
Abstract
Purpose of Review
Diabetic kidney disease (DKD) continues to be the primary cause of chronic kidney disease in the USA and around the world. The numbers of people with DKD also continue to rise despite current treatments. Certain newer hypoglycemic drugs offer a promise of slowing progression, but it remains to be seen how effective these will be over time. Thus, continued exploration of the mechanisms underlying the development and progression of DKD is essential in order to discover new treatments. Hyperglycemia is the main cause of the cellular damage seen in DKD. But, exactly how hyperglycemia leads to the activation of processes that are ultimately deleterious is incompletely understood.
Recent Findings
Studies primarily over the past 10 years have provided novel insights into the interplay of hyperglycemia, glucose metabolic pathways, mitochondrial function, and the potential importance of what has been called the Warburg effect on the development and progression of DKD.
Summary
This review will provide a brief overview of glucose metabolism and the hypotheses concerning the pathogenesis of DKD and then discuss in more detail the supporting data that indicate a role for the interplay of glucose metabolic pathways and mitochondrial function.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Diabetic kidney disease (DKD) has continued to be the main cause of chronic kidney disease and the leading cause of end-stage renal disease (ESRD) in the USA and throughout most of the world [1,2,3]. Current treatments consist of controlling the blood sugar and blood pressure as well as lowering the urine albumin level which have proven to be effective therapies for slowing the development and progression of DKD [1, 4]. But despite these proven treatments, the number of people who develop DKD and the number of people who progress to an end-stage renal disease continue to rise indicating that new medications need to be developed [2]. The health risk for people who develop DKD is not only the risk for developing ESRD but also, as kidney disease progresses, which is diagnosed as a decline in the estimated glomerular filtration rate and/or an increase in the urine albumin level, there is a highly significant increase risk of developing cardiovascular disease [5]. Indeed, this association is so close and important that there is a much higher likelihood of dying from cardiovascular disease than reaching ESRD [5]. Thus, there is a great need for developing new treatments for both preventing the development of DKD and for slowing the progression of DKD. Hope for new effective treatments has been provided by recent studies with the newer hypoglycemic agents, SGLT2 inhibitors and GLP-1 agonists [6,7,8]. Although the recent studies are very promising (especially for SGLT2 inhibitors), it remains to be seen how great an impact these medications will have on changing the course of DKD and how safe they will be over time (especially for SGLT2 inhibitors). Thus, there is still an urgent need to continue to discover new reno-protective drugs.
Critical to finding new drugs is a clear mechanistic understanding of the genetic and biochemical pathways that are responsible for the development and progression of DKD. Over many years, the risk for developing DKD and risk for progression of DKD have been associated with particular genes and with a number of biochemical processes including, but not limited to, excessive reactive oxygen species formation (ROS), development of advanced glycation end products, increased activity of the aldose reductase pathway, activation of isoforms of protein kinase C, development of endoplasmic reticulum stress, and abnormal autophagy pathways [9,10,11,12,13,14]. A central feature of all of these pathological pathways is that hyperglycemia is the stimulant that leads to the activation of these deleterious processes. Hence, there have been many studies exploring whether altered glucose metabolism per se may play a role in the development of DKD. In this article, an overview of glucose metabolism and mitochondrial function will be provided followed by an in-depth discussion about findings from the past 10 years that incorporate both old and new ideas on the interplay of glucose metabolic pathways and the development of DKD.
Glucose Metabolic Pathways
Glucose Metabolism
Glucose enters most cells via facilitated diffusion transport through a class of glucose transporters called GLUT1-GLUT14 [15, 16]. The expression of the specific class of transporters is tissue and cell specific. Class I transporters (GLUT1–GLUT4 and GLUT14) are the most common. In humans, for example, GLUT1 is found primarily in beta cells of the pancreas, GLUT3 is expressed in the brain, and GLUT4 is expressed in the brain, skeletal muscle, heart, and adipose tissue [15, 16]. Glucose transporters may act as both a transporter of glucose and as a glucose sensor [17]. The classification of the other glucose transporters, class II (GLUTs 5, 7, 9, 11) and class III (GLUTs 6, 8, 10, 12, 13) based on sequence similarities. The evolutionary and physiological reasons for so many glucose transporters remain unexplained. In addition, there are sodium linked glucose cotransporters (SGLT1-SGLT6) [15, 18]. These proteins also may function as either glucose transporters or as glucose sensors as demonstrated, for example, in a recent study using rat mesangial cells which demonstrated that SGLT2 could regulate mesangial cell contractility [19]. Of this class, the two that are most highly expressed are SGLT1 which is found primarily in the small intestine and SGLT2 which is found mostly in the proximal tubule of the kidney [18].
Following the entry into the cell, glucose will have different metabolic fates which, under normal circumstances, is regulated by cellular needs (Fig. 1). The ultimate fate of glucose is determined by well-known biochemical processes such as enzyme affinity, enzyme location, and expression. Typically, glucose is rapidly phosphorylated to glucose-6-phosphate by hexokinase [9]. Of note prior to being phosphorylated, under conditions where hexosamine is saturated, glucose may be reduced by aldose reductase to sorbitol and in the process, NADPH is converted to NADP+ [20]. Sorbitol is then converted to fructose by the enzyme sorbitol dehydrogenase [20]. But, the main pathway for glucose is conversion to glucose-6-phosphate (G6P) at which multiple fates are possible. Following phosphorylation of glucose, G6P may be metabolized by the following pathways glycolysis, the pentose phosphate pathway (PPP), glycogen synthesis pathway, or hexosamine pathway (Fig. 1) [9]. The oxidation of glucose via glycolysis ultimately leads to the production of pyruvate which can then either be converted to lactate or enter mitochondria upon which it provides fuel for the Krebs cycle also called the tricarboxylic acid cycle (TCA) in mitochondria or for gluconeogenesis (which primarily occurs in the liver and kidney) [9, 21]. Glucose can also enter the PPP through the rate-limiting enzyme, glucose-6-phosphate dehydrogenase (G6PD). The major products of the PPP are NADPH which is required for the proper function of the antioxidant cellular enzymes as it is the main intracellular antioxidant and ribose-5-phosphate which is required for nucleic acid biosynthesis [22,23,24]. Glucose may also be converted to glycogen via the actions of phosphoglucomutase that converts glucose-6-phosphate to glucose-1-phosphate followed by the rate-limiting enzyme glycogen synthase kinase 3 which ultimately leads to the production of polysaccharides, a storage form of glucose [9, 25]. Lastly, glucose may be converted to hexosamine via the initial action of glucose-6-phosphate isomerase which converts glucose-6-phosphate to fructose-6-phosphate and is ultimately converted into glycoproteins and glycolipids [9, 13, 26].

Schematic representation of the fate of glucose. See text for discussion. G6PD, glucose-6-phosphate dehydrogenase; GFAT, glutamine:fructose-6-phosphate amidotransferase; G6P isomerase, glucose-6-phosphate isomerase
Proposed Mechanisms Responsible for the Development and Progression of DKD Associated with Glucose Metabolism
To date, the principal pathophysiologic pathways considered to underlie DKD include both glucose-associated pathways and hemodynamic pathways. Hence, the currently accepted approach to the prevention of DKD and slowing progression involves primarily blood sugar control and blood pressure control. In addition, if a person with DKD has significantly elevated urine albumin levels, then efforts to lower these levels will likely slow the progression of DKD [1, 4]. The main agents used to lower urine albumin levels are almost all inhibitors of the renin-angiotensin-aldosterone pathway which appear to exert a protective effect, at least in part, via a decrease in glomerular pressures [27]. Whether these agents’ beneficial effects are due primarily to lowering of glomerular pressure is debatable as the principal targets of these medications, angiotensin II and aldosterone, have many other potentially deleterious effects such as stimulation of inflammatory cytokines, activation of processes that increase ROS, and more [27,28,29]. As previously mentioned, in addition to the glomerular hemodynamic changes that predispose to development and progression of DKD, there are a number of hyperglycemia-induced biochemical processes that have been shown to be of pathophysiological mechanistic importance.
The following processes have been directly linked to the pathogenesis of DKD via hyperglycemia. The overall increase in ROS (which leads to oxidation of proteins, lipids, and nucleic acids causing impaired cellular function and cell survival) is initially stimulated by hyperglycemia both by activating ROS-producing processes (for example by activating NADPH oxidase) and by impairing antioxidant function (for example by impairing critical antioxidant enzymes such as glutathione peroxidase, catalase, and glucose-6-phosphate dehydrogenase) [22, 23, 30]. In addition to the direct actions of hyperglycemia on increasing ROS, hyperglycemia stimulates many cellular processes that also increase ROS, some of which are discussed below. Activation of aldose reductase occurs also via hyperglycemia and may cause osmotic stress as a result of the increased sorbitol levels and activation of aldose reductase may also contribute to increased ROS by lowering NADPH levels (NADPH is the principal cellular antioxidant) as NADPH is converted to NADP+ by aldose reductase [20]. Advanced glycation end products (AGEs) are produced at higher levels which is directly related to the level of hyperglycemia as a result of a non-enzymatic glycation process that happens when certain amino acids are exposed to glucose over a long enough period of time [14]. AGEs are produced normally but are greatly increased in people with diabetes [14]. Consequences of AGEs include cellular membrane defects, impaired protein function, increased ROS, inflammation, and other deleterious cellular events [14]. Furthermore, hyperglycemia may lead to increased hexosamine formation and higher levels of hexosamines have been linked to insulin resistance and activation of deleterious factors such as protein kinase C and transforming growth factor β [13].
Increased Mitochondrial Activity and DKD
In the early 2000s, Michael Brownlee proposed a unifying hypothesis that potentially explained how increased hyperglycemia could directly lead to a number of the proposed pathophysiologic consequences that have been associated with the development of diabetic complications via a central instigating process. The hypothesis stated that hyperglycemia would stimulate an increase in aerobic glycolysis with the resultant increase in substrate delivery to the mitochondria and a subsequent increase in mitochondrial activity [31]. Increased TCA activity would produce more NADH and FADH2 which would then be used in the electron transport chain in the mitochondria. But, according to the hypothesis and supporting data, a threshold is reached in the electron transport chain that ultimately results in a block at Complex III and subsequent increased production of superoxide via coenzyme Q [31]. According to this hypothesis, the increased superoxide ultimately leads to inhibition of the glycolytic enzyme, glyceraldehyde-3-phosphate dehydrogenase (according to the hypothesis and supporting data, the mitochondrial-produced superoxide did not directly inhibit G3PD but rather activated poly(ADP-ribose) polymerase which then modified G3PD and led to enzyme inhibition), and a resultant inhibition or slowing of glycolysis which then would lead to accumulation of upstream substrates. These upstream substrates were then shunted into other metabolic pathways that have been shown to be of pathophysiological importance in DKD: the polyol pathway (aldose reductase), the hexosamine pathway, the diacylglycerol pathway (which is a stimulator of protein kinase C), and the advanced glycation end product pathway. This research provided a very interesting theory and stimulated many subsequent studies including a potential treatment with benfotiamine (a form of vitamin B1 or thiamine), a cofactor for the enzyme transketolase which would shift substrates into the non-oxidative branch of the PPP with the effect of bypassing the inhibition of G3PD and lowering of the substrates that were activating the deleterious pathways [31]. But although benfotiamine appeared to show efficacy in animal studies [32], to date, benfotiamine has not been shown to especially effective as a treatment for DKD [33]. Also, there have been persistent questions surrounding the hypothesis. For example, would the process be self-limiting as when G3PD was inhibited, in addition to there being an accumulation of upstream substrates would there also be a decrease in downstream substrates and therefore a decrease in substrate delivery to the mitochondria, decreased mitochondrial production of superoxide, and decreased inhibition of G3PD? Furthermore, there has been much debate over whether mitochondria activity is increased or decreased in affected tissues in people with diabetes mellitus.
Decreased Mitochondrial Activity and DKD
In 2013, Sharma and colleagues reported a metabolomic analysis of 94 urine metabolites from people with diabetes and kidney disease as compared to other control groups [34••]. Thirteen metabolites were determined to be significantly decreased in those with diabetes and chronic kidney disease as compared to healthy controls (and 12 of 13 remained significantly decreased when compared to diabetic participants without chronic kidney disease). Moreover, 12 of the 13 metabolites were directly linked to mitochondrial metabolism via network analysis of possible biochemical pathways. These results suggested that mitochondrial function was decreased in all of these patients as compared to those without chronic kidney disease. Further analysis from archived kidney biopsy samples from people with diabetes or chronic kidney disease as well as from normal kidneys was studied for signs of decreased mitochondrial function. Using an antibody to cytochrome C oxidase (complex IV), the authors demonstrated a reduction in staining in those with diabetic kidney disease as compared to the normal kidney biopsies. They also looked for a reduction in mitochondrial DNA from exosomes in the urine in those with DKD and also found lower levels providing further evidence for decreased mitochondrial function in DKD. Lastly, they evaluated gene expression for PGC1α, a major regulator of mitochondrial biogenesis, and found that there was a decrease in expression as compared to biopsies from other non-DKD people [34••]. The arguments underlying a more central role for lower rather than higher mitochondrial function in the pathogenesis of DKD are well presented in two articles [35, 36]. The authors make a case for excessive mitochondrial superoxide production as being more a reflection of healthy mitochondrial function rather than pathologic mitochondrial function. Moreover, further studies also suggest that mitochondrial dysfunction rather than overactivity is more likely in the pathogenesis of DKD. Sas and colleagues did an extensive transcriptomic and metabolomic analysis as well as a metabolic flux analysis from kidneys in a mouse model of diabetes and from human biopsy tissue [37]. The studies determined that tissue from DKD had increased TCA cycle activity but impaired mitochondrial transport chain function consistent with the concept that had been recently proposed by Sharma and colleagues. Other researchers have found similar findings [38, 39]. A recent study using kidney biopsy samples and metabolomics determined that DKD patients had increased mitochondrial DNA damage as compared to controls which was associated with apoptosis and loss of mitochondrial membrane potential in tubules [39]. The differences in findings between the more recent studies suggesting low mitochondrial function versus the previous studies reporting high mitochondrial function may reflect cell type, when measurements were done, and how measurements were done.
Warburg Effect
The issue as to whether DKD is associated with increased or decreased mitochondrial function has certainly not been fully resolved, but considering the recent findings, it is reasonable to consider the concept that mitochondria are indeed less active in diabetic kidney disease (for an excellent in-depth discussion of mitochondrial function, actually dysfunction, in DKD, please read the recent review by Forbes and Thorburn) [40]. Then, the obvious research questions are why are mitochondria less active, can this be corrected, and does improving mitochondrial function lead to reno-protection? To understand this more, it is necessary to go back to the 1920s and Otto Warburg’s laboratory in Berlin, Germany. Professor Warburg was studying oxygen consumption in tissue from tumors versus normal tissue. He hypothesized that the tumor tissue would utilize oxygen at a much higher rate than the normal tissue [21, 22, 41]. Warburg measured oxygen consumption (respiration) in tissue slices from rat liver and kidney and from transplanted seminal vesicle tumors, and found very little difference in the respiration of tumor tissue slices compared to that of normal tissue slices. He then quantified lactate production as a measure of fermentation rate and found that under anaerobic conditions, all tissues exhibited increased production of lactate. In contrast, under aerobic conditions, the behavior of tumor tissue and normal tissue differed. While the tumor tissue continued to produce elevated levels of lactate, the normal tissue did not. He initially thought that possibly the glucose concentration in the culture media was rate limiting. But when he added glucose, the effect was even more pronounced; the tumor cells produced significantly more lactate as compared to the normal tissue [21, 22, 41]. The combination of less oxygen consumption and increased lactate production suggested that there is decreased mitochondrial activity in cancer cells (it turns out that not all cancer cells show this effect). Is there a physiological advantage to the Warburg effect? An intriguing possible reason for the Warburg effect is that excessive lactate production has a number of beneficial effects for the cancer cell such as enhancement of angiogenesis, metastatic spread, and escape from host immune responses among others [42, 43]. Of course, what may be beneficial for a cancer cell appears to be deleterious for people with DKD. And, although there are similarities between the Warburg effect in cancer cells and DKD, there are also differences. For example, a hallmark of cancer is the activation of the PPP via its rate-limiting enzyme G6PD [44, 45]. Indeed, the Warburg effect in cancer cells has been shown to lead to increased PPP activity leading to increased production of the cell growth substrates NADPH (via activation of the enzymes G6PD and 6-phosphogluconate dehydrogenase) and ribose-5-phosphate [46]. But, G6PD activity is decreased in tissues from diabetic kidneys and many other tissues [22, 44, 47, 48]. Certainly, there are likely many other differences as well in this complex metabolic event which suggests that the mechanisms responsible for the Warburg effect in cancer and in diabetes are not necessarily the same. There have been many studies over the years aimed at elucidating the biochemical mechanisms responsible for the Warburg effect [21, 41, 49]. But for the development of potential treatments, the specific mechanisms underlying the Warburg effect pathophysiology in DKD need to be understood. And in this regard, of particular interest was an important discovery in cancer cells that provided at least one mechanistic explanation for the Warburg effect that also has been discovered to play a role in the Warburg effect seen in DKD [50].
PKM2 and the Warburg Effect
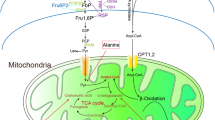
Pyruvate kinase catalyzes the last and physiologically irreversible step in glycolysis which involves the conversion of phosphoenolpyruvate (PEP) to pyruvate. There are four isoforms—PKR (expressed in red blood cells), PKL (expressed primarily in the liver), PKM1 (expressed in muscle and some other tissues), PKM2 (expressed primarily in embryonic tissue and cancer cells but also expressed in some normal adult tissues) [51]. In 2008, Cantley and colleagues demonstrated that cancer cells had increased expression of PKM2 as compared to PKM1 [46, 50]. They also demonstrated that PKM2 was much less active than PKM1 and this low activity likely contributed to the development of the Warburg effect by slowing the transit of glucose metabolites into the mitochondria and shifting them to lactate production. Indeed, when PKM1 was expressed in the cancer cells (following knockdown of PKM2), the Warburg effect was not observed [50]. Further work has determined that a number of inhibitory processes can lead to inhibition of PKM2 including oxidation at a critical cysteine residue (cys358), by acetylation and by other inhibitors [46]. It is important to note that pyruvate kinase is active in a tetrameric configuration but not as a dimer [52•] (Fig. 2). Many of the inhibitors appear to primarily interfere with tetramer formation and lead to preferential expression of the inactive dimer [52•].

Schematic representation of the Warburg effect as mediated by pyruvate kinase (PK) M2 isoform. On the left is normal metabolism in which active pyruvate kinase (tetrameric form) allows the normal mitochondrial function to occur. On the right, the dimeric form of PKM2 is inactive and pyruvate is preferentially shifted to lactate production rather than being utilized by mitochondria. See text for details
PKM2 and Diabetic Kidney Disease
A role for the Warburg effect in the pathogenesis of DKD was determined by Qi et al. [53••] by evaluating kidney tissue samples from participants enrolled in the 50-Year Medalist Study at the Joslin Diabetes Center [54]. The goal of this project has been to understand how people with type 1 diabetes mellitus for 50 years or more have been so healthy for so long. These participants have been identified at the Joslin Diabetes Center as they have been awarded a 50-year and sometimes 75-year medal for living with type 1 diabetes mellitus for that length of time to recognize their achievement of self-management. The hypothesis for the study is that some of these people may well have protective factors that have allowed them to survive this long with either no or relatively few complications [55]. Many participants in this study have no signs of DKD but a subset of participants does have signs of DKD as determined by lower estimated glomerular filtration rate or increased urine albumin excretion [53••, 56]. Moreover, some of the 50-year medalist participants’ kidney tissue was evaluated by biopsy or post-mortem. The participants were then separated into 2 groups called protected (n = 11) or nonprotected (n = 7). Participants were assessed for the severity of DKD by pathology analysis of tissue samples and protected individuals were those with class 0 or I DKD and nonprotected individuals were those with class IIb or III DKD. Proteomics evaluation revealed 88 proteins were upregulated in the protected cohort as compared to the nonprotected cohort including aldose reductase, PKM1, PKM2, enolase, glyoxalase 1, and mitochondrial-encoded cytochrome C oxidase II. Evaluation of enzyme activities from isolated human glomeruli showed lowered pyruvate kinase activity in the nonprotected group as compared to the protected group. In mouse studies of DKD, similar findings were found and in addition, the DKD mice had higher levels of the dimeric form of PKM2 as compared to the non-diabetic mice. Moreover, evaluation of PKM2 from the glomeruli of DKD mice showed oxidation of cys358. These results are very similar to those observed in cancer cells and provide a rationale for focusing on the activation of pyruvate kinase as a treatment for DKD. Recent reports have both confirmed these findings and added more mechanistic insights. For example, a recent publication showed that Smad4 binds to PKM2 decreasing the tetrameric form of PKM2 and this decrease contributed to the development of DKD in mice [57]. And, another mouse study determined that suppression of SIRT3 protein was associated with fibrosis in DKD via activation of transforming growth factorβ/smad signaling pathways and associated with increased dimeric PKM2 [58]. These results suggest that DKD is associated with Warburg effect-like pathophysiology and that PKM2 may be the target for treatment.
Conclusions
The landmark Diabetes Control and Complications Trial and the follow-up, Epidemiology of Diabetes Interventions and Complications clearly established that hyperglycemia is the major factor in the development and progression of DKD in people with type 1 diabetes [59]. It is also clear that there is a complex interplay of all of the downstream pathways that are stimulated by hyperglycemia. Of course, there are many questions and challenges. The following series of questions could be applied to all complications of diabetes but, due to the scope of this article, are aimed at DKD. These questions include the following: (1) How relevant are these mechanisms in different cell types? The kidney is comprised of 3 cell types in glomeruli, cell types in the tubules, as well as vascular cells, and the interstitial area in which inflammatory cells may migrate. (2) Is it possible to target a specific enzyme or pathway involved with as central a process as glucose metabolism without disrupting the normal function of the pathway or associated pathways that may have adverse consequences? With respect to the Warburg effect, in cancer, the goal is to eliminate all of the cells. In DKD, the goal would be to change the pathophysiology from a pathologic pattern to a normal pattern. This will likely be much more of a therapeutic challenge. (3) Are various mechanisms for targeting therapeutic interventions relevant at different time points of the disease process? For example, do the development of DKD and the progression of DKD share similar mechanistic pathways? And after the development of DKD, are the disease mechanisms responsible for progression the same at different time points? For example, it is quite possible that mitochondrial activity will not only vary based on cell type but possibly by disease time point. Hence, the predominant pathophysiologic mechanism(s) that should be targeted at a particular time point may well be different. In addition, it is likely that epigenetic processes will play a role in both the development and progression of DKD, thus adding another layer of complexity in determining the best treatments for individual patients. Nevertheless, the discoveries highlighted in this article concerning the central role of glucose metabolism per se in the development and progression of DKD do provide the necessary impetus to continue research that will hopefully lead to new, effective, and safe treatments.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Alicic RZ, Rooney MT, Tuttle KR. Diabetic kidney disease: challenges, progress, and possibilities. Clin J Am Soc Nephrol. 2017;12(12):2032–45.
Saran R, Robinson B, Abbott KC, Bragg-Gresham J, Chen X, Gipson D, et al. US renal data system 2019 annual data report: epidemiology of kidney disease in the United States. Am J Kidney Dis. 2020;75(1 Suppl 1):A6–7.
Thomas MC, Cooper ME, Zimmet P. Changing epidemiology of type 2 diabetes mellitus and associated chronic kidney disease. Nat Rev Nephrol. 2016;12(2):73–81.
Stanton RC. Clinical challenges in diagnosis and management of diabetic kidney disease. Am J Kidney Dis. 2014;63(2 Suppl 2):S3–21.
Palsson R, Patel UD. Cardiovascular complications of diabetic kidney disease. Adv Chronic Kidney Dis. 2014;21(3):273–80.
Gerstein HC, Colhoun HM, Dagenais GR, Diaz R, Lakshmanan M, Pais P, et al. Dulaglutide and renal outcomes in type 2 diabetes: an exploratory analysis of the REWIND randomised, placebo-controlled trial. Lancet. 2019;394(10193):131–8.
Mann JFE, Orsted DD, Brown-Frandsen K, Marso SP, Poulter NR, Rasmussen S, et al. Liraglutide and renal outcomes in type 2 diabetes. N Engl J Med. 2017;377(9):839–48.
Perkovic V, Jardine MJ, Neal B, Bompoint S, Heerspink HJL, Charytan DM, et al. Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. N Engl J Med. 2019;380(24):2295–306.
Bouche C, Serdy S, Kahn CR, Goldfine AB. The cellular fate of glucose and its relevance in type 2 diabetes. Endocr Rev. 2004;25(5):807–30.
Ding Y, Choi ME. Autophagy in diabetic nephropathy. J Endocrinol. 2015;224(1):R15–30.
Fan Y, Lee K, Wang N, He JC. The role of endoplasmic reticulum stress in diabetic nephropathy. Curr Diab Rep. 2017;17(3):17.
Noh H, King GL. The role of protein kinase C activation in diabetic nephropathy. Kidney Int Suppl. 2007;106:S49–53.
Schleicher ED, Weigert C. Role of the hexosamine biosynthetic pathway in diabetic nephropathy. Kidney Int Suppl. 2000;77:S13–8.
Vlassara H, Uribarri J. Advanced glycation end products (AGE) and diabetes: cause, effect, or both? Curr Diab Rep. 2014;14(1):453.
Deng D, Yan N. GLUT, SGLT, and SWEET: structural and mechanistic investigations of the glucose transporters. Protein Sci. 2016;25(3):546–58.
Thorens B, Mueckler M. Glucose transporters in the 21st century. Am J Physiol Endocrinol Metab. 2010;298(2):E141–5.
Thorens B. GLUT2, glucose sensing and glucose homeostasis. Diabetologia. 2015;58(2):221–32.
Ferrannini E. Sodium-glucose co-transporters and their inhibition: clinical physiology. Cell Metab. 2017;26(1):27–38.
Wakisaka M, Nagao T, Yoshinari M. Sodium glucose cotransporter 2 (SGLT2) plays as a physiological glucose sensor and regulates cellular contractility in rat mesangial cells. PLoS One. 2016;11(3):e0151585.
Tang WH, Martin KA, Hwa J. Aldose reductase, oxidative stress, and diabetic mellitus. Front Pharmacol. 2012;3:87.
Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441–64.
Spencer NY, Stanton RC. Glucose 6-phosphate dehydrogenase and the kidney. Curr Opin Nephrol Hypertens. 2017;26(1):43–9.
Stanton RC. Glucose-6-phosphate dehydrogenase, NADPH, and cell survival. IUBMB Life. 2012;64(5):362–9.
Stincone A, Prigione A, Cramer T, Wamelink MM, Campbell K, Cheung E, et al. The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biol Rev Camb Philos Soc. 2015;90(3):927–63.
Sullivan MA, Forbes JM. Glucose and glycogen in the diabetic kidney: heroes or villains? EBioMedicine. 2019;47:590–7.
Akella NM, Ciraku L, Reginato MJ. Fueling the fire: emerging role of the hexosamine biosynthetic pathway in cancer. BMC Biol. 2019;17(1):52.
Remuzzi G, Perico N, Macia M, Ruggenenti P. The role of renin-angiotensin-aldosterone system in the progression of chronic kidney disease. Kidney Int Suppl. 2005;99:S57–65.
Benigni A, Cassis P, Remuzzi G. Angiotensin II revisited: new roles in inflammation, immunology and aging. EMBO Mol Med. 2010;2(7):247–57.
Briet M, Schiffrin EL. Aldosterone: effects on the kidney and cardiovascular system. Nat Rev Nephrol. 2010;6(5):261–73.
Forbes JM, Coughlan MT, Cooper ME. Oxidative stress as a major culprit in kidney disease in diabetes. Diabetes. 2008;57(6):1446–54.
Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54(6):1615–25.
Babaei-Jadidi R, Karachalias N, Ahmed N, Battah S, Thornalley PJ. Prevention of incipient diabetic nephropathy by high-dose thiamine and benfotiamine. Diabetes. 2003;52(8):2110–20.
Alkhalaf A, Klooster A, van Oeveren W, Achenbach U, Kleefstra N, Slingerland RJ, et al. A double-blind, randomized, placebo-controlled clinical trial on benfotiamine treatment in patients with diabetic nephropathy. Diabetes Care. 2010;33(7):1598–601.
Sharma K, Karl B, Mathew AV, Gangoiti JA, Wassel CL, Saito R, et al. Metabolomics reveals signature of mitochondrial dysfunction in diabetic kidney disease. J Am Soc Nephrol. 2013;24(11):1901–12 Very Important. Excellent, detailed analysis demonstrating mitochondrial dysfunction in animal models of diabetes mellitus and in hiuman kidney samples.
Coughlan MT, Sharma K. Challenging the dogma of mitochondrial reactive oxygen species overproduction in diabetic kidney disease. Kidney Int. 2016;90(2):272–9.
Sharma K. Mitochondrial hormesis and diabetic complications. Diabetes. 2015;64(3):663–72.
Sas KM, Kayampilly P, Byun J, Nair V, Hinder LM, Hur J, et al. Tissue-specific metabolic reprogramming drives nutrient flux in diabetic complications. JCI Insight. 2016;1(15):e86976.
Czajka A, Malik AN. Hyperglycemia induced damage to mitochondrial respiration in renal mesangial and tubular cells: implications for diabetic nephropathy. Redox Biol. 2016;10:100–7.
Jiang H, Shao X, Jia S, Qu L, Weng C, Shen X, et al. The mitochondria-targeted metabolic tubular injury in diabetic kidney disease. Cell Physiol Biochem. 2019;52(2):156–71.
Forbes JM, Thorburn DR. Mitochondrial dysfunction in diabetic kidney disease. Nat Rev Nephrol. 2018;14(5):291–312.
Zhang G, Darshi M, Sharma K. The Warburg effect in diabetic kidney disease. Semin Nephrol. 2018;38(2):111–20.
San-Millan I, Brooks GA. Reexamining cancer metabolism: lactate production for carcinogenesis could be the purpose and explanation of the Warburg effect. Carcinogenesis. 2017;38(2):119–33.
San-Millan I, Julian CG, Matarazzo C, Martinez J, Brooks GA. Is lactate an oncometabolite? Evidence supporting a role for lactate in the regulation of transcriptional activity of cancer-related genes in MCF7 breast cancer cells. Front Oncol. 2019;9:1536.
Ge T, Yang J, Zhou S, Wang Y, Li Y, Tong X. The role of the pentose phosphate pathway in diabetes and cancer. Front Endocrinol (Lausanne). 2020;11:365.
Yang HC, Wu YH, Yen WC, Liu HY, Hwang TL, Stern A, et al. The redox role of G6PD in cell growth, cell death, and cancer. Cells. 2019;8(9).
Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang JK, Shen M, et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science. 2011;334(6060):1278–83.
Xu Y, Zhang Z, Hu J, Stillman IE, Leopold JA, Handy DE, et al. Glucose-6-phosphate dehydrogenase-deficient mice have increased renal oxidative stress and increased albuminuria. FASEB J. 2010;24(2):609–16.
Zhang Z, Liew CW, Handy DE, Zhang Y, Leopold JA, Hu J, et al. High glucose inhibits glucose-6-phosphate dehydrogenase, leading to increased oxidative stress and beta-cell apoptosis. FASEB J. 2010;24(5):1497–505.
Spencer NY, Stanton RC. The Warburg effect, lactate, and nearly a century of trying to cure cancer. Semin Nephrol. 2019;39(4):380–93.
Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452(7184):230–3.
Schormann N, Hayden KL, Lee P, Banerjee S, Chattopadhyay D. An overview of structure, function, and regulation of pyruvate kinases. Protein Sci. 2019;28(10):1771–84.
Dayton TL, Jacks T, Vander Heiden MG. PKM2, cancer metabolism, and the road ahead. EMBO Rep. 2016;17(12):1721–30 Important. Excellent review of glucose metabolism, PKM, and the Warburg Effect.
Qi W, Keenan HA, Li Q, Ishikado A, Kannt A, Sadowski T, et al. Pyruvate kinase M2 activation may protect against the progression of diabetic glomerular pathology and mitochondrial dysfunction. Nat Med. 2017;23(6):753–62 Very Important. Detailed analysis of animal models and human kidney samples demonstrating a potential central role for PKM2 in the development and progression of diabetid kidney disease.
Sun JK, Keenan HA, Cavallerano JD, Asztalos BF, Schaefer EJ, Sell DR, et al. Protection from retinopathy and other complications in patients with type 1 diabetes of extreme duration: the joslin 50-year medalist study. Diabetes Care. 2011;34(4):968–74.
Keenan HA, Costacou T, Sun JK, Doria A, Cavellerano J, Coney J, et al. Clinical factors associated with resistance to microvascular complications in diabetic patients of extreme disease duration: the 50-year medalist study. Diabetes Care. 2007;30(8):1995–7.
Gordin D, Harjutsalo V, Tinsley L, Fickweiler W, Sun JK, Forsblom C, et al. Differential association of microvascular attributions with cardiovascular disease in patients with long duration of type 1 diabetes. Diabetes Care. 2018;41(4):815–22.
Li J, Sun YBY, Chen W, Fan J, Li S, Qu X, et al. Smad4 promotes diabetic nephropathy by modulating glycolysis and OXPHOS. EMBO Rep. 2020;21(2):e48781.
Srivastava SP, Li J, Kitada M, Fujita H, Yamada Y, Goodwin JE, et al. SIRT3 deficiency leads to induction of abnormal glycolysis in diabetic kidney with fibrosis. Cell Death Dis. 2018;9(10):997.
Nathan DM, Group DER. The diabetes control and complications trial/epidemiology of diabetes interventions and complications study at 30 years: overview. Diabetes Care. 2014;37(1):9–16.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Robert C. Stanton declares no potential conflicts of interest.
Human and Animal Rights
All reported studies/experiments with human or animal subjects performed by the authors have been previously published and complied with all applicable ethical standards (including the Helsinki declaration and its amendments, institutional/national research committee standards, and international/national/institutional guidelines).
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Microvascular Complications—Nephropathy
Rights and permissions
About this article

Cite this article
Stanton, R.C. Role of Glucose Metabolism and Mitochondrial Function in Diabetic Kidney Disease. Curr Diab Rep 21, 6 (2021). https://doi.org/10.1007/s11892-020-01372-2
Accepted:
Published:
DOI: https://doi.org/10.1007/s11892-020-01372-2