, Charpentier J, Branchereau M, et al. A 3-week nonalcoholic steatohepatitis mouse model shows elafibranor benefits on hepatic inflammation and cell death. Clin Transl Sci. 2020. https://doi.org/10.1111/cts.12735.
Roth JD, Veidal SS, Fensholdt LKD, Rigbolt KTG, Papazyan R, Nielsen JC, et al. Combined obeticholic acid and elafibranor treatment promotes additive liver histological improvements in a diet-induced ob/ob mouse model of biopsy-confirmed NASH. Sci Rep. 2019;9:9046. https://doi.org/10.1038/s41598-019-45178-z.
Li TH, Yang YY, Huang CC, Liu CW, Tsai HC, Lin MW, et al. Elafibranor interrupts adipose dysfunction-mediated gut and liver injury in mice with alcoholic steatohepatitis. Clin Sci (Lond). 2019;133:531–44. https://doi.org/10.1042/CS20180873.
Tolbol KS, Kristiansen MN, Hansen HH, Veidal SS, Rigbolt KT, Gillum MP, et al. Metabolic and hepatic effects of liraglutide, obeticholic acid and elafibranor in diet-induced obese mouse models of biopsy-confirmed nonalcoholic steatohepatitis. World J Gastroenterol. 2018;24:179–94. https://doi.org/10.3748/wjg.v24.i2.179.
•• Ratziu V, Harrison SA, Francque S, Bedossa P, Lehert P, Serfaty L, et al. Elafibranor, an agonist of the peroxisome proliferator-activated receptor-alpha and -delta, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology. 2016;150:1147–1159.e5. https://doi.org/10.1053/j.gastro.2016.01.038The GOLDEN-505 trial demonstrated the efficacy of elafibranor in improving NASH histology, though the study did not meet its primary endpoint of NASH resolution.
Eichenbaum N, Lavin H. GENFIT: Announces results from interim analysis of RESOLVE-IT phase 3 trial of elafibranor in adults with NASH and fibrosis. https://ir.genfit.com/news-releases/news-release-details/genfit-announces-results-interim-analysis-resolve-it-phase-3. Updated 2020.
Jain MR, Giri SR, Bhoi B, Trivedi C, Rath A, Rathod R, et al. Dual PPARalpha/gamma agonist saroglitazar improves liver histopathology and biochemistry in experimental NASH models. Liver Int. 2018;38:1084–94. https://doi.org/10.1111/liv.13634.
Gawrieh S, Noureddin M, Loo N, Mohseni R, Awasty V, Kowdley K, et al. A phase 2, prospective, multicenter, double-blind, randomized study of saroglitazar magnesium 1 MG, 2 MG, or 4 MG versus placebo in patients with nonalcoholic fatty liver disease and/or nonalcoholic steatohepatitis (EVIDENCES IV). Hepatology. 2019;10.
Nawrat A. Zydus’ saroglitazar becomes first NASH drug approved globally. https://www.pharmaceutical-technology.com/news/saroglitazar-nash-first/. Updated 2020.
Boubia B, Poupardin O, Barth M, Binet J, Peralba P, Mounier L, et al. Design, synthesis, and evaluation of a novel series of indole sulfonamide peroxisome proliferator activated receptor (PPAR) alpha/gamma/delta triple activators: Discovery of lanifibranor, a new antifibrotic clinical candidate. J Med Chem. 2018;61:2246–65. https://doi.org/10.1021/acs.jmedchem.7b01285.
Sinha R, Yen PM. Cellular action of thyroid hormone. 2000 doi: NBK285568.
Sinha RA, Bruinstroop E, Singh BK, Yen PM. Nonalcoholic fatty liver disease and hypercholesterolemia: Roles of thyroid hormones, metabolites, and agonists. Thyroid. 2019;29:1173–91. https://doi.org/10.1089/thy.2018.0664.
Zucchi R. Thyroid hormone analogues: An update. Thyroid. 2020. https://doi.org/10.1089/thy.2020.0071.
Vatner DF, Weismann D, Beddow SA, Kumashiro N, Erion DM, Liao XH, et al. Thyroid hormone receptor-beta agonists prevent hepatic steatosis in fat-fed rats but impair insulin sensitivity via discrete pathways. Am J Physiol Endocrinol Metab. 2013;305:89. https://doi.org/10.1152/ajpendo.00573.2012.
Cable EE, Finn PD, Stebbins JW, Hou J, Ito BR, van Poelje PD, et al. Reduction of hepatic steatosis in rats and mice after treatment with a liver-targeted thyroid hormone receptor agonist. Hepatology. 2009;49:407–17. https://doi.org/10.1002/hep.22572.
Martagon AJ, Lin JZ, Cimini SL, Webb P, Phillips KJ. The amelioration of hepatic steatosis by thyroid hormone receptor agonists is insufficient to restore insulin sensitivity in ob/ob mice. PLoS One. 2015;10:e0122987. https://doi.org/10.1371/journal.pone.0122987.
Perra A, Simbula G, Simbula M, Pibiri M, Kowalik MA, Sulas P, et al. Thyroid hormone (T3) and TRbeta agonist GC-1 inhibit/reverse nonalcoholic fatty liver in rats. FASEB J. 2008;22:2981–9. https://doi.org/10.1096/fj.08-108464.
• Loomba R, Neutel J, Mohseni R, Bernard D, Severance R, Dao M, et al. LBP-20-VK2809, a novel liver-directed thyroid receptor beta agonist, significantly reduces liver fat with both low and high doses in patients with non-alcoholic fatty liver disease: A phase 2 randomized, placebo-controlled trial. J Hepatol:e150–1. https://doi.org/10.1016/S0618-8278(19)30266-XThis study demonstrated the association between reduction in hepatic steatosis and treatment with resmetirom.
Harrison SA, Bashir MR, Guy CD, Zhou R, Moylan CA, Frias JP, et al. Resmetirom (MGL-3196) for the treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet. 2019;394:2012–24. https://doi.org/10.1016/S0140-6736(19)32517-6.
Zarei M, Pizarro-Delgado J, Barroso E, Palomer X, Vazquez-Carrera M. Targeting FGF21 for the treatment of nonalcoholic steatohepatitis. Trends Pharmacol Sci. 2020;41:199–208.
Tucker B, Li H, Long X, Rye KA, Ong KL. Fibroblast growth factor 21 in non-alcoholic fatty liver disease. Metabolism. 2019;101:153994.
Yilmaz Y, Eren F, Yonal O, Kurt R, Aktas B, Celikel CA, et al. Increased serum FGF21 levels in patients with nonalcoholic fatty liver disease. Eur J Clin Investig. 2010;40:887–92. https://doi.org/10.1111/j.1365-2362.2010.02338.x.
Wu G, Li H, Fang Q, Zhang J, Zhang M, Zhang L, et al. Complementary role of fibroblast growth factor 21 and cytokeratin 18 in monitoring the different stages of nonalcoholic fatty liver disease. Sci Rep. 2017;7:5095. https://doi.org/10.1038/s41598-017-05257-5.
Yin J, Bao L, Chen R, Gao W, Gao X, Yao W. Enhanced expression and distinctive characterization of a long-acting FGF21 and its potential to alleviate nonalcoholic steatohepatitis. Biochimie. 2018;151:166–75.
Lee JH, Kang YE, Chang JY, Park KC, Kim HW, Kim JT, et al. An engineered FGF21 variant, LY2405319, can prevent non-alcoholic steatohepatitis by enhancing hepatic mitochondrial function. Am J Transl Res. 2016;8:4750–63.
Bao L, Yin J, Gao W, Wang Q, Yao W, Gao X. A long-acting FGF21 alleviates hepatic steatosis and inflammation in a mouse model of non-alcoholic steatohepatitis partly through an FGF21-adiponectin-IL17A pathway. Br J Pharmacol. 2018;175:3379–93. https://doi.org/10.1111/bph.14383.
• Charles ED, Neuschwander-Tetri BA, Pablo Frias J, Kundu S, Luo Y, Tirucherai GS, et al. Pegbelfermin (BMS-986036), PEGylated FGF21, in patients with obesity and type 2 diabetes: Results from a randomized phase 2 study. Obesity (Silver Spring). 2019;27:41–9. https://doi.org/10.1002/oby.22344This study demonstrated the efficacy of pegbelfermin in the improvement of hepatic steatosis.
Sanyal A, Charles ED, Neuschwander-Tetri BA, Loomba R, Harrison SA, Abdelmalek MF, et al. Pegbelfermin (BMS-986036), a PEGylated fibroblast growth factor 21 analogue, in patients with non-alcoholic steatohepatitis: a randomised, double-blind, placebo-controlled, phase 2a trial. Lancet. 2019;392:2705–17.
Hawley C. All AKR-001 dose groups met week 12 efficacy endpoints in NASH phase 2a BALANCED study. https://ir.akerotx.com/news-releases/news-release-details/all-akr-001-dose-groups-met-week-12-efficacy-endpoints-nash. Updated 2020.
Rakipovski G, Rolin B, Nohr J, Klewe I, Frederiksen KS, Augustin R, et al. The GLP-1 analogs liraglutide and semaglutide reduce atherosclerosis in ApoE(−/−) and LDLr(−/−) mice by a mechanism that includes inflammatory pathways. JACC Basic Transl Sci. 2018;3:844–57. https://doi.org/10.1016/j.jacbts.2018.09.004.
Armstrong MJ, Gaunt P, Aithal GP, Barton D, Hull D, Parker R, et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet. 2016;387:679–90. https://doi.org/10.1016/S0140-6736(15)00803-X.
Hsia DS, Grove O, Cefalu WT. An update on sodium-glucose co-transporter-2 inhibitors for the treatment of diabetes mellitus. Curr Opin Endocrinol Diabetes Obes. 2017;24:73–9. https://doi.org/10.1097/MED.0000000000000311.
Seko Y, Nishikawa T, Umemura A, Yamaguchi K, Moriguchi M, Yasui K, et al. Efficacy and safety of canagliflozin in type 2 diabetes mellitus patients with biopsy-proven nonalcoholic steatohepatitis classified as stage 1–3 fibrosis. Diabetes Metab Syndr Obes. 2018;11:835–43. https://doi.org/10.2147/DMSO.S184767.
Kuchay MS, Krishan S, Mishra SK, Farooqui KJ, Singh MK, Wasir JS, et al. Effect of empagliflozin on liver fat in patients with type 2 diabetes and nonalcoholic fatty liver disease: A randomized controlled trial (E-LIFT trial). Diabetes Care. 2018;41:1801–8. https://doi.org/10.2337/dc18-0165.
Lai LL, Vethakkan SR, Nik Mustapha NR, Mahadeva S, Chan WK. Empagliflozin for the treatment of nonalcoholic steatohepatitis in patients with type 2 diabetes mellitus. Dig Dis Sci. 2020;65:623–31. https://doi.org/10.1007/s10620-019-5477-1.
Tobita H, Sato S, Miyake T, Ishihara S, Kinoshita Y. Effects of dapagliflozin on body composition and liver tests in patients with nonalcoholic steatohepatitis associated with type 2 diabetes mellitus: A prospective, open-label, uncontrolled study. Curr Ther Res Clin Exp. 2017;87:13–9. https://doi.org/10.1016/j.curtheres.2017.07.002.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Dr. Alkhouri has served on advisory boards for Allergan, Gilead, Intercept, Pfizer, and Zydus; he has served as a speaker for AbbVie, Alexion, Gilead, Intercept, and Simply Speaking; and has received research support from Akero, Albireo, Allergan, Axcella, BI, BMS, Celgene, Gilead, Galmed, Galectin, Genfit, Enanta, Enyo, Hanmi, Inventiva, Madrigal, Merck, Novartis, Novo Nordisk, Pfizer, Poxel and Zydus.
Human and Animal Rights and Informed Consent
All reported studies/experiments with human or animal subjects performed by the authors have been previously published and complied with all applicable ethical standards (including the Helsinki declaration and its amendments, institutional/national research committee standards, and international/national/institutional guidelines).
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Fatty Liver Disease
Rights and permissions
About this article

Cite this article
Aggarwal, P., Singh, T. & Alkhouri, N. Metabolic Targets in Nonalcoholic Steatohepatitis: Treating the Disease at the Metabolic Root. Curr Hepatology Rep 19, 302–314 (2020). https://doi.org/10.1007/s11901-020-00533-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11901-020-00533-x