
Abstract
Purpose of Review
ATL is a rare and highly aggressive T cell malignancy caused by HTLV-1. We will review the state of the art of ATL epidemiology, pathogenesis, diagnosis, and treatment.
Recent Findings
Because of population migration, cases of ATL in non-HTLV-1 endemic countries including North America and Europe are increasingly recognized. ATL has diverse clinical manifestations, limited treatment options not widely available globally, and poor prognosis despite therapy. A small subset of patients may achieve prolonged survival with antiviral therapy or allogeneic stem cell transplant. Mogamulizumab, an anti-CCR4 monoclonal antibody, and lenalidomide, an immunomodulatory drug, are approved for ATL in Japan. Molecular studies have identified key alterations in T cell signaling and DNA methylation that may further guide drug development. Patients should be encouraged to enroll in prospective clinical trials when possible.
Summary
Ongoing, collaborative, international research continues to elucidate disease pathogenesis and contribute to an evolving therapeutic landscape for ATL.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
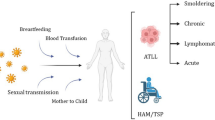
Adult T cell lymphoma/leukemia (ATL) is a peripheral T cell neoplasm caused by a chronic infection with human T-lymphotropic virus, type 1 (HTLV-1). ATL is endemic to several regions owing to the epidemiology of HTLV-1, with the highest rates found in southwest Japan, the Caribbean basin, and areas of South America and sub-Saharan Africa [1, 2]. According to the International Peripheral T-cell Lymphoma (PTCL) Project, ATL is rare in North America and Europe (≤ 2% of cases); however, because of population migration, sporadic case series are increasingly documenting ATL across the world, including in non-endemic areas [3,4,5]. The origin of this geographic distribution remains puzzling but is likely due to a founder effect in certain populations [6].
HTLV-1 infects an estimated 5 to 10 million people worldwide, and a small fraction (4–5%) develop ATL after several decades of infection [1]. Routes of HTLV-1 transmission are breast feeding, sexual intercourse, and blood transfusion; however, ATL preferentially develops after four to five decades in individuals infected during childhood, and seldom occurs in those infected with the virus as an adult [1]. Similar to other retroviruses, HTLV-1 integrates into infected T cells in a random fashion in the asymptomatic carrier state. Patients with ATL however have monoclonal integration of a single viral copy in the majority of cases [7, 8••]. The HTLV-1 genome expresses two oncogenic proteins: the transactivator protein (Tax) that is critical for T cell proliferation mainly via activation of NFκB and AP-1 pathways, and the HTLV-1 basic leucine zipper protein (HBZ) that may serve as a tumor promoter [8••]. Given the long latency from HTLV-1 infection to onset of ATL, other genomic events are likely important in disease pathogenesis. A recent large-scale genomic analysis of over 400 cases of ATL found a number of alterations, including mutated genes implicated in T cell receptor-NFκB signaling (e.g., PLCG1, PRKCB, CARD11, and STAT3), T cell trafficking (e.g., CCR4), and immunosurveillance [9••].
ATL is characterized by marked diversity in its clinical manifestations with frequent blood and lymph node involvement, as well as infiltration of other organs such as the skin, central nervous system, and gastrointestinal tract. Hypercalcemia and a predilection for opportunistic infection are also common features. The Shimoyama Classification requires confirmation of HTLV-1 infection with serologic testing and proposes four clinical subtypes based on presence and degree of leukemic T cells, additional sites of infiltration, lactate dehydrogenase level (LDH), and hypercalcemia (Table 1); however, there may be overlap between subtypes [10]. The acute and lymphoma subtypes (aggressive ATL) have the poorest prognosis with median survivals of only 6–10 months even with intensive chemotherapy [11]. Much of the clinical data on ATL derives from the Japanese population where subtype compositions and outcomes may differ from those of other regions, notably the Caribbean. Caribbean ATL patients with primarily aggressive subtypes have an even worse survival among patients diagnosed in the USA [4, 5].
There is no standard treatment for ATL outside of Japan and most guidelines recommend patients enroll in clinical trials when possible. Recent work suggests a subset of patients may benefit from antiviral therapy alone and that allogeneic stem cell transplant can prolong survival [12,13,14,15]. New agents have received approval in Japan, and clinical trials are ongoing in other populations. This review will summarize the state of the art of ATL pathogenesis, diagnosis, and treatment approach.
How Does HTLV-1 Cause ATL?
Since its discovery and isolation from a patient with T cell malignancy by Gallo and coworkers in the late 1970s, the molecular pathophysiology of ATL has been the subject of fascinating and ongoing research in virally induced T cell transformation [16]. HTLV-1 is a complex deltaretrovirus and its genome includes structural genes (gag, pol, and env) and accessory genes (tax, rex, p12, p21, p30, p13, and HBZ) that play an important role in the regulation of viral replication, persistence, and leukemogenesis [17]. Both Tax and HBZ are implicated specifically in oncogenesis, with models implicating Tax in tumor initiation and HBZ in maintenance [17].
ATL develops after a prolonged latency in approximately 5% of individuals infected almost exclusively from breastfeeding [1]. The virus is spread between cells via cell-to cell transmission, rather than by free virion, as no free virion is detected in plasma of HTLV-1 infected individuals, and the infectivity of free virions is poor compared with that of infected cells [18]. The proviral integration site of HTLV-1 into the host genome is strongly biased towards certain transcription factor binding sites, notably STAT1, TP53, and HDAC6 [19, 20]. HTLV-1 proviral load measurement in peripheral blood mononuclear cells (PBMC) by quantitative PCR has been used to study in vivo infection burden, and in few prospective studies of asymptomatic carriers, higher proviral loads predict the development of ATL [21, 22•, 23].
HTLV-1 causes a clonal expansion of infected T cells and elicits a high titer of specific antibody as well as activated CD8+ cytotoxic T lymphocytes (CTLs) [22•]. In non-malignant HTLV-1 infection, thousands of small clones of HTLV-1 infected cells exist; however, a single dominant clone is found among a background of hundreds of clones in patients with ATL [22•, 23]. Factors that may determine the risk of developing ATL in an asymptomatic HTLV-1 carrier are divided into host- and viral-specific factors. Host-specific factors include host genotype, age at infection, and the presence of certain co-infections, especially Strongyloides stercocarlis [22•]. Viral-specific factors include the proviral integration site and the development of secondary mutations that increase the proliferative or survival advantage of an HTLV-1+ clone [22•]. Recent whole genome sequencing provides evidence that mutations in three key signaling pathways—NFκB, T cell receptor, and CCR4—are associated with ATL [9••]. In addition, abnormalities in DNA methylation in 53 genes, including many involved in apoptosis, were described [9••]. These findings will open a new direction for drug development in ATL.
Recognizing ATL and Pitfalls of the Shimoyama Classification
ATL cells in the majority of cases express CD3, CD4, and CD25, but lack CD7; in ~ 10–15% of cases, cells express both CD4 and CD8 [24]. ATL cells are thought to be the malignant counterpart of regulatory T cells based on the expression of FoxP3 in some tumor cells; however, this remains a subject of debate [25]. There is no difference in immunophenotype of ATL cells by disease subtype. CCR4 however is expressed in more than 90% of patients, and is associated with frequent skin involvement and poor outcome [26]. ATL cells often have characteristic multilobed nuclei leading to the name “flower cells,” which supports the diagnosis in leukemic ATL where patients have circulating clonal T cells; in the modern day, flow cytometry is used [10, 24]. ATL cells have varied and complex genomic changes, and no characteristic translocation contributes to the diagnosis. Detection of HTLV-1 positivity by ELISA and western blot confirmation is essential to the diagnosis [10].
Once a pathologic diagnosis of ATL is made, the disease can have a variable clinical course ranging from an asymptomatic blood test abnormality to a rapidly fatal multisystem disease, and patients should be classified according to the Shimoyama Classification (Table 1) [10]. Shimoyama first proposed classification of ATL into four subtypes based on circulating cell counts, sites of involvement, calcium, and LDH levels; however, there are difficulties in certain cases with intermediate cell counts where the classification can become complex. For example, the acute subtype with bulky lymphadenopathy may behave more clinically like the lymphoma subtype, and the smoldering subtype with only circulating abnormal lymphocytes may overlap with a non-malignant HTLV-1 carrier state [27•]. In fact, a borderline state between healthy carriers and ATL patients has been described [28]. The classification of chronic ATL into favorable and unfavorable groups has also been proposed on the basis of elevated LDH, blood urea nitrogen, and low albumin, which predicts the risk of progression [23].
The variable presentation of ATL requires specific staging studies at diagnosis. ATL has a predilection for extranodal sites of disease, thus a full skin exam should be performed, as well as a skeletal survey for select patients with bone symptoms, fractures, or an unexplained elevation in alkaline phosphatase [29]. Computed tomography scans of the neck, chest, abdomen, and pelvis are used to detect nodal and extranodal lesions, as well as potential opportunistic infections [27•]. PET/CT is used routinely for the initial staging of non-Hodgkin lymphoma and may be useful to stage ATL; however, the significance of a negative interim PET/CT is not yet known. A bone marrow biopsy and aspirate is not always performed if the diagnosis of ATL is established by peripheral blood flow cytometry or nodal mass biopsy. Approximately 10–20% of patients with aggressive ATL will experience CNS progression, thus sampling of CSF and incorporation of intrathecal prophylaxis are necessary in the management of aggressive ATL, even in patients without CNS lesions or symptoms [27•].
Additional laboratory studies may be considered at ATL diagnosis including a baseline assessment for CMV which is useful to monitor for CMV reactivation during therapy [29]. Factors that may predict poor prognosis include poor performance status, elevated LDH level, at least four total involved lesions, hypercalcemia, age greater than 40 years, thrombocytopenia, eosinophilia, bone marrow involvement, high interleukin-5 serum level, CCR4 expression, and presence of lung resistance-related protein, p53 mutation, and/or p16 deletion. Risk models incorporating combinations of these factors have been developed; however, long-term overall survival (OS) in these studies is poor, even in the lowest risk groups [4, 10, 30,31,32,33]. Soluble IL2 receptor (sIL2R) may prognosticate alloSCT outcome [30].
Risk-Adapted Treatment Strategies
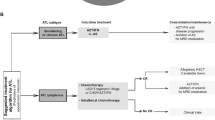
Current treatment regimens for ATL are unsatisfactory and treatment is based on clinical subtype, prognostic factors, and response to initial therapy. International consensus varies as many therapeutic agents used in the treatment of ATL are not available in all countries, and strategies differ across the world. Table 2 summarizes a recommended treatment strategy by ATL subtype; however, it is important to recognize that few large-scale, prospective studies have been performed. All patients should be referred to participate in a prospective clinical trial if available. Outside of a clinical trial, current upfront treatment options for ATL include multi-agent chemotherapy, zidovudine plus interferon α (AZT/IFN), alloSCT, or watchful waiting for indolent subtypes.
Multi-agent chemotherapy options include the following combinations: VCAP-AMP-VECP (vincristine, cyclophosphamide, doxorubicin, and prednisone; doxorubicin, ranimustine, and prednisolone; and vindesine, etoposide, carboplatin, and prednisolone); CHOP (cyclophosphamide, vincristine, doxorubicin, and prednisolone); etoposide, prednisolone, vincristine, cyclophosphamide, doxorubicin (EPOCH); CHOEP (cyclophosphamide, vincristine, doxorubicin, etoposide, and prednisolone); dose-adjusted EPOCH; or hyper CVAD (cyclophosphamide, vincristine, doxorubicin, and dexamethasone, alternating with high-dose methotrexate and cytarabine) [34,35,36,37]. The selection of specific agents or combinations may vary geographically. Outcome, regardless of choice of multi-agent chemotherapy, is frequently poor, particularly for aggressive ATL, with 2–3 year survival ranging from 11 to 31.3% [34, 37]. Patients typically have either primary refractory disease or relapse early. Rarely, long-term responders have been reported [33]. In addition, there is a high incidence of infectious complications, especially opportunistic infection due to the additive effect of disease- and treatment-associated immunosuppression. Several prospective studies have evaluated the role of adding novel agents to combination regimens, including a phase II trial conducted by the AIDS Malignancy Consortium of dose-adjusted EPOCH in combination with bortezomib and antiviral therapy (raltegravir) in aggressive ATL. The overall response rate (ORR) was 67%; however, after a follow-up of over 2 years, the median progression-free survival (PFS) and (OS) were both only 6 months [38].
The activity of AZT/IFN has been reported in a number of small studies and case reports [39,40,41,42]. A meta-analysis of retrospectively studied cases indicated that outcomes with AZT/IFN were more successful in leukemic disease, and the outcome was particularly good in chronic ATL (100% 5-year survival) [12]. In acute ATL, only a third of patients respond; however, those that do may have a better prognosis than with chemotherapy (22% 5-year survival) [12]. While encouraging, this approach will need evaluation in a prospective clinical trial. Though AZT/IFN is not approved for ATL in Japan, it is being studied in a randomized controlled trial compared with chemotherapy. Multi-agent chemotherapy remains the therapy of choice for lymphoma subtype ATL; however, a UK-based study of aggressive ATL concluded chemotherapy followed by concurrent or sequential AZT/IFN prolonged survival for lymphoma subtype patients [43].
For chronic or smoldering ATL, watchful waiting may be appropriate for asymptomatic patients. For patients who are symptomatic (e.g., skin lesions, opportunistic infections), treatment options include participation in a prospective clinical trial, skin-directed therapies, or AZT/IFN [12, 35, 36].
AlloSCT may be the only curative option for aggressive ATL; however, it is not without substantial challenges. In the latest update of the nationwide retrospective study of alloSCT for ATL in Japan, the median OS was 9.9 months and 3-year OS was 36% in 586 transplanted ATL patients [44••]. Although no significant difference in OS between recipients of myeloablative conditioning (MAC) and reduced intensity conditioning (RIC) was observed, there was a trend indicating RIC contributed to better OS in older patients. TRM at 1 year after transplantation was 32.7% for MAC recipients and 29.2% in RIC recipients. Based on donor source, early retrospective analyses report 3-year OS is highest for patients with related HLA-matched donors of bone marrow or peripheral blood stem cells (41%, 95% CI 33–49), followed by those with unrelated bone marrow (39%, 95% CI 29–49) [45]. Single-unit unrelated cord blood transplantation (CBT) carried a relatively low 2-year OS (20%) and high TRM (46.1%) [45]. Grade I/II acute graft versus host disease (GVHD) and limited or extensive chronic GVHD were associated with improved OS, perhaps demonstrating the existence of a graft versus ATL effect [46]. The European Group for Blood and Marrow Transplantation’s (EBMT) Lymphoma Working Party has confirmed alloSCT may also salvage ATL patients in Western countries [47]. For patients with aggressive ATL who achieve an initial response to primary therapy, consolidation with early alloSCT is recommended [35, 36, 48]. Unfortunately, many patients never make it to transplant because of refractoriness to induction therapy, poor performance status, severe immunosuppression, and a low probability of finding a donor [48, 49]. When evaluating donors for ATL patients, HTLV-1 seronegative donors are preferred to avoid the risk of donor-derived ATL [50]. If only an HTLV-1 seropositive related donor is available, the presence of monoclonal bands and HTLV-1 related disease should be excluded [51].
Recent insight into the molecular biology of ATL has provided ideas for novel targets for therapy, and two agents, mogamulizumab, an anti-CCR4 monoclonal antibody, and lenalidomide, an immunomodulatory drug, are approved for ATL in Japan following pivotal trials for relapsed aggressive ATL [52, 53]. In a phase II study by Ishida et al., mogamulizumab had an objective response rate of 50% and a median PFS and OS of 5.2 and 13.7 months, respectively [52]. Toxicities were manageable and included infusion reactions and skin rash [52]. The depletion of T regulatory cells may explain why mogamuluzimab use prior to alloSCT is associated with an increased risk of steroid refractory GVHD and GVHD-related mortality [54, 55]. Mogamulizumab has been also approved in Japan for newly diagnosed aggressive ATL in combination with VCAP-AMP-VECP chemotherapy on the basis of higher response rates compared to intensive chemotherapy alone (52 vs 33%); however, PFS and OS were identical in both arms [56]. A recent phase II study in Europe, the US, and South America of single-agent mogamulizumab versus investigator’s choice of salvage chemotherapy (pralatrexate, gemcitabine/oxaliplatin, and DHAP) for relapsed aggressive ATL revealed a confirmed objective response rate of 15% in the mogamulizumab arm versus 0% in the investigator’s choice [57]. In a phase II study of lenalidomide monotherapy in Japanese patients with relapsed or recurrent aggressive ATL, objective responses were noted in 11 of 26 patients (objective response rate 42%), including four complete responses (CRs) and one unconfirmed CR. The median PFS and OS were 3.8 and 20.3 months, respectively. The most frequent grade ≥ 3 adverse events were neutropenia (65%), leukopenia (38%), lymphopenia (38%), and thrombocytopenia (23%), which were all manageable and reversible [42]. Lenalidomide was not effective in four patients treated with ATL in the USA in a study that closed early due to poor patient accrual [58].
Outside of Japan, there are limited effective therapies for ATL because patients have been excluded or underrepresented in clinical trials of novel agents. Other drugs available for relapsed/refractory peripheral T cell lymphoma (PTCL) have been used for ATL in the USA, including the antifolate agent pralatrexate, histone deacetylase (HDAC) inhibitors belinostat and romidepsin, and the antibody drug conjugate, brentuximab vedotin in CD30+ cases. Much of the data supporting the use of these agents in ATL is from small case series or single-case reports [35, 59, 60]. Arsenic trioxide in combination with IFN- α may be effective, despite significant toxicity [61, 62]. Alemtuzumab may also have single-agent activity. In a phase II study of 29 patients, alemtuzumab resulted in an ORR of 52% with a median duration of response of 14.5 months in responders; however, CMV reactivation was observed in all patients [63]. A summary of recently completed and/or ongoing studies of novel therapies in ATL is reported by Hermine and colleagues, and will hopefully expand available treatment options [64•].
Therapeutic vaccines are in early clinical development and may offer yet another novel mechanism for treating ATL. The Tax peptide-pulsed dendritic cell (Tax-DC) vaccine was designed to augment an HTLV-1 Tax-specific CTL response and has been studied in three previously treated ATL patients [65]. THV02 is a therapeutic vaccine candidate comprised of two lentiviral vectors encoded with a unique polypeptide derived from Tax, HBZ, p12I, and p30II involved in HTLV pathogenesis and recognized by the immune system. Preclinical results demonstrate THV02 can induce an immune response in animal models and, clinical trials in ATL are planned [66].
Conclusion
Since the initial case reports of ATL in Japan and the Caribbean, cases of ATL are now diagnosed across the world. Though recent advances in our understanding of the molecular pathogenesis have improved our knowledge and identified new therapeutic targets, ATL is rarely cured with available therapies. Only those patients with favorable, indolent subtypes experience an extended survival. All other patients should be encouraged to participate in prospective clinical trials and if not available, multi-agent chemotherapy followed by early alloSCT is recommended. AZT/IFN may be effective for leukemic and indolent subtypes without bulky adenopathy but this has not been studied in a prospective, randomized fashion. New agents offer increased response rates; however, it is not yet clear if this translates into an improvement in overall survival. Collaborative research is urgently needed to improve outcomes for patients around the world.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Iwanaga M, Watanabe T, Yamaguchi K. Adult T-cell leukemia: a review of epidemiological evidence. Front Microbiol. 2012;3:322.
Ishitsuka K, Tamura K. Human T-cell leukaemia virus type I and adult T-cell leukemia-lymphoma. Lancet Oncol. 2014;15(11):e517–26.
Yoshida N, Chihara D. Incidence of adult T-cell leukemia/lymphoma in nonendemic areas. Curr Treat Options in Oncol. 2015;16(2):7.
Phillips AA, Shapira I, Willim RD. A critical analysis of prognostic factors in North American patients with human T-cell lymphotropic virus type-1-associated adult T-cell leukemia/lymphoma: a multicenter clinicopathologic experience and new prognostic score. Cancer. 2010;116(14):3438–46.
Zell MI, Assal A, Konda B, Braunschweig I, Derman O, Kornblum N, et al. Analysis of large cohort shows that Caribbean adult T-cell leukemia/lymphoma is a chemotherapy refractory disease with very poor prognosis that behaves distinctly from Japanese subtypes. Blood. 2014;124:1685.
Gessain A, Cassar O. Epidemiological aspects and world distribution on HTLV-1 infection. Front Microbiol. 2012;3:388.
Firouzi S, Farmanbar A, Nakai K, Iwanaga M, Uchimaru K, Utsunomiya A, et al. Clonality of HTLV-1-infected T cells as a risk indicator for development and progression of adult T-cell leukemia. Blood Adv. 2017;1(15):1195–205.
•• Watanabe T. Adult T-cell leukemia: molecular basis for clonal expansion and transformation of HTLV-1-infected T cells. Blood. 2017;129(9):1071–81. This is a comprehensive overview of recently uncovered information on the molecular basis of leukemogenesis in ATL.
•• Kataoka K, Nagata Y, Kitanaka A, Shiraishi Y, Shimamura T, Yasunaga J, et al. Integrated molecular analysis of adult T-cell leukemia/lymphoma. Nat Genet. 2015;47:1304–15. This is an integrated molecular study of 426 ATL cases which identifies alterations in T cell receptor-NFκB signaling, T cell trafficking, and other T cell related pathways which will guide the development of new diagnostics and therapeutics in ATL.
Shimoyama M. Diagnostic criteria and classification of clinical subtypes of adult T-cell leukemia-lymphoma. A report from the Lymphoma Study Group (1984–87). Br J Haemtol. 1991;79(3):428–37.
Yamada Y, Tomonaga M, Fukuda H, Hanada S, Utsunomiya A, Tara M, et al. A new GCSF-supported combination chemotherapy, LSG15, for adult T-cell leukemia-lymphoma: Japan Clinical Oncology Group Study 9303. Br J Haemtol. 2001;113:375–82.
Bazarbachi A, Plumelle Y, Carlos Ramos J, Tortevoye P, Otrock Z, Taylor G, et al. Meta-analysis on the use of zidovudine and interferon- alfa in adult T-cell leukemia/lymphoma showing improved survival in the leukemic subtypes. J Clin Oncol. 2010;28(27):4177–83.
Okamura J, Utsunomiya A, Tanosaki R, Uike N, Sonoda S, Kannagi M, et al. Allogeneic stem-cell transplantation with reduced conditioning intensity as a novel immunotherapy and antiviral therapy for adult T-cell leukemia/lymphoma. Blood. 2005;105:4143–5.
Tanosaki R, Uike N, Utsunomiya A, Saburi Y, Masuda M, Tomonaga M, et al. Allogeneic hematopoietic stem cell transplantation using reduced-intensity conditioning for adult T cell leukemia/lymphoma: impact of antithymocyte globulin on clinical outcome. Biol Blood Marrow Transplant. 2008;14:702–8.
Choi I, Tanosaki R, Uike N, Utsunomiya A, Tomonaga M, Harada M, et al. ATLL allo-HSCT Study Group. Long-term outcomes after hematopoietic SCT for adult T-cell leukemia/lymphoma: results of prospective trials. Bone Marrow Transplant. 2011;46:116–8.
Gallo RC. The discovery of the first human retrovirus: HTLV-1 and HTLV-2. Retrovirology. 2005;2:17.
Matsuoka M. Human T-cell leukemia virus type I and adult T-cell leukemia. Oncogene. 2003;22:5131–40.
Demontis MA, Saidq MT, Golz S, Taylor GP. HTLV-1 viral RNA is detected rarely in plasma of HTLV-1-infected subjects. J Med Virol. 2015;87(12):2130–4.
Melamed A, Laydon DJ, Gillet NA, Tanaka Y, Taylor GT, Bangham CR. Genome-wide determinants of proviral targeting clonal abundance and expression in natural HTLV-1 infection. PLoS Pathog. 2013;9:e1003271.
Gillet NA, Malani N, Melamed A, Gormley N, Carter R, Bentley D, et al. The host genomic environment of the provirus determines the abundance of HTLV-1-infected T-cell clones. Blood. 2011;117(11):3113–22.
Cook LB, Melamed A, Niederer H, Valganon M, Laydon D, Foroni L, et al. The role of HTLV-1 clonality, proviral structure, and genomic integration site in adult T-cell leukemia/lymphoma. Blood. 2014;123:3925–31.
• Bangham CR, Ratner L. How does HTLV-1 cause adult T-cell leukemia/lymphoma (ATL)? Curr Opin Virol. 2015;14:93–100. This reviews the host and viral factors that are implicated in HTLV-1 oncogenesis.
Iwanaga M, Watanabe T, Utsunomiya A, Okayama A, Uchimaru K, Koh KR, et al. Human T-cell leukemia virus type I (HTLV-1) proviral load and disease progression in asymptomatic HTLV-1 carriers: a nationwide prospective study in Japan. Blood. 2010;116:1211–9.
Dahmoush L, Hijazi Y, Barnes E, Stetler-Stevenson M, Abati A. Adult T-cell leukemia/lymphoma: a cytopathologic, immunocytochemical and flow cytometric study. Cancer. 2002;96(2):110–6.
Toulza F, Nosaka K, Takiguchi M, Pagliuca T, Mitsuya H, Tanaka Y, et al. FoxP3+ regulatory T cells are distinct from leukemia cells in HTLV-1 associated adult T-cell leukemia. Int J Cancer. 2009;125(10):2375–82.
Yoshie O, Fujisawa R, Nakayama T, Harasawa H, Tago H, Izawa D, et al. Frequent expression of CCR4 in adult T-cell leukemia and human T-cell leukemia virus type 1- transformed cells. Blood. 2002;99(5):1505–11.
• Bazarbachi A, Suarez F, Fields P, Hermine O. How I treat adult T-cell leukemia/lymphoma. Blood. 2011;118:1736–45. This is a clinical review of the presentation, diagnosis, and treatment approach to ATL.
Wattel E, Vartanian JP, Pannetier C, Wain-Hobson S. Clonal expansion of human T-cell leukemia virus type I-infected cells in asymptomatic and symptomatic carriers without malignancy. J Virol. 1995;69(5):2863–8.
Yared JA, Kimball AS. Optimizing management of patients with adult T-cell leukemia-lymphoma. Cancers. 2015;7:2318–29.
Tokunaga M, Uto H, Takeuchi S, Nakano N, Kubota A, Tokunaga M, et al. Newly identified poor prognostic factors for adult T-cell leukemia-lymphoma treated with allogeneic hematopoietic stem cell transplantation. Leuk Lymphoma. 2017;58(1):37–44.
Tsukasaki K, Hermine O, Bazarbachi A, Ratner L, Ramos JC, Harrington W Jr, et al. Definition, prognostic factors, treatment, and response criteria of adult T-cell leukemia-lymphoma: a proposal from an international consensus meeting. J Clin Oncol. 2009;27(3):453–9.
Suzumiya J, Ohshima K, Tamura K, Karube K, Uike N, Tobinai K, et al. The international prognostic index predicts outcome in aggressive adult T-cell leukemia/lymphoma: analysis of 126 patients from the International Peripheral T-Cell Lymphoma Project. Ann Oncol. 2009;20:715–21.
Fukushima T, Nomura S, Shimoyama M, Shibata T, Imaizumi Y, Moriuchi Y, et al. Japan Clinical Oncology Group (JCOG) prognostic index and characterization of long-term survivors of aggressive adult T-cell leukaemia-lymphoma (JCOG0902A). Br J Haematol. 2014;166:739–48.
Yamada Y, Tomonaga M, Fukuda H, Hanada S, Utsunomiya A, Tara M, et al. A new G-CSF-supported combination chemotherapy, LSG15, for adult T-cell leukaemia-lymphoma: Japan Clinical Oncology Group Study 9303. Br J Haematol. 2001;113(2):375–82.
Zelenetz AD, Abramson JS, Advani RH, Andreadis CB, Byrd JC, Czuczman MS, et al. Clinical practice guidelines in oncology: non-Hodgkin’s lymphomas. J Natl Compr Cancer Netw. 2010 Mar;8(3):288–334.
Dearden CE, Johnson R, Pettengell R, Devereux S, Cwynarski K, Whittaker S, et al. Guidelines for the management of mature T-cell and NK-cell neoplasms (excluding cutaneous T-cell lymphoma). Br J Haematol. 2011 May;153(4):451–85.
Tsukasaki K, Utsunomiya A, Fukuda H, Shibata T, Fukushima T, Takatsuka Y, et al. VCAP-AMP-VECP compared with biweekly CHOP for adult T-cell leukemia-lymphoma: Japan Clinical Oncology Group Study JCOG9801. J Clin Oncol. 2007 Dec 1;25(34):5458–64.
Ratner L, Rauch D, Abel H, Caruso B, Noy A, Barta SK, et al. Dose-adjusted EPOCH chemotherapy with bortezomib and raltegravir for human T-cell leukemia virus-associated adult T-cell leukemia lymphoma. Blood Cancer J. 2016;6:e408.
Gill PS, Harrington W Jr, Kaplan MH, Ribeiro RC, Bennett JM, Liebman HA, et al. Treatment of adult T-cell leukemia-lymphoma with zidovudine and interferon alfa. N Engl J Med. 1995;332:1744–8.
Hermine O, Bouscary D, Gessain A, Turlure P, Leblond V, Franck N, et al. Brief report. Treatment of adult T-cell leukemia-lymphoma with zidovudine and interferon alfa. N Engl J Med. 1995;332:1749–51.
Matutes E, Taylor GP, Cavenagh J, Pagliuca A, Bareford D, Domingo A, et al. Interferon alpha and zidovudine therapy in adult T-cell leukemia lymphoma: response and outcome in 15 patients. Br J Haemtol. 2001;113:779–84.
Ramos JC, Ruiz P Jr, Ratner L, et al. IRF4- and c-Rel expression in antiviral-resistant adult T-cell leukemia/lymphoma. Blood. 2007;109:3060–8.
Hodson A, Crichton S, Montoto S, Mir N, Matutes E, Cwynarski K, et al. Use of zidovudine and interferon alfa with chemotherapy improves survival in both acute and lymphoma subtypes of adult T-cell leukemia/lymphoma. J Clin Oncol. 2001;29(35):4696–701.
•• Ishida T, Hishizawa M, Kato K, Tanosaki R, Fukuda T, Taniguchi S, et al. Allogeneic hematopoietic stem cell transplantation for adult T-cell leukemia-lymphoma with special emphasis on preconditioning regimen: a nationwide retrospective study. Blood. 2012;120(8):1734–41. This is the largest study of ATL patients receiving hematopoietic stem cell transplantation
Hishizawa M, Kanda J, Utsunomiya A, Taniguchi S, Eto T, Moriuchi Y, et al. Transplantation of allogeneic stem cells for adult T-cell leukemia: a nationwide retrospective study. Blood. 2011;116:1369–76.
Kurata M, Yanagisawa A, Atsuta Y, Sakamaki H, Kato K, Ichinohe T, et al. Hematopoietic cell transplantation in Japan, Clinical blood. 2014;55(12):2381–2399, https://doi.org/10.11406/rinketsu.55.2381, https://www.jstage.jst.go.jp/article/rinketsu/55/12/55_2381/_article/-char/ja
Bazarbachi A, Cwynarski K, Boumendil A, Finel H, Fields P, Raj K, et al. Outcome of patients with HTLV-1-associated adult T-cell leukemia/lymphoma after SCT: a retrospective study by the EBMT LWP. Bone Marrow Transplant. 2014;49(10):1266–8.
Phillips AA. Hematopoeitic stem cell transplantation for adult T cell leukemia-lymphoma: who is the best candidate? New 46 Leuk Lymphoma. 2017;58(1):1–3.
Marcais A, Suarez F, Sibon D, Bazarbachi A, Hermine O. Clinical trials of adult T-cell leukaemia/lymphoma treatment. Leuk Res Treatment. 2012;2012:932175. 8 pages
Tamaki H, Matsuoka M. Donor-derived T-cell leukemia after bone marrow transplantation. N Engl J Med. 2006;354:1758–9.
Okayama A, Stuver S, Matsuoka M, Ishizaki J, Tanaka GI, Kubuki Y, et al. Role of HTLV-1 proviral DNA load and clonality in the development of adult T-cell leukemia/lymphoma in asymptomatic carriers. Int J Cancer. 2004;110:621–5.
Ishida T, Joh T, Uike N, Yamamoto K, Utsunomiya A, Yoshida S, et al. Defucosylated anti-CCR4 monoclonal antibody (KW-0761) for relapsed adult T-cell leukemia-lymphoma: a multicenter phase II study. J Clin Oncol. 2012;30(8):837–42.
Ishida T, Fujiwara H, Nosaka K, Taira N, Abe Y, Imaizumi Y, et al. Multicenter phase II study of lenalidomide in relapsed or recurrent adult T-cell leukemia/lymphoma: ATLL-002. J Clin Oncol. 2016;34:4086–93.
Fuji S, Inoue Y, Utsunomiya A, Moriuchi Y, Uchimaru K, Choi I, et al. Pretransplantation anti-CCR4 antibody mogamulizumab against adult T-cell leukemia/lymphoma is associated with significantly increase risks of severe and corticosteroid-refractory graft-versus-host disease, nonrelapse mortality, and overall mortality. J Clin Oncol. 2016;34:3426–33.
Sugio T, Kato K, Aoki T, Ohta T, Saito N, Yoshida S, et al. Mogamulizimab treatment prior to allogeneic hematopoetic stem cell transplantation induces severe acute graft-versus-host disease. Biol Blood Marrow Transplant. 2016;22:1608–14.
Ishida T, Jo T, Takemoto S, Suzushima H, Uozumi K, Yamamoto K, et al. Dose-intensified chemotherapy alone or in combination with mogamulizumab in newly diagnosed aggressive adult T-cell leukaemia-lymphoma: a randomized phase II study. Br J Haematol. 2015;169:672–82.
Phillips AA, Fields P, Hermine O, Ramos JC, Beltran BE, Pereira P, et al. A prospective, multicenter, randomized study of anti-CCR4 monoclonal antibody mogamulizumab (moga) vs investigator's choice (IC) in the treatment of patients (pts) with relapsed/refractory (R/R) adult T-cell leukemia-lymphoma (ATL). J Clin Oncol. 2016;34:abstr 7501.
Phillips AA, Giddings J, Horwitz SM. A phase II study of lenalidomide in patients with relapsed or refractory ATLL. Int J Blood Res Disorders. 2015;2:010.
Mukhi N, Verma V, Ahmed A, et al. Romidepsin in relapsed/refratcory HTLV-1 associated adult T-cell lymphoma/leukemia: a case series. Blood. 2015;126:5113.
Lunning MA, Gonsky J, Ruan J, Phillips AA, Pulitzer M, Moskowitz A, et al. Pralatrexate in relapsed/refractory HTLV-1 associated adult T-cell lymphoma/leukemia: a New York City multi-institutional experience. Blood. 2012;120:2735.
Hermine O, Dombret H, Poupon J, Arnulf B, Lefrère F, Rousselot P, et al. Phase II trial of arsenic trioxide and alpha interferon in patients with relapsed/refractory adult T-cell leukemia/lymphoma. Hematol J. 2004;5:130–4.
Ishitsuka K, Suzumiya J, Aoki M, Ogata K, Hara S, Tamura K. Therapeutic potential of arsenic trioxide with or without interferon-alpha for relapsed adult T-cell leukemia/lymphoma. Haematologica. 2007;92:719–20.
Sharma K, Janik JE, O’Mahony D, et al. Phase II study of alemtuzumab (CAMPATH-1) in patients with HTLV-1-associated adult T-cell leukemia/lymphoma. Clin Cancer Res. 2017;23:35–42.
• Hermine O, Ramos JC, Tobinai K. A review of new findings in adult T-cell leukemia-lymphoma: a focus on current and emerging treatment strategies. Adv Ther. 2018;35:135–52. A summary of recently completed and/or ongoing studies of novel therapies in ATL.
Suehiro Y, Hasegawa A, Iino T, Sasada A, Watanabe N, Matsuoka M, et al. Clinical outcomes of a novel therapeutic vaccine with Tax peptide-pulsed dendritic cells for adult T cell leukaemia/lymphoma in a pilot study. Br J Haematol. 2015;169(3):356–67. https://doi.org/10.1111/bjh.13302.
Revaud D, Bejanariu A, Loussaief L, et al. Development of an anti-HTLV-1 vaccine for the treatment of adult T-cell leukemia/lymphoma. Presented at: 57th Annual Meeting and Exposition of the American Society of Hemtology, December 5–8, 2015, Orlando, FL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Adrienne Phillips is a principal investigator for a prospective, multicenter, randomized study of anti-CCR4 monoclonal antibody mogamulizumab (moga) vs investigator’s choice (IC) in the treatment of patients (pts) with relapsed/refractory (R/R) adult T cell leukemia-lymphoma (ATL) and received research funding. Janine Harewood declares that she has no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on T-Cell and Other Lymphoproliferative Malignancies
Rights and permissions
About this article

Cite this article
Phillips, A.A., Harewood, J.C.K. Adult T Cell Leukemia-Lymphoma (ATL): State of the Art. Curr Hematol Malig Rep 13, 300–307 (2018). https://doi.org/10.1007/s11899-018-0458-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11899-018-0458-6