
Abstract
Purpose of Review
Apoptosis results from the interaction between pro- and anti-apoptotic proteins, mediated by BCL-2 homology 3 (BH3) proteins. B cell lymphoma-2 (BCL-2) is an inhibitor of apoptosis which stabilizes the mitochondria, resulting in the prevention of activation of the pro-apoptotic proteins. In addition, BCL-2 is overexpressed in the leukemic stem cell (LSC) population, and its inhibition may lead to selective LSC eradication. Herein, we will discuss the mechanism and rationale of BCL-2 inhibition in acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS) with an overview of the selective BCL-2 inhibitor venetoclax.
Recent Findings
Venetoclax has activity against AML and has displayed synergistic activity with hypomethylating agents in the preclinical setting. In the clinical setting, although it has only modest activity as a single agent in relapsed and refractory AML, in the older, treatment-naïve population, in combination with either a hypomethylator or low-dose cytarabine, it is well tolerated with impressive efficacy. In addition, BCL-2 inhibition may also have activity in MDS, and although clinical trials are in their early phases, this may be an effective strategy in both the up-front and relapsed setting.
Summary
BCL-2 inhibition with venetoclax is well tolerated and active in older patients with newly diagnosed AML and in the relapsed setting has activity that may be improved in combination with other therapies. It may prove to be effective in MDS and is an exciting treatment strategy for myeloid malignancies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Myelodysplastic syndromes (MDS) are clonal hematopoietic stem-cell disorders mostly affecting the elderly (median age at diagnosis is 70–75 years), characterized by dysplastic morphological changes, ineffective erythropoiesis, variable propensity to progress to acute myeloid leukemia (AML), and associated with recurrent somatic mutations [1,2,3]. There are no curative Food and Drug Administration (FDA)-approved drugs, and patients with MDS have a variable prognosis based on risk stratification ranging from several years in low-risk patients to several months in those with high-risk disease [4, 5]. Hematopoietic cell transplantation (HCT) is the only potentially curative option, but is often not tolerated due to prohibitive transplant-related side effects or is not feasible due to age or comorbidities [2].
AML, the most common acute leukemia in adults, is a heterogeneous hematopoietic stem-cell disorder which also predominantly affects the elderly (median age at diagnosis of 69 years) [6], and is characterized by uncontrolled clonal proliferation of myeloid precursor cells resulting in impaired production of the normal blood cells; AML is universally fatal if untreated [7]. Older patients with AML are more likely to have poor prognostic features, adverse cytogenetics and have a higher chance of having AML from an antecedent hematological malignancy—all of which result in a worse prognosis [7,8,9,10]. In addition, patients, especially the elderly, tolerate induction chemotherapy or HCT poorly, and in the relapsed/refractory setting, there are limited effective therapies [7, 9, 10].
Due to the poor outcomes with current standard-of-care therapies in the majority of patients with AML and higher-risk MDS, novel approaches to treatment are needed. B cell lymphoma 2 (BCL-2) family of proteins are overexpressed in various malignancies including AML [11] and MDS [12, 13] and are critical for cell survival; inhibition of BCL-2 protein, a member of the BCL-2 family of proteins, may be a novel and effective target for the treatment of these conditions. In addition, BCL-2 inhibition has activity against the leukemic stem cell (LSC) population in AML and MDS [12, 14, 15, 16••]. Here, we discuss the mechanism of BCL-2 inhibition, the rationale for targeting BCL-2 in AML and MDS, and provide an overview of the potent and selective BCL-2 inhibitor venetoclax.
BCL-2 Family of Proteins and Apoptosis
BCL-2, a member of the BCL-2 gene family, was first discovered at the t(14;18) breakpoint in follicular lymphoma [17], resulting in BCL-2 overexpression [17, 18]. BCL-2 has been shown to be aberrantly overexpressed in nearly all B cell malignancies, and variably in AML and MDS. [11,12,13]
The BCL-2 family of proteins consists of more than 20 proteins; based on their function, they can be divided into three groups [19]:
-
i.
Multi-domain pro-apoptotic proteins—also called the apoptotic effectors, this group consists of BCL-2 antagonist/killer (BAK) and BCL-2 associated X protein (BAX). The BAK and BAX contain three BCL-2 homology (BH) domains, BH1, BH2, and BH3.
-
ii.
Multi-domain anti-apoptotic proteins—also called the pro-survival proteins, this group inhibits apoptosis. They include BCL-2, BCL-X large (BCLXL), BCL-2-like protein 10 (BCLB), BCL-2-like protein 2 (BCLW), myeloid cell leukemia sequence 1 (MCL1), and BCL-2- related protein (BFL1). They contain four BH domains, BH1, BH2, BH3, and BH4.
-
iii.
BH3-only proteins—these are important for interaction between the pro- and anti-apoptotic BCL-2 proteins and play an important role in apoptosis. The group includes proteins like PUMA, BIM, tBID, NOXA, and BAD.
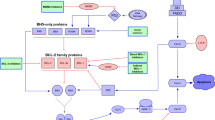
Apoptosis, a type I programmed cell death, is recognized to play an important role in maintaining tissue homeostasis in normal cells, while neoplastic cells are characterized by the ability to avert apoptosis [20]. Apoptosis can be triggered by two pathways (see Fig. 1), the extrinsic (extracellular or death receptor) pathway and the intrinsic (intracellular or mitochondrial) pathway [21,22,23]. The extrinsic pathway is triggered when a member of the tumor necrosis factor superfamily (death-inducing ligand), bind to the cell surface (death) receptors, resulting in the activation of the caspase cascade via the formation of an intracellular death-inducing signaling complex (DISC), leading to cell death. [22,23,24]. The intrinsic pathway, triggered by cellular stress (e.g., growth factor deprivation, DNA damage, oxidative stress), is controlled by members of the BCL-2 protein family [21, 23].

The apoptotic pathways. The extrinsic apoptotic pathway is triggered by the binding of death receptor to the death receptor ligand which activates caspases cascade. The activated caspases-8 and -10 in turn activate effector caspases-3 and -7 leading to apoptosis. The intrinsic apoptotic pathway is triggered when cellular stress leads to BH3-only protein activation which in turn leads to activation of BAX and BAK resulting in mitochondrial outer membrane permeabilization (MOMP) causing release of cytochrome-c and SMAC (second mitochondria-derived activator of caspase). Cytochrome-c causes apoptosome-mediated activation of caspase-9 which in turn activates caspase-3 and -7, leading to apoptosis. SMAC blocks the caspase inhibitor, XIAP (X-linked inhibitor of apoptosis protein) and facilitates apoptosis. Caspase-8 cleavage of the BID enables interaction between the extrinsic and intrinsic pathways
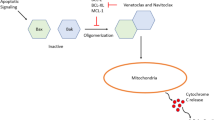
Apoptosis occurs due to an interaction between the multi-domain anti-apoptotic proteins and the pro-apoptotic proteins via the BH3-only proteins which mediate their interaction by either inhibiting the anti-apoptotic proteins or directly activating the pro-apoptotic proteins, BAX and BAK [19]. BCL-2 dysregulation results in the overexpression of the anti-apoptotic BCL-2 protein, leading to an alteration in the equilibrium between pro- and anti-apoptotic members of the BCL-2 family [19, 25]. When the pathway is activated, the BH3-only proteins inhibit the anti-apoptotic proteins BCL-2, BCL-XL, MCL1, and BCLW, and activate the pro-apoptotic proteins BAK and BAX, resulting in the release of mitochondrial cytochrome-c. This causes activation of caspase 9, leading to activation of procaspase 3 and procaspase 7, resulting in cell death [19, 25].
BCL-2 is an inhibitor of apoptosis [26], and stabilizes the mitochondria, leading to the prevention of activation of the pro-apoptotic proteins [27]. Overexpression of BCL-2 promotes tumorigenesis by inhibiting cell death rather than by promoting cell proliferation, a mechanism distinct from several other previously identified oncogenes [19, 28].
Rationale of BCL-2 Inhibition in AML
As discussed, the roles of the BCL-2 family of proteins in apoptosis and oncogenesis have been studied extensively. The known function of BH3 proteins in promoting apoptosis with resultant leukemic cell death [19, 25] was the basis for the initial development of BCL-2 inhibitors as a potential therapy in patients with AML.
Despite advances in treatments for other hematological malignancies, the standard of care therapy of intensive chemotherapy for AML patients has remained unchanged for over 40 years, without significant improvements in outcomes. The high risk of relapse after standard cytotoxic chemotherapy is at least partially due to the persistence of the “rare and quiescent” [29] LSCs that resist such treatments [29,30,31]. Therefore, specifically targeting the LSC population is a potential treatment approach for patients with AML.
It has been demonstrated that high levels of BCL-2 are expressed in LSCs [16••]. BCL-2 inhibition may modulate mitochondrial activity, leading to the selective eradication of the LSC population [16••]. In addition, in AML, BCL-2 overexpression can lead to therapeutic resistance [32], and predicts for worse responses to chemotherapy [11, 33]. These findings suggest that BCL-2 inhibition may specifically target LSCs, and BCL-2 inhibitors may represent a novel therapeutic approach for patients with AML.
Inhibition of BCL-2 in the Pre-Venetoclax Era
In preclinical studies, it was shown that downregulation of BCL-2 by an antisense oligodeoxynucleotide (BCL-2-AS) led to the selective inhibition of leukemic cells by apoptosis and established the preclinical foundation supporting the potential role of BCL-2 inhibitors in AML [34]. Based on this work, a BCL-2 antisense oligonucleotide, oblimersen (G3139), was combined with chemotherapy to treat relapsed/refractory AML in various studies (Table 1) [35,36,37,38]. In a phase 1 study which included 17 patients with relapsed/refractory AML, oblimersen in combination with FLAG (fludarabine, cytarabine, and ganulocyte colony-stimulating factor) showed an overall response rate (ORR) of 41% [35]. In another phase 1 study of untreated older (≥ 60 years) AML patients, oblimersen showed an encouraging ORR of 48%; responses were durable, with 50% of patients who achieved complete remission remaining disease free at a median follow-up time of 12.6 months [36]. However, in a phase 3 trial of induction chemotherapy with or without oblimersen in newly diagnosed AML patients (CALGB 10201), oblimersen did not show a benefit with respect to complete response (CR), overall survival (OS), and event free-survival (EFS) rates [38].
Obatoclax, a BH3-mimetic and pan-BCL-2 inhibitor, showed limited clinical activity as a single agent (Table 1) in patients with relapsed or untreated AML [39, 40] and MDS [13, 39]. In a phase 1 clinical trial in patients with hematological malignancies including AML (n = 25) and MDS (n = 14), the best responses were hematological improvement in one AML and three MDS patients [39]. In a follow-up phase 1/2 clinical trial of elderly (≥ 70 years) AML patients, there were no objective responses [40].
ABT-737 and ABT-263 (navitoclax), BH3 mimetic agents that inhibit both BCL-2 and BCL-XL, were also studied in preclinical and clinical settings [41,42,43,44,45]. However, they were noted to have a dose-dependent severe thrombocytopenia due to the on-target inhibition of BCL-XL, limiting their clinical utility in patients with AML or MDS.
Venetoclax and Other BCL-2 Family Protein Modulation in AML
Following the experience with ABT-737 and navitoclax, it was hypothesized that a more selective BCL-2 inhibitor would be able to alleviate the dose-limiting side effects while maintaining efficacy; this led to the development of the first highly selective and potent BCL-2 inhibitor, ABT-199 (venetoclax).
The first effort to study venetoclax in AML was as a single agent in the relapsed/refractory setting. When studied in a phase 2, single-arm study, venetoclax had an ORR of 19% [46]. Those with isocitrate dehydrogenase (IDH 1/2) mutations showed better CR rates (4/12, 33%) substantiating preclinical data that AML with IDH1/2 mutations are sensitive to BCL-2 inhibition and may activate apoptosis at a lower threshold [47].
If a major mechanism of action of venetoclax in AML is through inhibition of LSCs, the modest response rate observed in the relapsed/refractory study and subsequently observed when used off label in various combinations [48] might be explained by the impact of chemotherapy on LSCs. Ho et al. reported the characteristics of LSCs in vivo at the time of diagnosis compared with relapse after exposure to induction chemotherapy; this revealed significant increases in LSC frequency and heterogeneity [49•], suggesting LSC-directed therapies might be more effective in the up-front treatment setting (Fig. 2).

Impact of intensive chemotherapy on acute myeloid leukemia (AML) that eventually relapses: With induction chemotherapy, although there is initial decrease in leukemia stem cells (LSCs), at the time of relapse, the quantity and diversity of LSCs increases supporting the hypothesis that intensive induction chemotherapy subsequently results worsening of AML (Figure adopted with permission, courtesy of Shanshan Pei, PhD)
Preclinical data suggests the potential for synergistic activity between venetoclax and backbone therapies [50,51,52], and these combinations are being explored, mostly in the up-front treatment setting. When combined with either azacitidine or decitabine in the front-line setting in older (≥ 60 years) AML patients unsuitable for standard induction chemotherapy (NCT02203773), venetoclax was well tolerated with a reported complete-response/complete response with incomplete hematologic recovery (CR/CRi) rate of 61% [53•]. Updated results of the trial from 145 patients were reported at the 2017 American Society of Hematology annual meeting [54] and were consistent with the published data from the dose escalation/ expansion study [53•]. The CR + CRi + partial response (PR) rate was 67% (97/145), and the ORR was 83% (120/145) [54]. At a median time on study of 7 months (range < 1 to 27 months), the median OS was 17.5 months (95% CI, 12.3—upper limit not reached). Subgroup analysis by cytogenetic risk showed that patients with intermediate-risk AML achieved a CR/CRi rate of 74%, whereas the response rate in the poor-risk group was 60%. Patients with secondary AML achieved a 67% CR + CRi + PR and those with IDH1/2, FLT3, and TP53 mutations achieved CR/CRi rates of 68, 64, and 56%, respectively [54]. These results are very promising with better response rates than historically seen with conventional chemotherapy in this population [55], particularly in those with adverse disease biology. This combination is being studied in a phase 3 trial comparing venetoclax plus azacitadine with azacitadine alone in AML patients not eligible for standard induction chemotherapy (NCT02993523). Low-dose cytarabine (LDAC) has also been explored as a backbone therapy with venetoclax in the same population [56]. This combination similarly resulted in a CR/CRi rate of 62% and a median OS of 11.4 months (95% CI, 5.7–15.7). Patients with DNMT3A, FLT3-ITD, and SRSF2 mutations had CR/CRi rates of ≥ 75%, whereas those with TP53 mutations had the CR/CRi rates of 44%. Notably, in contrast to the hypomethylator backbone study, the LDAC study allowed patients who had received a prior hypomethylator for MDS, suggesting that this population was perhaps more challenging to treat. Venetoclax combined with LDAC versus LDAC alone is currently being studied in an ongoing phase 3 trial (NCT03069352).
Preclinical study in venetoclax-resistant AML cell lines suggested that venetoclax decreased the association of BIM (a pro-apoptotic protein) with BCL-2 and increased the association of BIM with MCL-1 (pro-survival protein), leading to the stabilization of MCL-1 and resistance to venetoclax [57]. In such resistant cells, venetoclax showed no effect on intrinsic apoptosis, suggesting an altered balance of BCL-2 family members led to venetoclax resistance [57]. Based on this observation, inhibition of MCL-1 was suggested as a way to improve responses to venetoclax and/or overcome resistance. Multiple avenues to inhibit MCL-1 are being explored including: (1) small molecules (AMG 176 [Amgen], NCT02675452; S64315 [Servier] NCT02979366; AZD5991 [AstraZeneca], NCT03218683), (2) BH3 mimetics [58], (3) therapies that have off-target MCL-1 inhibition, such as the cyclin-dependent kinase-9 (CDK9) inhibitor alvocidib (Tolero), which via inhibition of CDK9 downregulates MCL1, leading to cell death in MCL-1 dependent AML [59], and (4) other indirect methods via concomitant inhibition of MDM2 and MEK [60, 61].
Venetocalx as a single agent in the relapsed/ refractory AML population appears to be more active than other less selective BH3 mimetic agents that target BCL-2 [38, 40]. The explanation for this observation is not necessarily obvious given the hypothesis that MCL-1 and BCL-XL upregulation may provide a mechanism of resistance to venetoclax [62]. However, decreased toxicity form more selective BCL-2 inhibition could partially explain this observation and an improved understanding of the mechanism of venetocalx in AML may help better understand this finding.
BCL-2 Inhibition and Potential Role of Venetoclax in MDS
In MDS, cytopenias coexist with bone marrows that typically show hypercellularity, an incongruence that has been attributed to apoptosis leading to ineffective erythropoiesis. [63, 64] In high-risk MDS patients, there is higher BCL-2 expression compared to low-risk MDS, suggesting that MDS acquires apoptotic resistance as the disease progresses, [64, 65] an observation also supported by a significant decrease in the ratio of pro-apoptotic versus anti-apoptotic BCL-2 family proteins at disease progression [66]. Furthermore, in a preclinical study, inhibition of BCL-2 has been shown to be specifically toxic to the MDS stem cells from high-risk MDS patients [12]. These findings support the use of BCL-2 inhibitors in MDS (Table 1). However, in the clinical setting, obatoclax, a pan-BCL-2 inhibitor, was largely ineffective, with hematological improvement seen in only 3 of 14 MDS patients in the phase 1 setting [39]. When studied as a first-line treatment in MDS patients in a phase 2 study, obatocalx again showed a response rate of only 8% [13]. It may be that just as venetoclax outperformed its less specific BCL-2 family protein inhibitors in AML, the same will be seen in MDS. These studies are ongoing (Table 2), with venetoclax being studied in combination with azacitidine as a front-line therapy in higher-risk MDS patients in a phase 1 study (NCT02942290), as well as in higher-risk MDS patients as a single agent or in combination with azacitidine (NCT02966782).
Future Directions
Some believe venetoclax may be beneficial when combined with intensive chemotherapy regimens in the untreated setting, and those studies are planned or ongoing (NCT03214562). FLT3-ITD activation may mediate resistance to venetoclax [67], providing a mechanistic rationale for combining venetoclax with FLT-3 inhibitors in FLT-3 positive patients. Telomerase is known to be overexpressed in AML cells and its inhibition may target LSCs; [68] the telomerase inhibitor imetelstat in combination with venetoclax enhanced apoptosis in vitro and increased survival in vivo in AML [69], establishing a rationale for combination studies in the future. Glasdegib, an inhibitor of Smoothened in the Hedghehog signaling pathway, was recently studied in combination with venetoclax [70], suggesting this may be of benefit. Recently, mononuclear cells obtained from patients with AML were evaluated for sensitivity to drug combinations that target non-overlapping biological pathways. Several combinations of kinase inhibitors with venetoclax were noted to be effective and such novel combination therapies could be avenues for future clinical trials [71•].
Finally, BCL-2 inhibitors other than ventetoclax are being developed. The small molecule BCL-2 inhibitor S055746 is currently being studied in phase 1 clinical trial (NCT02920541), and PNT2258, which causes BCL-2 targeted DNA damage [72], warrant further study in myeloid malignancies.
Conclusions
BCL-2 inhibits apoptosis leading to disease progression and chemotherapy resistance in AML and its inhibitor, venetoclax, modulates mitochondrial activity leading to apoptotic cell death. Venetoclax has a better tolerability profile than traditional chemotherapy, allowing for the treatment of elderly or frail individuals. Various venetoclax combination studies are either ongoing, planned, or have sound mechanistic basis with the potential to advance the therapeutic landscape for AML. BCL-2 inhibition also has promising activity in MDS and is being explored in clinical trials.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance•• Of major Importance
Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013;122(22):3616–27.
Gangat N, Patnaik MM, Tefferi A. Myelodysplastic syndromes: contemporary review and how we treat. Am J Hematol. 2016;91(1):76–89.
Sekeres MA. The epidemiology of myelodysplastic syndromes. Hematol Oncol Clin North Am. 2010;24(2):287–94.
Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Sole F, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120(12):2454–65.
Schanz J, Tuchler H, Sole F, Mallo M, Luno E, Cervera J, et al. New comprehensive cytogenetic scoring system for primary myelodysplastic syndromes (MDS) and oligoblastic acute myeloid leukemia after MDS derived from an international database merge. J Clin Oncol. 2012;30(8):820–9.
SEER. Acute Myeloid Leukemia - Cancer Stat Facts 2017 [Available from: https://seer.cancer.gov/statfacts/html/amyl.html.
De Kouchkovsky I, Abdul-Hay M. Acute myeloid leukemia: a comprehensive review and 2016 update. Blood Cancer J. 2016;6(7):e441.
Juliusson G, Antunovic P, Derolf A, Lehmann S, Mollgard L, Stockelberg D, et al. Age and acute myeloid leukemia: real world data on decision to treat and outcomes from the Swedish Acute Leukemia Registry. Blood. 2009;113(18):4179–87.
Estey EH. Therapeutic options for acute myelogenous leukemia. Cancer. 2001;92(5):1059–73.
Appelbaum FR, Gundacker H, Head DR, Slovak ML, Willman CL, Godwin JE, et al. Age and acute myeloid leukemia. Blood. 2006;107(9):3481–5.
Campos L, Rouault JP, Sabido O, Oriol P, Roubi N, Vasselon C, et al. High expression of bcl-2 protein in acute myeloid leukemia cells is associated with poor response to chemotherapy. Blood. 1993;81(11):3091–6.
Jilg S, Reidel V, Muller-Thomas C, Konig J, Schauwecker J, Hockendorf U, et al. Blockade of BCL-2 proteins efficiently induces apoptosis in progenitor cells of high-risk myelodysplastic syndromes patients. Leukemia. 2016;30(1):112–23.
Arellano ML, Borthakur G, Berger M, Luer J, Raza A. A phase II, multicenter, open-label study of obatoclax mesylate in patients with previously untreated myelodysplastic syndromes with anemia or thrombocytopenia. Clin Lymphoma Myeloma Leuk. 2014;14(6):534–9.
Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 2006;10(5):375–88.
Baev DV, Krawczyk J, M OD, Szegezdi E. The BH3-mimetic ABT-737 effectively kills acute myeloid leukemia initiating cells. Leukemia Research Reports. 2014;3(2):79–82.
•• Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. 2013;12(3):329–41. This study demonstrated that LSCs were characterized by low levels of reactive oxygen species (ROS-low), that ROS-low LSCs aberrantly overexpressed BCL-2, and that BCL-2 inhibition reduced oxidative phosphorylation and selectively eradicated LSCs.
Tsujimoto Y, Finger LR, Yunis J, Nowell PC, Croce CM. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science (New York, NY). 1984;226(4678):1097–9.
Cleary ML, Smith SD, Sklar J. Cloning and structural analysis of cDNAs for bcl-2 and a hybrid bcl-2/immunoglobulin transcript resulting from the t(14;18) translocation. Cell. 1986;47(1):19–28.
Dai H, Meng XW, Kaufmann SH. BCL2 Family, Mitochondrial Apoptosis, and Beyond. Cancer Transl Med. 2017;2(1):7–20.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116(2):205–19.
Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science (New York, NY). 1998;281(5381):1305–8.
Jin Z, El-Deiry WS. Overview of cell death signaling pathways. Cancer Biol Ther. 2005;4(2):139–63.
Yan N, Shi Y. Mechanisms of apoptosis through structural biology. Annu Rev Cell Dev Biol. 2005;21:35–56.
Ashkenazi A, Fairbrother WJ, Leverson JD, Souers AJ. From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat Rev Drug Discov. 2017;16(4):273–84.
Reed JC. Bcl-2 family proteins: regulators of apoptosis and chemoresistance in hematologic malignancies. Semin Hematol. 1997;34(4 Suppl 5):9–19.
Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9(1):47–59.
Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335(6189):440–2.
Pollyea DA, Jordan CT. Therapeutic targeting of acute myeloid leukemia stem cells. Blood. 2017;129(12):1627–35.
Jordan CT, Guzman ML, Noble M. Cancer stem cells. N Engl J Med. 2006;355(12):1253–61.
Warner JK, Wang JC, Hope KJ, Jin L, Dick JE. Concepts of human leukemic development. Oncogene. 2004;23(43):7164–77.
Wang HG, Reed JC. Mechanisms of Bcl-2 protein function. Histol Histopathol. 1998;13(2):521–30.
Banker DE, Radich J, Becker A, Kerkof K, Norwood T, Willman C, et al. The t(8;21) translocation is not consistently associated with high Bcl-2 expression in de novo acute myeloid leukemias of adults. Clin Cancer Res. 1998;4(12):3051–62.
Konopleva M, Tari AM, Estrov Z, Harris D, Xie Z, Zhao S, et al. Liposomal Bcl-2 antisense oligonucleotides enhance proliferation, sensitize acute myeloid leukemia to cytosine-arabinoside, and induce apoptosis independent of other antiapoptotic proteins. Blood. 2000;95(12):3929–38.
Marcucci G, Byrd JC, Dai G, Klisovic MI, Kourlas PJ, Young DC, et al. Phase 1 and pharmacodynamic studies of G3139, a Bcl-2 antisense oligonucleotide, in combination with chemotherapy in refractory or relapsed acute leukemia. Blood. 2003;101(2):425–32.
Marcucci G, Stock W, Dai G, Klisovic RB, Liu S, Klisovic MI, et al. Phase I study of oblimersen sodium, an antisense to Bcl-2, in untreated older patients with acute myeloid leukemia: pharmacokinetics, pharmacodynamics, and clinical activity. J Clin Oncol Off J Am Soc Clin Oncol. 2005;23(15):3404–11.
Moore J, Seiter K, Kolitz J, Stock W, Giles F, Kalaycio M, et al. A phase II study of Bcl-2 antisense (oblimersen sodium) combined with gemtuzumab ozogamicin in older patients with acute myeloid leukemia in first relapse. Leuk Res. 2006;30(7):777–83.
Marcucci G, Moser W, Blum W, Stock M, Wetzler J, Kolitz JE, et al. A phase III randomized trial of intensive induction and consolidation chemotherapy ± oblimersen, a pro-apoptotic Bcl-2 antisense oligonucleotide in untreated acute myeloid leukemia patients > 60 years old. J Clin Oncol Off J Am Soc Clin Oncol. 2007;25:7012.
Schimmer AD, O'Brien S, Kantarjian H, Brandwein J, Cheson BD, Minden MD, et al. A phase I study of the pan bcl-2 family inhibitor obatoclax mesylate in patients with advanced hematologic malignancies. Clin Cancer Res. 2008;14(24):8295–301.
Schimmer AD, Raza A, Carter TH, Claxton D, Erba H, DeAngelo DJ, et al. A multicenter phase I/II study of obatoclax mesylate administered as a 3- or 24-hour infusion in older patients with previously untreated acute myeloid leukemia. PLoS One. 2014;9(10):e108694.
Zhang H, Nimmer PM, Tahir SK, Chen J, Fryer RM, Hahn KR, et al. Bcl-2 family proteins are essential for platelet survival. Cell Death Differ. 2007;14(5):943–51.
Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68(9):3421–8.
Mason KD, Carpinelli MR, Fletcher JI, Collinge JE, Hilton AA, Ellis S, et al. Programmed anuclear cell death delimits platelet life span. Cell. 2007;128(6):1173–86.
Wilson WH, O'Connor OA, Czuczman MS, LaCasce AS, Gerecitano JF, Leonard JP, et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010;11(12):1149–59.
Schoenwaelder SM, Jarman KE, Gardiner EE, Hua M, Qiao J, White MJ, et al. Bcl-xL-inhibitory BH3 mimetics can induce a transient thrombocytopathy that undermines the hemostatic function of platelets. Blood. 2011;118(6):1663–74.
Konopleva M, Pollyea DA, Potluri J, Chyla B, Hogdal L, Busman T, et al. Efficacy and Biological Correlates of Response in a Phase II Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer Discov. 2016;6(10):1106–17.
Chan SM, Thomas D, Corces-Zimmerman MR, Xavy S, Rastogi S, Hong WJ, et al. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat Med. 2015;21(2):178–84.
DiNardo CD, et al. Clinical experience with the BCL2-inhibitor venetoclax in combination therapy for relapsed and refractory acute myeloid leukemia and related myeloid malignancies. Am J Hematol. 2018;93(3):401–7.
• Ho TC, LaMere M, Stevens BM, Ashton JM, Myers JR, O'Dwyer KM, et al. Evolution of acute myelogenous leukemia stem cell properties after treatment and progression. Blood 2016;128(13):1671–8. This study showed the evolution of the LSCs showing increases in LSC frequency and heterogeneity at relapse. This study showed that LSC-directed therapies, like with venetoclax, would be more effective in up-front setting rather than after disease relapse.
Tsao T, Shi Y, Kornblau S, Lu H, Konoplev S, Antony A, et al. Concomitant inhibition of DNA methyltransferase and BCL-2 protein function synergistically induce mitochondrial apoptosis in acute myelogenous leukemia cells. Ann Hematol. 2012;91(12):1861–70.
Bogenberger JM, Delman D, Hansen N, Valdez R, Fauble V, Mesa RA, et al. Ex vivo activity of BCL-2 family inhibitors ABT-199 and ABT-737 combined with 5-azacytidine in myeloid malignancies. Leuk Lymphoma. 2015;56(1):226–9.
Bogenberger JM, Kornblau SM, Pierceall WE, Lena R, Chow D, Shi CX, et al. BCL-2 family proteins as 5-Azacytidine-sensitizing targets and determinants of response in myeloid malignancies. Leukemia. 2014;28(8):1657–65.
• DiNardo CD, Pratz KW, Letai A, Jonas BA, Wei AH, Thirman M, et al. Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: a non-randomised, open-label, phase 1b study. Lancet Oncol. 2018;19(2):216–28. This ongoing study showed that venetoclax plus hypomethylating agent therapy is well-tolerated regimen with promising activity in elderly patient population in upfront setting.
DiNardo CD, Pollyea DA, Jonas BA, Konopleva M, Pullarkat V, Wei A, et al. Updated Safety and Efficacy of Venetoclax with Decitabine or Azacitidine in Treatment-Naive, Elderly Patients with Acute Myeloid Leukemia. Blood. 2017;130(Suppl 1):2628.
Almeida AM, Ramos F. Acute myeloid leukemia in the older adults. Leuk Res Rep. 2016;6:1–7.
Wei A, Strickland SA, Roboz GJ, Hou J-Z, Fiedler W, Lin TL, et al. Phase 1/2 Study of Venetoclax with Low-Dose Cytarabine in Treatment-Naive, Elderly Patients with Acute Myeloid Leukemia Unfit for Intensive Chemotherapy: 1-Year Outcomes. Blood. 2017;130(Suppl 1):890.
Niu X, Zhao J, Ma J, Xie C, Edwards H, Wang G, et al. Binding of released Bim to Mcl-1 is a mechanism of intrinsic resistance to ABT-199 which can be overcome by combination with daunorubicin or cytarabine in AML cells. Clin Cancer Res. 2016;22(17):4440–51.
Pan R, Ruvolo VR, Wei J, Konopleva M, Reed JC, Pellecchia M, et al. Inhibition of Mcl-1 with the pan-Bcl-2 family inhibitor (-)BI97D6 overcomes ABT-737 resistance in acute myeloid leukemia. Blood. 2015;126(3):363–72.
Bogenberger J, Whatcott C, Hansen N, Delman D, Shi CX, Kim W, et al. Combined venetoclax and alvocidib in acute myeloid leukemia. Oncotarget. 2017;8:107206–22.
Kojima K, Konopleva M, Samudio IJ, Schober WD, Bornmann WG, Andreeff M. Concomitant inhibition of MDM2 and Bcl-2 protein function synergistically induce mitochondrial apoptosis in AML. Cell Cycle. 2006;5(23):2778–86.
Daver N, Pollyea DA, Yee KWL, Fenaux P, Brandwein JM, Vey N, et al. Preliminary Results from a Phase Ib Study Evaluating BCL-2 Inhibitor Venetoclax in Combination with MEK Inhibitor Cobimetinib or MDM2 Inhibitor Idasanutlin in Patients with Relapsed or Refractory (R/R) AML. Blood. 2017;130(Suppl 1):813.
Bose P, Gandhi V, Konopleva M. Pathways and mechanisms of venetoclax resistance. Leuk Lymphoma. 2017;58:1–17.
Rajapaksa R, Ginzton N, Rott LS, Greenberg PL. Altered oncoprotein expression and apoptosis in myelodysplastic syndrome marrow cells. Blood. 1996;88(11):4275–87.
Corey SJ, Minden MD, Barber DL, Kantarjian H, Wang JC, Schimmer AD. Myelodysplastic syndromes: the complexity of stem-cell diseases. Nat Rev Cancer. 2007;7(2):118–29.
Invernizzi R, Pecci A, Bellotti L, Ascari E. Expression of p53, bcl-2 and ras oncoproteins and apoptosis levels in acute leukaemias and myelodysplastic syndromes. Leuk Lymphoma. 2001;42(3):481–9.
Parker JE, Mufti GJ, Rasool F, Mijovic A, Devereux S, Pagliuca A. The role of apoptosis, proliferation, and the Bcl-2-related proteins in the myelodysplastic syndromes and acute myeloid leukemia secondary to MDS. Blood. 2000;96(12):3932–8.
Mali RS, Lasater EA, Doyle K, Malla R, Boghaert E, Souers A, et al. FLT3-ITD activation mediates resistance to the BCL-2 selective antagonist, venetoclax, in FLT3-ITD mutant AML models. 2017.
Bruedigam C, Bagger FO, Heidel FH, Kuhn CP, Guignes S, Song A, et al. Telomerase inhibition effectively targets mouse and human AML stem cells and delays relapse following chemotherapy. Cell Stem Cell. 2014;15(6):775–90.
Rusbuldt JJ, Luistro L, Chin D, Smith M, Wong A, Romero M, et al. Telomerase inhibitor imetelstat in combination with the BCL-2 inhibitor venetoclax enhances apoptosisin vitro and increases survival in vivo in acute myeloid leukemia. Cancer Res. 2017;77(Suppl 13):1101.
Tauchi T, Okabe S, Katagiri S, Tanaka Y, Ohyashiki K. Combining Effects of the SMO Inhibitor and BCL-2 Inhibitor in MDS-Derived Induced Potent Stem Cells (iPSC). Blood. 2017;130(Suppl 1):1249.
• Kurtz SE, Eide CA, Kaempf A, Khanna V, Savage SL, Rofelty A, et al. Molecularly targeted drug combinations demonstrate selective effectiveness for myeloid- and lymphoid-derived hematologic malignancies. Proc Natl Acad Sci U S A. 2017;114(36):E7554–e63. In this study, several combinations of kinase inhibitors with venetoclax were effective in inhibiting myeloid-derived cells showing that novel combination therapies with venetoclax could be avenues for future AML clinical trials.
Ebrahim AS, Kandouz M, Liddane A, Sabbagh H, Hou Y, Li C, et al. PNT2258, a novel deoxyribonucleic acid inhibitor, induces cell cycle arrest and apoptosis via a distinct mechanism of action: a new class of drug for non-Hodgkin’s lymphoma. Oncotarget. 2016;7(27):42374–84.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Daniel A Pollyea served as an advisory board member for Celyad, Agios, Celgene, Abbvie, Argenx, Pfizer, Curis, Takeda, and Servier, and received research funding from Agios and Pfizer. Prashant Sharma declares no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Myelodysplastic Syndromes
Rights and permissions
About this article

Cite this article
Sharma, P., Pollyea, D.A. Shutting Down Acute Myeloid Leukemia and Myelodysplastic Syndrome with BCL-2 Family Protein Inhibition. Curr Hematol Malig Rep 13, 256–264 (2018). https://doi.org/10.1007/s11899-018-0464-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11899-018-0464-8