
Abstract
Recognition of the molecular heterogeneity of colorectal cancer (CRC) has led to the classification of CRC based on a variety of clinical and molecular characteristics. Although the clinical significance of the majority of these molecular alterations is still being ascertained, it is widely anticipated that these characteristics will improve the accuracy of our ability to determine the prognosis and therapeutic response of CRC patients. A few of these markers, such as microsatellite instability and the CpG island methylator phenotype (CIMP), show promise as predictive markers for cytotoxic chemotherapy. KRAS is a validated biomarker for epidermal growth factor receptor (EGFR)-targeted therapy, while NRAS and PI3KCA are evolving markers for targeted therapies. Multiple new actionable drug targets and potential response biomarkers are being identified on a regular basis, but most are not ready for clinical use at this time. This review focuses on key molecular features of CRCs and the application of these molecular alterations as predictive biomarkers for CRC.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Historically, the use of surgery, chemotherapy, and/or radiation therapy for colorectal cancer (CRC) has been guided by the TNM stage alone [1]. However, it is well known that the TNM stage is, at best, a modestly accurate predictor for response to treatment. It has been increasingly recognized that there are tumor molecular characteristics that more accurately guide the management of CRC patients by improving our ability to identify individuals at high risk of disease recurrence and to determine therapies that will be most effective in specific patients. This review focuses on recent advances in the discovery and validation of predictive markers that can be used to guide the treatment of CRC patients. We will discuss both validated and emerging biomarkers for conventional cytotoxic chemotherapy as well as for targeted therapies, such as anti-EGFR therapy. Mutant KRAS has served as the paradigm for predictive biomarkers for anti-EGFR therapy. However, the complexity of the molecular genetics and epigenetics of CRC and of the molecular biology of signaling pathways has revealed that the development and validation of other biomarkers for targeted therapy will be challenging. Many potential candidate genes for targeted therapy have been recently identified, creating opportunities for companion predictive marker assays. Predictive gene signature platforms are also under active investigation. In just the last few years, this area has greatly expanded as researchers and clinicians attempt to develop CRC treatments that will be optimally effective for specific patients.
Molecular Characterization
The identification of specific chromosomal abnormalities and gene mutations in CRC over 30 years ago provided the first glimpse into the potential for these molecular alterations to be used to guide therapy for CRC. Since that time, tremendous advances have been made in our understanding of the molecular pathology of CRC. There are three commonly recognized molecular subclasses of CRC that are characterized by their forms of genomic and epigenomic instability: chromosome unstable (CIN; also referred to as microsatellite stable (MSS) CRC), microsatellite unstable (MSI), and the CpG island methylator phenotype (CIMP) [2]. Although this classification scheme is believed to oversimplify the molecular complexity of CRCs, it has been shown that CIN, MSI, and CIMP CRCs each have characteristic clinical and molecular phenotypes.
Recent Advances in the Molecular Characterization of CRC
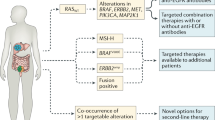
The Cancer Genome Atlas (TCGA) Network performed a genome-scale analysis of 276 CRC samples, noting heterogeneity in the gene expression signatures and mutation profiles of the different individuals’ tumors [3]. Of those that underwent whole genome sequencing, 16 % were found to be hypermutated, which meant that they had a substantially higher density of sequence mutations compared to the other CRCs. The vast majority of these hypermutated cases were also MSI and/or CIMP, although a previously unrecognized class of hypermutable CRCs was also observed. In addition to the common driver genes already known to occur in CRC (e.g., APC, KRAS, TP53), the non-hypermutated CRCs had a variety of gene mutations in other genes, including 24 that occurred with a reasonably high frequency. The findings from the TCGA and others have led multiple groups to develop new classification schemes that are more accurate and consistent than our current scheme based on CIN, MSI, and CIMP [4].
Gene Expression Signature Assays and Mutation Signature Assays
With the advent of next-generation sequencing and improving high-throughput technologies, gene signature assays have been developed for CRC. Two assays have been validated and approved for clinical use: Oncotype DX (Genomic Health, Inc.) and ColoPrint (Agendia) [5, 6]. These expression signature assay platforms appear to be best used for prognosis, rather than prediction of chemotherapy benefit [7, 8]. Multiple other assays, such as UW-OncoPlex (University of Washington), attempt to more broadly assay somatic alterations to identify clinically actionable and emerging somatic mutations [9]. The uptake of such assays into the clinic varies widely, but is likely to become a standard-of-care instrument in the future.
Cytotoxic Chemotherapy
Currently, CRC is predominantly managed with surgical resection for early stage CRC and with a combination of surgery and chemotherapy for advanced disease. 5-Fluorouracil (5-FU) is the backbone cytotoxic agent used in most regimens. While originally given as a single agent, 5-FU is now most commonly administered in combination with oxaliplatin (FOLFOX) for adjuvant therapy for stage III (and high-risk stage II) CRC and with either oxaliplatin (FOLFOX) or irinotecan (FOLFIRI) for metastatic CRC [10–13]. Consequently, predictive biomarkers for CRC have initially focused on these conventional cytotoxic chemotherapy agents.
Established Molecular Markers
There has been considerable interest in identifying predictive molecular markers for chemotherapy effect both in the adjuvant and metastatic settings. Candidate biomarkers that have been heavily scrutinized include mutant TP53, thymidylate synthase (TS) expression, and amplified ERCC1, among others [14–16]. Unfortunately, none of these markers has been established as a predictive marker that is ready to be used clinically. However, despite the lack of success so far, recent data has shown that MSI is a robust prognostic marker for survival and is also a promising marker for 5-FU responsiveness [17, 18].
Microsatellite Instability
MSI was discovered in the early 1990s during studies to identify potential tumor suppressor genes in CRC [17]. MSI is recognized by the presence of a high frequency of frameshift mutations in microsatellite DNA and results from inactivation of the DNA mismatch repair (MMR) system. A portion of MSI tumors are due to germline mutations in one of the MMR genes, MLH1, MSH2, MSH6, or PMS2, which cause Lynch syndrome. The majority (80 %) of MSI cases is sporadic and is due to hypermethylation of the MLH1 gene promoter [17, 19]. This is often associated with mutant BRAF V600E and CIMP [20].
A large number of studies have demonstrated that individuals with proximal MSI CRCs have a better prognosis than stage-matched MSS CRCs, particularly when no adjuvant therapy is given [18]. More recently, MSI has also been shown to predict lack of benefit of adjuvant 5-FU in stage II–III colon cancer patients (and possible harm in stage II patients) [21–23]. However, its role as a predictive marker with modern combination chemotherapy regimens, such as FOLFOX and FOLFIRI, is uncertain [24, 25]. This uncertainty is a consequence of differences in the studies’ chemotherapy regimens and assays for MSI, which make it difficult to compare studies and develop a robust and generalizable conclusion about the role of MSI as a predictive marker for conventional chemotherapy.
There is continued investigation of the use of MSI to predict response to chemotherapy for modern combination regimens. A recently published retrospective analysis of stage III patients treated with adjuvant FOLFOX in the N0147 trial demonstrated a predictive value for MSI [22]. This marked difference in chemotherapy response prediction capability between single-agent 5-FU and combination regimens may be partly due to variability in multivariate models, as newer models often account for KRAS and BRAF status [26]. The ability of MSI to predict response to oxaliplatin may reflect effects unrelated to inactivation of the MMR system, as was concluded by investigators in the NSABP-07 trial. The benefit from oxaliplatin in individuals with MSS tumors may attenuate the beneficial effect of MSI status on survival [27]. Although MSI was retrospectively shown to predict improved disease-free survival (DFS) with adjuvant irinotecan and 5-FU (IFL regimen) in the CALGB (Alliance) 89803 trial, MSI has not consistently served as a predictor of benefit from combination chemotherapy with 5-FU and irinotecan [28–30].
Interestingly, the underlying cause of MSI may affect whether MSI is predictive for chemotherapy responsiveness. In a retrospective analysis of stage II and III colon cancer patients that received adjuvant 5-FU or placebo, Sinicrope et al. compared individuals with MSI CRCs secondary to germline mutations (i.e., Lynch syndrome) to those with sporadic MSI tumors [31]. Individuals with germline mutations had improved DFS after 5-FU adjuvant chemotherapy, while patients with sporadic MSI CRCs did not receive benefit (p = 0.006). These findings suggest that recognizing both molecular features and their etiology is important for determining the utility of predictive biomarkers. Additional studies are needed to clarify whether the disparate MSI effect in single-agent 5-FU vs. in modern multi-agent therapies is also related to the etiology of MSI. Thus, MSI is largely accepted now as a prognostic marker, but its role as a predictive biomarker is more controversial, which has led to its continued scrutiny (Table 1).
Emerging Markers
CIMP
CIMP is found in 5–40 % of CRCs and is defined by an exceptionally high frequency of aberrantly methylated CpG dinucleotides in the genome [45]. In normal cells, the majority of DNA is methylated at CpG dinucleotides except in CG-rich regions (“CpG islands”). These CpG islands are found in approximately half of gene promoters and are usually unmethylated in normal cells. However, in the majority of CRCs, many of the CpG islands become aberrantly methylated. CIMP CRCs are distinct from other CRCs because they have an exceptionally high frequency of methylated CpG islands. CIMP is thought to promote carcinogenesis by silencing tumor suppressor genes via methylation-mediated transcriptional repression [2].
Since the initial discovery of CIMP in 1999, a number of assay panels have been developed to determine the CIMP status of CRCs. Unfortunately, there has been no consensus regarding which panel to use, which has, in turn, slowed the investigation and development of CIMP as a predictive biomarker [45]. Despite the lack of a “gold-standard” CIMP assay, there does appear to be an association between CIMP and poor prognosis and, albeit less consistently, between CIMP and a favorable response to 5-FU [45] (Table 2). Recently, in an exploratory retrospective analysis of a randomized trial of 5-FU/leucovorin (5-FU/LV) alone vs. with irinotecan (IFL regimen) in stage III colon cancer patients (CALGB/Alliance 89803), Shiovitz, et al. found that CIMP-positive individuals had worse overall survival [46•, 59]. These authors also found on sub-group analysis that patients with CIMP-positive colon cancers that were also MMR-intact (MMR-I) did better with IFL than with 5-FU/LV alone with a 5-year overall survival (OS) of 66 vs. 46 %, respectively. Furthermore, in this updated data analysis from this trial, CIMP was a stronger prognostic feature than MSI [28, 46•]. Other studies assessing the use of CIMP to predict response to FOLFOX therapy did not observe CIMP to be a significant predictor [60]. At this time, CIMP appears to have a strong potential to be a prognostic marker for CRC, but its use as a predictive marker for conventional chemotherapy will require further investigation.
BRAF
There is substantial evidence from the retrospective analysis of cohort and randomized clinical trials that mutant BRAF is a marker of poor prognosis [26, 36]. There is also some data to suggest that individuals with BRAF-mutant metastatic CRC may benefit from aggressive first-line therapy (Table 1). When the chemotherapy triplet regimen of 5-FU, oxaliplatin, and irinotecan (FOLFOXIRI) was compared to FOLFIRI, significantly improved response rate (RR; 60 vs. 34 %, respectively), progression-free survival (PFS; 9.8 vs. 6.9 months), and OS (22.6 vs. 16.7 months) were noted in unselected stage IV patients [32]. The clinical importance of these results is not clear as this regimen is not standard-of-care. Also, of note are results from the analysis of a small phase 2 trial suggesting improved PFS with the use of FOLFOXIRI + bevacizumab in patients with metastatic BRAF-mutant CRCs [42•]. In these patients, median PFS was 9.2 months and OS was 24.1 months. For approximate comparison, median PFS for BRAF-mutant CRCs was 5.6 months with FOLFIRI and 8.0 months with FOLFIRI + cetuximab [37]. However, in the MRC FOCUS trial of advanced CRC, mutant BRAF was not shown to be a significant predictive marker for any 5-FU-based chemotherapy regimen (5-FU/LV, 5-FU + oxaliplatin, or 5-FU + irinotecan) [61]. Thus, BRAF is best considered as an emerging marker for predicting response to conventional chemotherapy, which is still in need of further assessment.
Anti-angiogenic Therapy
In the decade since the FDA approval of the vascular endothelial growth factor (VEGF)-targeted agent bevacizumab for CRC, multiple studies have demonstrated superiority of bevacizumab-based chemotherapy to cytotoxic chemotherapy alone for the treatment of metastatic CRC [62, 63]. Notably, bevacizumab-related hypertension occurs more often in individuals who respond to treatment [64, 65]. Consequently, hypertension is considered a pharmacodynamic biomarker, but lacks the predictive capability needed to guide the selection of optimized individual therapeutic regimens. Overall, to date, no reliable biomarkers for prediction of response to anti-angiogenic therapy have been found, although there are some promising potential biomarkers, which will be discussed below.
Multiple retrospective studies have attempted to identify predictive markers of bevacizumab response in CRC [66]. Recently, retrospective studies have suggested an association between carcinoembryonic antigen (CEA) levels and response to bevacizumab-based chemotherapy. Lower baseline CEA levels suggest a higher likelihood of treatment response and longer median PFS and OS [67•, 68]. Prager et al. assessed baseline CEA levels in a cohort of patients receiving first-line therapy with chemotherapy (FOLFOX, XELOX, FOLFIRI, or XELIRI) and bevacizumab and compared them to patients who received a cetuximab-based regimen. The CEA classification of high vs. low was somewhat arbitrarily derived as above or below the study median value, questioning the use of this threshold in other populations, but the results were validated in an independent cohort. Arguing against the use of CEA, a similar unrelated retrospective analysis did not observe that CEA predicted response to either bevacizumab or cediranib [69•]. Additional suggested predictive biomarkers for bevacizumab treatment response and/or efficacy include: level of tumor VEGF-A expression, germline genetic variations in pericyte maturation genes, phosphatase and tensin homolog (PTEN) loss, and levels of phosphorylated AMP-activated protein kinase (pAMPK) [47, 70–73]. However, despite the identification of a number of plausible predictive markers for responsiveness to bevacizumab, none of these markers has been validated as a clinically useful biomarker. Baseline serum VEGF and CEA levels appear to have the highest potential to be used as predictive markers to date, but will require additional studies using stringent thresholds and validated assays before they are ready to be used clinically [68, 74].
Similar to the situation for bevacizumab the newer anti-angiogenic agents regorafenib and aflibercept lack predictive markers [75]. Thus, with a deficiency of predictive biomarkers, the selection of anti-angiogenic therapy should be guided by other factors at this time, including potential side effects and tumor molecular features that predict benefit or harm from other agents.
EGFR-Targeted Therapy
Two targeted anti-epidermal growth factor receptor (EGFR) monoclonal antibodies have been approved in combination with chemotherapy for the treatment of metastatic CRC. While rare reports note EGFR mutations that preferentially affect cetuximab vs. panitumumab responsiveness [76•], generally, these agents are thought to have equivalent efficacy [77]. The addition of panitumumab in the first line in unselected stage IV patients showed a 1.4-month increase in PFS compared to FOLFOX alone (PRIME trial [38•]), while cetuximab combined with FOLFIRI in the CRYSTAL trial added a 0.9-month PFS benefit [33]. The OPUS study, which combined cetuximab and FOLFOX in unselected metastatic patients, failed to demonstrate a survival benefit [34].
Established predictive markers
KRAS exon 2
Shortly after the clinical benefit of cetuximab and panitumumab was shown in patients with metastatic CRC, it was noted that anti-EGFR therapy efficacy was confined to individuals with KRAS wild-type tumors, with no response or even harm to patients with KRAS-mutant tumors [78, 79]. For example, stratified analysis in the OPUS study noted improved survival in individuals with KRAS wild-type CRCs (7.7 vs. 7.2 months in cetuximab + FOLFOX vs. FOLFOX alone, p = 0.02), but worse survival in individuals with KRAS-mutant tumors (5.5 vs. 8.6 months, p = 0.02) [34]. Importantly, this original definition of mutation status was restricted to KRAS exon 2, and codons 12 and 13. Interestingly, EGFR expression does not appear to correlate with response to either cetuximab or panitumumab [80, 81]. KRAS mutation status is now clearly established as the first clinically useful biomarker to predict response to anti-EGFR therapy.
Expanded RAS testing
Multiple retrospective studies have now shown that KRAS testing restricted to exon 2 misses cases resistant to anti-EGFR therapy. “Expanded RAS” testing is now advocated, which also tests exons 3 (codon 61) and 4 (codons 117, 146) of KRAS and exons 2–4 of NRAS [1]. In the PRIME trial, 17 % of patients with CRCs that were wild type for KRAS exon 2 were found to have a mutation in another RAS exon: 4 % KRAS exon 3, 6 % KRAS exon 4, 3 % NRAS exon 2, and 4 % NRAS exon 3 (and 0 % NRAS exon 4) [40••]. While the numbers were small, any RAS mutation was associated with a statistically significant worse PFS (hazard ratio (HR) 1.31, 95 % confidence interval (CI) = 1.07–1.60). Those patients with no RAS mutations detected using the expanded RAS protocol (“expanded wild type”) were most likely to receive benefit (HR 0.72, 95 % CI = 1.04–1.62). Similarly, in the FIRE-3 study, 16 % of individuals who were wild type at KRAS exon 2 were found to have a RAS mutation by expanded RAS testing [43••]. Comparing FOLFIRI + cetuximab to FOLFIRI + bevacizumab, PFS in the expanded wild-type population was 10.4 vs. 10.2 months (p = 0.54) and OS was 33.1 vs. 25.6 months (p = 0.011), respectively. These absolute differences were greater than when evaluating the entire intent-to-treat population, which included a more limited assessment of KRAS mutation status.
Anti-EGFR therapy has been evaluated beyond its use with doublet chemotherapy in metastatic CRC. Expansion of anti-EGFR therapy to adjuvant therapy in non-metastatic CRC has failed to show a benefit, even within KRAS wild-type CRC [82, 83•]. This suggests a difference in tumor biology in stage III as compared to stage IV patients. The application of anti-EGFR therapy is also being explored with triplet therapy for metastatic CRC. There is phase 2 clinical trial data that first-line panitumumab + FOLFOXIRI is efficacious in patients with wild type KRAS, HRAS, NRAS, and BRAF with a median PFS of 11.3 months [44].
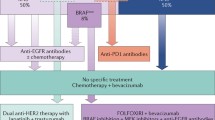
Based on the above information, the National Comprehensive Cancer Network (NCCN) currently advocates for full testing of KRAS and NRAS in all cases of metastatic CRC to guide therapy selection [1] (Table 1). KRAS, NRAS, and BRAF mutations appear to be mutually exclusive [39]. Retrospective studies suggest that the clinical outcomes predicted by KRAS mutations are irrespective of the mutated codon [84•, 85]. It remains controversial whether patients with KRAS G13D mutations might gain benefit from anti-EGFR therapy; the apparent responsiveness of CRCs with KRAS G13D mutations may be an effect of codon 13 mutations reflecting a worst prognosis in early stage and metastatic CRC [86, 87]. Currently, the role of EGFR pathway testing in non-metastatic CRC is not established since anti-EGFR therapy has failed to show efficacy in stage II or III CRC patients. As BRAF lacks the predictive value for anti-EGFR therapy, BRAF testing is currently considered optional for KRAS/NRAS wild-type tumors [1, 37]. Given the evolving knowledge about the RAS pathway, clinical trials that include anti-EGFR therapy should be critically evaluated in the context of their inclusion/exclusion criteria and RAS testing strategies.
Emerging Predictive Markers
PI3K/AKT/PTEN
Distinct from the RAS/RAF pathway, members of the phosphoinositide-3-kinase (PI3K), pathway, PIK3CA, AKT, and PTEN also are downstream effectors of EGFR signaling. Mutations in these genes may also affect anti-EGFR therapy responsiveness. In retrospective studies, patients with CRCs that have PIK3CA exon 20 (kinase domain) mutations have much worse outcomes with cetuximab compared to patients with PIK3CA wild-type CRCs [39]. PIK3CA exon 9 (helical domain) mutations do not seem to serve as a predictive marker for anti-EGFR therapy, which highlights the complexity of the effects of the specific mutations on the functions of the altered kinases. Of interest, PIK3CA exon 9 mutations are often seen with KRAS mutations and KRAS remains the stronger predictive biomarker. Collectively, approximately 70–80 % of cases unresponsive to EGFR-targeted therapy appear to be secondary to mutations in KRAS, NRAS, PI3KCA, and other proteins relevant to EGFR signaling [39, 53•, 57].
PIK3CA mutations may also be beneficial in guiding secondary prevention options. Large studies have demonstrated that aspirin reduces adenoma and CRC formation [88]. More recent observational data suggests that the aspirin benefit is limited to individuals with PIK3CA-mutant tumors [48••]. The use of aspirin in secondary prevention remains a complex issue because of the risks of aspirin use; further study is required.
TP53
TP53 is not in the EGFR pathway or traditionally thought of as a predictive biomarker for EGFR-targeted therapy. However, in the phase 2 EXPERT-C trial, which added cetuximab in the adjuvant setting for high-risk stage II rectal cancer patients, exploratory retrospective analysis suggested TP53 mutation status did predict benefit from cetuximab [89•, 90]. Patients with TP53 wild-type tumors who received cetuximab in addition to adjuvant capecitabine and oxaliplatin (CAPOX) had improved PFS and OS compared to CAPOX alone (Table 2). However, among individuals with TP53-mutant tumors, there was no difference in survival with the addition of cetuximab. Of note, only 60 % of patients were wild type for KRAS (exons 2 and 3) and BRAF, but the interaction between TP53 and treatment arm remained significant when adjusting for KRAS mutation status. At this time, TP53 mutation status is not routinely assessed in rectal cancer patients.
Emerging Mechanism-Driven Therapy
Several hallmark cellular changes necessary for carcinogenesis are now recognized, including the induction of angiogenesis, evading growth suppressors, and activation of invasion and metastasis [91] The development of newer agents for CRC has focused on key pathways in CRC that mediate these hallmark behaviors of cancer cells [92], with the aim of counteracting the cellular signaling changes that result from driver gene mutations in these central signaling pathways. Beyond the EGFR pathway, none of these agents are currently ready for clinical use in patients with CRC. However, several agents are on the horizon and show promise for future implementation. Ideally, these agents will have companion diagnostic assays that will be used to select individuals who have the highest likelihood of response.
PI3K Inhibitors
PI3K pathway inhibitors have been developed, but their role in the treatment of CRC is currently unclear. Newer investigational agents have shown some promise in the preclinical setting for PI3KCA-mutant tumors [49, 50]. However, once applied to the clinical setting, PI3K inhibitors have largely failed to show significant clinical benefit in CRC, despite the hypothesis that they would be effective given the high frequency of mutations in PIK3CA, PTEN, and other PI3K signaling pathway members in CRC [51, 52]. Further study of these agents is likely to focus on the efficacy of this class of agents in combination with targeted therapies, as guided by tumor molecular characteristics, such as concurrent KRAS and PIK3CA mutations [93].
MEK inhibitors
MEK inhibitors have been evaluated as MEK is downstream of EGFR in the RAS/RAF pathway. While responses were seen in melanoma and lung cancer, the pure MEK inhibitor RO4987655 failed to show benefit in KRAS-mutant CRC [94]. Despite the lack of efficacy of RO4987655, selumetinib (AZD6244) is a selective MEK-1/2 inhibitor that has shown in vitro and in vivo activity in CRC. It is currently being evaluated for the treatment of metastatic CRC in early phase clinical trials [55]. Selective MEK-1/2 inhibitors have shown in vitro efficacy in modulating sensitivity to cetuximab [56]. It remains to be seen whether MEK inhibitors will make it through multi-phase clinical trials and into clinical practice and whether specific biomarkers will be useful in selecting patients eligible for treatment.
Dual Pathway Blockade
Overcoming resistance to anti-EGFR therapies has been one focus of dual-targeted therapy trials. Amplification of the MET gene and increased expression of MET has been associated with both primary and secondary (acquired) resistance to cetuximab/panitumumab, distinct from the effect of mutations in KRAS, NRAS, BRAF, and PIK3CA [53•]. MJ-56 has shown in vitro efficacy in human CRC cell lines, attenuating signaling of EGFR and c-MET [54]. HER2 amplification as a potential resistance mechanism also has been identified in KRAS/NRAS/BRAF/PIK3CA wild-type (“quadruple negative”) tumors [57]. In preclinical studies, dual blockade of EGFR and HER2 in HER2-amplified colorectal xenografts was more effective than either agent alone suggesting that this regimen may be effective in patients with CRCs that have amplified HER2.
Some studies have used more than one targeted therapy or have used a multiple pathway agent in an attempt to block multiple key growth pathways in the tumor cell. Following the recognition of the frequency of BRAF V600E mutations in melanoma, BRAF inhibitors were developed and implemented in clinical practice [95]. However, the success seen in the treatment of melanoma with these agents was not paralleled in CRC [58]. This has led to the use of BRAF inhibitors in combination with other agents in early phase dual agent trials. Dual blockade of the MEK and PI3K/mTOR pathways in mouse CRC xenografts improved results over monotherapy, but did not induce tumor regression [96]. Although based on solid conceptual grounds as a way to overcome resistance to single targeted therapy regimens, the use of multiple pathway inhibitors has had suboptimal results when applied to the clinical setting, often increasing toxicity but not improving treatment outcomes [97••, 98]. This observation has led to the consideration of a strategy involving targeted therapy and cytotoxic chemotherapy, which may cause better tumor response. Along this vein, there is early preclinical data that the proteasome inhibitor carfilzomib may be synergistic with irinotecan in CRC cell lines through effects mediated on the MEK/ERK and PI3K/AKT pathways [99]. Thus, new therapeutic targets for CRC continue to be explored and optimized (Table 2), but are not ready for clinical practice.
Conclusions
The next era of CRC treatment trials involves focusing on tumor genetic signatures, rather than single-gene alterations. This includes identification of new targets, combination targeted therapies, and possibly immune modulation. There is clear lack of benefit from EGFR-targeted therapies in individuals with metastatic CRC harboring KRAS exon 2 mutations. In addition, mutations in KRAS exons 3 and 4, NRAS exons 2–4, PIK3CA exon 20, and possibly TP53 are predictive of lack of benefit. No clinically useful predictive biomarkers for anti-angiogenic therapy have been identified. Inhibitors of PI3K, MEK, MET, HER2, and other newer targets are under active investigation but are not yet ready for application to clinical practice. The increasing complexity of mutation profiling for therapies targeting EGFR and other endpoints suggests the need for greater molecular characterization of tumors before, and possibly after, starting treatment. Future trials will need to better determine eligibility based on who is predicted to respond to targeted therapy. In addition, combination targeted therapies and/or dual downstream inhibition of resistance mechanisms appear to be promising future approaches to CRC treatment.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Referenced with permission from The NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines(r)) for Colon Cancer V.3.2014. (c) National Comprehensive Cancer Network, Inc 2013. All rights reserved. Accessed June 16, 2014. To view the most recent and complete version of the guideline, go online to www.nccn.org. NATIONAL COMPREHENSIVE CANCER NETWORK(r), NCCN(r), NCCN GUIDELINES(r), and all other NCCN Content are trademarks owned by the National Comprehensive Cancer Network, Inc.
Grady WM, Carethers JM. Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology. 2008;135:1079–99.
Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7.
Dienstmann R, Guinney J, Delorenzi M et al. Colorectal Cancer Subtyping Consortium (CRCSC) identification of a consensus of molecular subtypes. In 2014 ASCO Annual Meeting. J Clin Oncol. 32:5 s, 2014 (suppl; abstr 3511).
O’Connell MJ, Lavery I, Yothers G, et al. Relationship between tumor gene expression and recurrence in four independent studies of patients with stage II/III colon cancer treated with surgery alone or surgery plus adjuvant fluorouracil plus leucovorin. J Clin Oncol. 2010;28:3937–44.
Salazar R, Roepman P, Capella G, et al. Gene expression signature to improve prognosis prediction of stage II and III colorectal cancer. J Clin Oncol. 2011;29:17–24.
Gray RG, Quirke P, Handley K, et al. Validation study of a quantitative multigene reverse transcriptase-polymerase chain reaction assay for assessment of recurrence risk in patients with stage II colon cancer. J Clin Oncol. 2011;29:4611–9.
Venook AP, Niedzwiecki D, Lopatin M, et al. Biologic determinants of tumor recurrence in stage II colon cancer: validation study of the 12-gene recurrence score in cancer and leukemia group B (CALGB) 9581. J Clin Oncol. 2013;31:1775–81.
Pritchard CC, Salipante SJ, Koehler K, et al. Validation and implementation of targeted capture and sequencing for the detection of actionable mutation, copy number variation, and gene rearrangement in clinical cancer specimens. J Mol Diagn. 2014;16:56–67.
Andre T, Boni C, Navarro M, et al. Improved overall survival with oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment in stage II or III colon cancer in the MOSAIC trial. J Clin Oncol. 2009;27:3109–16.
de Gramont A, Figer A, Seymour M, et al. Leucovorin and fluorouracil with or without oxaliplatin as first-line treatment in advanced colorectal cancer. J Clin Oncol. 2000;18:2938–47.
Kohne CH, van Cutsem E, Wils J, et al. Phase III study of weekly high-dose infusional fluorouracil plus folinic acid with or without irinotecan in patients with metastatic colorectal cancer: European Organisation for Research and Treatment of Cancer Gastrointestinal Group Study 40986. J Clin Oncol. 2005;23:4856–65.
Saltz LB, Cox JV, Blanke C, et al. Irinotecan plus fluorouracil and leucovorin for metastatic colorectal cancer. Irinotecan Study Group. N Engl J Med. 2000;343:905–14.
Huh JW, Lee JH, Kim HR. Pretreatment expression of 13 molecular markers as a predictor of tumor responses after neoadjuvant chemoradiation in rectal cancer. Ann Surg. 2014;259:508–15.
Noda E, Maeda K, Inoue T, et al. Predictive value of expression of ERCC 1 and GST-p for 5-fluorouracil/oxaliplatin chemotherapy in advanced colorectal cancer. Hepatogastroenterology. 2012;59:130–3.
Viguier J, Boige V, Miquel C, et al. ERCC1 codon 118 polymorphism is a predictive factor for the tumor response to oxaliplatin/5-fluorouracil combination chemotherapy in patients with advanced colorectal cancer. Clin Cancer Res. 2005;11:6212–7.
Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology. 2010;138:2073–87. e2073.
Laghi L, Malesci A. Microsatellite instability and therapeutic consequences in colorectal cancer. Dig Dis. 2012;30:304–9.
Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med. 2003;348:919–32.
Lynch HT, Lynch JF, Lynch PM. Toward a consensus in molecular diagnosis of hereditary nonpolyposis colorectal cancer (Lynch syndrome). J Natl Cancer Inst. 2007;99:261–3.
Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol. 2005;23:609–18.
Sinicrope FA, Mahoney MR, Smyrk TC, et al. Prognostic impact of deficient DNA mismatch repair in patients with stage III colon cancer from a randomized trial of FOLFOX-based adjuvant chemotherapy. J Clin Oncol. 2013;31:3664–72.
Sargent DJ, Marsoni S, Monges G, et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J Clin Oncol. 2010;28:3219–26.
Des Guetz G, Uzzan B, Nicolas P, et al. Microsatellite instability does not predict the efficacy of chemotherapy in metastatic colorectal cancer. A systematic review and meta-analysis. Anticancer Res. 2009;29:1615–20.
Vilar E, Gruber SB. Microsatellite instability in colorectal cancer-the stable evidence. Nat Rev Clin Oncol. 2010;7:153–62.
Roth AD, Tejpar S, Delorenzi M, et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60–00 trial. J Clin Oncol. 2010;28:466–74.
Gavin PG, Colangelo LH, Fumagalli D, et al. Mutation profiling and microsatellite instability in stage II and III colon cancer: an assessment of their prognostic and oxaliplatin predictive value. Clin Cancer Res. 2012;18:6531–41.
Bertagnolli MM, Niedzwiecki D, Compton CC, et al. Microsatellite instability predicts improved response to adjuvant therapy with irinotecan, fluorouracil, and leucovorin in stage III colon cancer: Cancer and Leukemia Group B Protocol 89803. J Clin Oncol. 2009;27:1814–21.
Van Cutsem E, Labianca R, Bodoky G, et al. Randomized phase III trial comparing biweekly infusional fluorouracil/leucovorin alone or with irinotecan in the adjuvant treatment of stage III colon cancer: PETACC-3. J Clin Oncol. 2009;27:3117–25.
Fallik D, Borrini F, Boige V, et al. Microsatellite instability is a predictive factor of the tumor response to irinotecan in patients with advanced colorectal cancer. Cancer Res. 2003;63:5738–44.
Sinicrope FA, Foster NR, Thibodeau SN, et al. DNA mismatch repair status and colon cancer recurrence and survival in clinical trials of 5-fluorouracil-based adjuvant therapy. J Natl Cancer Inst. 2011;103:863–75.
Falcone A, Ricci S, Brunetti I, et al. Phase III trial of infusional fluorouracil, leucovorin, oxaliplatin, and irinotecan (FOLFOXIRI) compared with infusional fluorouracil, leucovorin, and irinotecan (FOLFIRI) as first-line treatment for metastatic colorectal cancer: the Gruppo Oncologico Nord Ovest. J Clin Oncol. 2007;25:1670–6.
Van Cutsem E, Kohne CH, Hitre E, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360:1408–17.
Bokemeyer C, Bondarenko I, Makhson A, et al. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J Clin Oncol. 2009;27:663–71.
Masi G, Loupakis F, Salvatore L, et al. Bevacizumab with FOLFOXIRI (irinotecan, oxaliplatin, fluorouracil, and folinate) as first-line treatment for metastatic colorectal cancer: a phase 2 trial. Lancet Oncol. 2010;11:845–52.
Therkildsen C, Bergmann TK, Henrichsen-Schnack T, et al. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: a systematic review and meta-analysis. Acta Oncol. 2014;53:852–64.
Van Cutsem E, Kohne CH, Lang I, et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol. 2011;29:2011–9.
Douillard JY, Siena S, Cassidy J et al. Final results from PRIME: randomized phase 3 study of panitumumab with FOLFOX4 for first-line treatment of metastatic colorectal cancer. Ann Oncol. 2014. Final results from the PRIME trial showed a 1.4 month benefit in progression-free survival from the addition of panitumumab to FOLFOX in unselected stage IV patients.
De Roock W, Claes B, Bernasconi D, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11:753–62.
Douillard JY, Oliner KS, Siena S, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369:1023–34. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer.
Irahara N, Baba Y, Nosho K, et al. NRAS mutations are rare in colorectal cancer. Diagn Mol Pathol. 2010;19:157–63.
Loupakis F, Cremolini C, Salvatore L, et al. FOLFOXIRI plus bevacizumab as first-line treatment in BRAF mutant metastatic colorectal cancer. Eur J Cancer. 2014;50:57–63. FOLFOXIRI plus bevacizumab as first-line treatment in BRAF mutant metastatic colorectal cancer.
Heinemann V, von Weikersthal LF, Decker T et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): a randomised, open-label, phase 3 trial. Lancet Oncol. 2014. The FIRE-3 study found 16 % of individuals who had wild-type KRAS exon 2 tumors had a mutation on expanded RAS testing. The absolute differences in treatment arms were greater when using the expanded definition of RAS wild type.
Fornaro L, Lonardi S, Masi G, et al. FOLFOXIRI in combination with panitumumab as first-line treatment in quadruple wild-type (KRAS, NRAS, HRAS, BRAF) metastatic colorectal cancer patients: a phase II trial by the Gruppo Oncologico Nord Ovest (GONO). Ann Oncol. 2013;24:2062–7.
Juo YY, Johnston FM, Zhang DY et al. Prognostic value of CpG island methylator phenotype among colorectal cancer patients: a systematic review and meta-analysis. Ann Oncol. 2014.
Shiovitz S, Bertagnolli MM, Renfro LA, et al. CpG island methylator phenotype is associated with response to adjuvant irinotecan-based therapy for stage 3 colon cancer. Gastroenterology. 2014;147:637–45. CpG island methylator phenotype is associated with response to adjuvant irinotecan-based therapy for stage 3 colon cancer.
Sclafani F, Rimassa L, Colombo P et al. An exploratory biomarker study in metastatic tumors from colorectal cancer patients treated with bevacizumab. Int J Biol Markers. 2014; 0.
Liao X, Lochhead P, Nishihara R, et al. Aspirin use, tumor PIK3CA mutation, and colorectal-cancer survival. N Engl J Med. 2012;367:1596–606. Aspirin use, tumor PIK3CA mutation, and colorectal-cancer survival.
D’Amato V, Rosa R, D’Amato C, et al. The dual PI3K/mTOR inhibitor PKI-587 enhances sensitivity to cetuximab in EGFR-resistant human head and neck cancer models. Br J Cancer. 2014;110:2887–95.
Fang DD, Zhang CC, Gu Y, et al. Antitumor efficacy of the dual PI3K/mTOR inhibitor PF-04691502 in a human xenograft tumor model derived from colorectal cancer stem cells harboring a mutation. PLoS One. 2013;8:e67258.
Rodon J, Brana I, Siu LL, et al. Phase I dose-escalation and -expansion study of buparlisib (BKM120), an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. Invest New Drugs. 2014;32:670–81.
Ganesan P, Janku F, Naing A, et al. Target-based therapeutic matching in early-phase clinical trials in patients with advanced colorectal cancer and PIK3CA mutations. Mol Cancer Ther. 2013;12:2857–63.
Bardelli A, Corso S, Bertotti A, et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013;3:658–73. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer.
Chen HJ, Jiang YL, Lin CM, et al. Dual inhibition of EGFR and c-Met kinase activation by MJ-56 reduces metastasis of HT29 human colorectal cancer cells. Int J Oncol. 2013;43:141–50.
Yeh TC, Marsh V, Bernat BA, et al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res. 2007;13:1576–83.
Troiani T, Napolitano S, Vitagliano D, et al. Primary and acquired resistance of colorectal cancer cells to anti-EGFR antibodies converge on MEK/ERK pathway activation and can be overcome by combined MEK/EGFR inhibition. Clin Cancer Res. 2014;20:3775–86.
Bertotti A, Migliardi G, Galimi F, et al. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011;1:508–23.
Falchook GS, Long GV, Kurzrock R, et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet. 2012;379:1893–901.
Saltz LB, Niedzwiecki D, Hollis D, et al. Irinotecan fluorouracil plus leucovorin is not superior to fluorouracil plus leucovorin alone as adjuvant treatment for stage III colon cancer: results of CALGB 89803. J Clin Oncol. 2007;25:3456–61.
Han SW, Lee HJ, Bae JM, et al. Methylation and microsatellite status and recurrence following adjuvant FOLFOX in colorectal cancer. Int J Cancer. 2013;132:2209–16.
Richman SD, Seymour MT, Chambers P, et al. KRAS and BRAF mutations in advanced colorectal cancer are associated with poor prognosis but do not preclude benefit from oxaliplatin or irinotecan: results from the MRC FOCUS trial. J Clin Oncol. 2009;27:5931–7.
Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–42.
Saltz LB, Clarke S, Diaz-Rubio E, et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol. 2008;26:2013–9.
Martin LK, Li X, Kleiber B, et al. VEGF remains an interesting target in advanced pancreas cancer (APCA): results of a multi-institutional phase II study of bevacizumab, gemcitabine, and infusional 5-fluorouracil in patients with APCA. Ann Oncol. 2012;23:2812–20.
Pant S, Martin LK, Geyer S et al. Treatment-related hypertension as a pharmacodynamic biomarker for the efficacy of bevacizumab in advanced pancreas cancer: a pooled analysis of 4 prospective trials of gemcitabine-based therapy with bevacizumab. Am J Clin Oncol. 2014.
Luo HY, Xu RH. Predictive and prognostic biomarkers with therapeutic targets in advanced colorectal cancer. World J Gastroenterol. 2014;20:3858–74.
Prager GW, Braemswig KH, Martel A et al. Baseline carcinoembryonic antigen (CEA) serum levels predict bevacizumab-based treatment response in metastatic colorectal cancer. Cancer Sci. 2014. Baseline CEA may predict bevacizumab responsiveness.
Spencer SK, Pommier AJ, Morgan SR, et al. Prognostic/predictive value of 207 serum factors in colorectal cancer treated with cediranib and/or chemotherapy. Br J Cancer. 2013;109:2765–73.
Jurgensmeier JM, Schmoll HJ, Robertson JD, et al. Prognostic and predictive value of VEGF, sVEGFR-2 and CEA in mCRC studies comparing cediranib, bevacizumab and chemotherapy. Br J Cancer. 2013;108:1316–23. Prognostic and predictive value of VEGF, sVEGFR-2 and CEA in mCRC studies comparing cediranib, bevacizumab and chemotherapy.
Mok T, Gorbunova V, Juhasz E, et al. A correlative biomarker analysis of the combination of bevacizumab and carboplatin-based chemotherapy for advanced nonsquamous non-small-cell lung cancer: results of the phase II randomized ABIGAIL study (BO21015). J Thorac Oncol. 2014;9:848–55.
Volz NB, Stintzing S, Zhang W et al. Genes involved in pericyte-driven tumor maturation predict treatment benefit of first-line FOLFIRI plus bevacizumab in patients with metastatic colorectal cancer. Pharmacogenomics J. 2014.
Bruhn MA, Townsend AR, Khoon Lee C, et al. Proangiogenic tumor proteins as potential predictive or prognostic biomarkers for bevacizumab therapy in metastatic colorectal cancer. Int J Cancer. 2014;135:731–41.
Zulato E, Bergamo F, De Paoli A, et al. Prognostic significance of AMPK activation in advanced stage colorectal cancer treated with chemotherapy plus bevacizumab. Br J Cancer. 2014;111:25–32.
Jubb AM, Harris AL. Biomarkers to predict the clinical efficacy of bevacizumab in cancer. Lancet Oncol. 2010;11:1172–83.
Kopetz S. “Right drug for the right patient”: hurdles and the path forward in colorectal cancer. Am Soc Clin Oncol Educ Book. 2013.
Montagut C, Dalmases A, Bellosillo B, et al. Identification of a mutation in the extracellular domain of the epidermal growth factor receptor conferring cetuximab resistance in colorectal cancer. Nat Med. 2012;18:221–3. Identification of a mutation in the extracellular domain of the epidermal growth factor receptor conferring cetuximab resistance in colorectal cancer.
Price TJ, Peeters M, Kim TW, et al. Panitumumab versus cetuximab in patients with chemotherapy-refractory wild-type KRAS exon 2 metastatic colorectal cancer (ASPECCT): a randomised, multicentre, open-label, non-inferiority phase 3 study. Lancet Oncol. 2014;15:569–79.
Lievre A, Bachet JB, Boige V, et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol. 2008;26:374–9.
Amado RG, Wolf M, Peeters M, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626–34.
Cunningham D, Humblet Y, Siena S, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351:337–45.
Hecht JR, Mitchell E, Neubauer MA, et al. Lack of correlation between epidermal growth factor receptor status and response to panitumumab monotherapy in metastatic colorectal cancer. Clin Cancer Res. 2010;16:2205–13.
Alberts SR, Sargent DJ, Nair S, et al. Effect of oxaliplatin, fluorouracil, and leucovorin with or without cetuximab on survival among patients with resected stage III colon cancer: a randomized trial. JAMA. 2012;307:1383–93.
Taieb J, Tabernero J, Mini E et al. Oxaliplatin, fluorouracil, and leucovorin with or without cetuximab in patients with resected stage III colon cancer (PETACC-8): an open-label, randomised phase 3 trial. Lancet Oncol. 2014. Cetuximab currently has no role in non-metastatic CRC.
Imamura Y, Lochhead P, Yamauchi M, et al. Analyses of clinicopathological, molecular, and prognostic associations of KRAS codon 61 and codon 146 mutations in colorectal cancer: cohort study and literature review. Mol Cancer. 2014;13:135. Analyses of clinicopathological, molecular, and prognostic associations of KRAS codon 61 and codon 146 mutations in colorectal cancer: cohort study and literature review.
Modest DP, Stintzing S, Laubender RP, et al. Clinical characterization of patients with metastatic colorectal cancer depending on the KRAS status. Anticancer Drugs. 2011;22:913–8.
De Roock W, Jonker DJ, Di Nicolantonio F, et al. Association of KRAS p.G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. JAMA. 2010;304:1812–20.
Lee DW, Kim KJ, Han SW et al. KRAS mutation is associated with worse prognosis in stage iii or high-risk stage ii colon cancer patients treated with adjuvant FOLFOX. Ann Surg Oncol. 2014.
Chan AT, Ogino S, Fuchs CS. Aspirin use and survival after diagnosis of colorectal cancer. JAMA. 2009;302:649–58.
Sclafani F, Gonzalez D, Cunningham D et al. TP53 mutational status and cetuximab benefit in rectal cancer: 5-year results of the EXPERT-C trial. J Natl Cancer Inst. 2014; 106. TP53 may serve as a predictor of cetuximab benefit among high-risk early stage rectal cancer patients. In this analysis, wild-type TP53 CRCs received benefit from cetuximab, even after adjusting for KRAS status.
Dewdney A, Cunningham D, Tabernero J, et al. Multicenter randomized phase II clinical trial comparing neoadjuvant oxaliplatin, capecitabine, and preoperative radiotherapy with or without cetuximab followed by total mesorectal excision in patients with high-risk rectal cancer (EXPERT-C). J Clin Oncol. 2012;30:1620–7.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Zoratto F, Rossi L, Verrico M, et al. Focus on genetic and epigenetic events of colorectal cancer pathogenesis: implications for molecular diagnosis. Tumour Biol. 2014;35:6195–206.
Kim A, Lee JE, Lee SS, et al. Coexistent mutations of KRAS and PIK3CA affect the efficacy of NVP-BEZ235, a dual PI3K/MTOR inhibitor, in regulating the PI3K/MTOR pathway in colorectal cancer. Int J Cancer. 2013;133:984–96.
Zimmer L, Barlesi F, Martinez-Garcia M et al. Phase I expansion and pharmacodynamic study of the Oral MEK inhibitor RO4987655 (CH4987655) in selected patients with advanced cancer with RAS-RAF mutations. Clin Cancer Res. 2014.
Salama AK, Flaherty KT. BRAF in melanoma: current strategies and future directions. Clin Cancer Res. 2013;19:4326–34.
Migliardi G, Sassi F, Torti D, et al. Inhibition of MEK and PI3K/mTOR suppresses tumor growth but does not cause tumor regression in patient-derived xenografts of RAS-mutant colorectal carcinomas. Clin Cancer Res. 2012;18:2515–25.
Dienstmann R, Serpico D, Rodon J, et al. Molecular profiling of patients with colorectal cancer and matched targeted therapy in phase I clinical trials. Mol Cancer Ther. 2012;11:2062–71. Molecular profiling of patients with colorectal cancer and matched targeted therapy in phase I clinical trials.
Shimizu T, Tolcher AW, Papadopoulos KP, et al. The clinical effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clin Cancer Res. 2012;18:2316–25.
Turke AB, Song Y, Costa C, et al. MEK inhibition leads to PI3K/AKT activation by relieving a negative feedback on ERBB receptors. Cancer Res. 2012;72:3228–37.
Compliance with Ethics Guidelines
Conflict of Interest
Stacey Shiovitz has received financial support from the National Institutes of Health (NIH T32-CA009515) and a 2014 Young Investigator Award from the Conquer Cancer Foundation of the American Society for Clinical Oncology. William M. Grady has received financial support from the National Institutes of Health (NIH RO1CA115513, P30CA15704, UO1CA152756, U54CA143862, P01CA077852) and a Burroughs Wellcome Fund Translational Research Award for Clinician Scientist.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on GI Oncology
Rights and permissions
About this article

Cite this article
Shiovitz, S., Grady, W.M. Molecular Markers Predictive of Chemotherapy Response in Colorectal Cancer. Curr Gastroenterol Rep 17, 6 (2015). https://doi.org/10.1007/s11894-015-0431-7
Published:
DOI: https://doi.org/10.1007/s11894-015-0431-7