
Abstract
Purpose of Review
Diabetic nephropathy (DN) has become the leading cause of end-stage renal disease (ESRD) worldwide. Accumulating evidence suggests that endoplasmic reticulum (ER) stress plays a major role in the development and progression of DN. Recent findings suggested that many attributes of DN, such as hyperglycemia, proteinuria, and increased advanced glycation end products and free fatty acids, can all trigger unfolded protein response (UPR) in kidney cells. Herein, we review the current knowledge on the role of ER stress in the setting of kidney injury with a specific emphasis on DN.
Recent Findings
As maladaptive ER stress response caused by excessively prolonged UPR will eventually cause cell death and increase kidney injury, several ER stress inhibitors have been shown to improve DN in animal models, albeit blocking both adaptive and maladaptive UPR. More recently, reticulon-1A (RTN1A), an ER-associated protein, was shown to be increased in both human and mouse diabetic kidneys. Its expression correlates with the progression of DN, and its polymorphisms are associated with kidney disease in people with diabetes. Increased RTN1A expression heightened the ER stress response and renal cell apoptosis, and conversely reduced RTN1A in renal cells decreased apoptosis and ameliorated kidney injury and DN progression, suggesting that RTN1A may be a novel target to specifically restrain the maladaptive UPR.
Summary
These findings suggest that ER stress response in renal cells is a key driver of progression of DN and that the inhibition of the unchecked ER stress response in DN, such as by inhibition of RTN1A function, may be a promising therapeutic approach against DN.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Diabetic nephropathy (DN) is the most common cause of end-stage renal disease (ESRD) worldwide, and the incidences of diabetes and DN have been increasing over the last decade [1]. Regardless of the recent advances, the incidence and prevalence of DN remains high due to limited therapeutic options. In the US, more than 30% of diabetic patients receive dialysis or kidney transplantation as a result of DN [2]. Clinically, DN is defined by the appearance of microalbuminuria at the early stage that progresses toward ESRD over time. Morphologically, DN is characterized by glomerular basement membrane (GBM) thickening, mesangial expansion, podocyte and tubular cell injury at the early stage and diffuse/nodular glomerulosclerosis and tubulointerstitial fibrosis with inflammation at the later stage of the disease.
Over the past decade, the underlying mechanisms predisposing to the development and progression of DN have been under active investigation. Several lines of evidence suggest that endoplasmic reticulum (ER) stress plays a major role in the development and progression of DN [3,4,5,6]. Hyperglycemia, proteinuria, advanced glycation end products (AGEs) and free fatty acids (FFA) have been reported as key inducers of ER stress and its downstream signaling pathway activation in diabetic kidneys [7, 8]. More recently, several novel ER-associated molecules have been described, contributing to ER stress-mediated kidney injury in DN [5, 9, 10••]. Herein, we present a review of recent studies on the role of ER stress in DN, with a highlight of new findings.
UPR and ER Stress
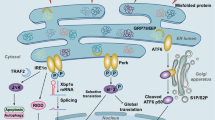
ER is an important organelle necessary for protein synthesis, folding, and maturation in the eukaryotic cell. Disruption of ER homeostasis leads to the accumulation of unfolded or misfolded proteins, resulting in ER stress and triggering the unfolded protein response (UPR) [11]. The ER chaperone protein glucose-regulated protein 78 (GRP78/BiP) serves as a master modulator for the UPR signaling network. Under ER stress, GRP78 preferentially binds to misfolded or unfolded proteins and dissociates from the 3 main ER sensors, namely protein kinase R-like ER kinase (PERK), inositol requiring-1α (IRE1α), and activating transcription factor 6 (ATF6), allowing their activation [12] (Fig. 1a). Dissociation of GRP78 from PERK allows its homodimerization and transphosphorylation that results in phosphorylation of eukaryotic translation initiation factor-2α (eIF2α), leading to the activation of activating downstream transcript factor 4 (ATF4) and UPR target genes such as C/EBP homologous protein (CHOP) and growth arrest and DNA damage inducible protein-34 (GADD34) [4, 13]. In parallel, ATF6 released from GRP78 moves to the Golgi where it is cleaved by site-1 and site-2 proteases (S1P/S2P), and phosphorylated IRE1α results in the splicing of X box binding protein-1 (XBP-1). As a result, both 50 kD ATF6 (p50ATF6) and spliced XBP-1 (XBP-1s) translocate to the nucleus to drive the activation of a group of transcriptional factors involved in ER maintenance, expansion, and ER-associated degradation (ERAD) proteins [14, 15].

ER stress response in DN. a Activation of UPR and ERAD in kidney cells in response to diabetic milieu. b Potential mechanism of RTN1A in ER stress and apoptosis induction in kidney cells. DN diabetic nephropathy, UPR unfolded protein response, ERAD endoplasmic reticulum-associated degradation, RTN1A reticulon-1A, ER endoplasmic reticulum
Adaptive phase of UPR is initiated to restore the homeostasis and protect cells against injury via increasing ER capacity and by enhancing the degradation of misfolded protein in ER by ERAD. Terminally misfolded proteins are deglycosylated by enzymes such as ER mannosidase I (Man I) and interact with ER degradation-enhancing α-mannosidase-like protein (EDEM) and other ERAD lectins. Substrates are then translocated into the cytosol, where they undergo ubiquitination that marks the protein for destruction by the proteasome [16•]. However, excessively prolonged UPR in ER will finally cause cell death by triggering pro-apoptotic signaling such as CHOP, TRB3, c-Jun N-terminal kinase (JNK), and caspase-12 pathways [3, 4, 17].
ER Stress and Kidney Diseases
ER stress has been shown to contribute to the development and progression of chronic kidney disease (CKD) [16•, 18]. Increased expression of the ER stress markers was observed in both glomeruli and tubular interstitium in human or murine model of kidney diseases. Heightened ER stress was observed in podocyte of proteinuric rats with passive Heymann nephritis [19]. Activation of GRP78 and eIF2α phosphorylation in glomeruli was also evidenced in a model of experimental glomerulonephritis [20]. Nakajo et al. [21] demonstrated the presence of ER stress in the rat model of minimal-change nephrotic syndrome by puromycin aminonucleoside (PAN), suggesting a critical role of ER stress in podocyte injury that is likely induced by albuminuria. In humans and experimental mouse models with nephrotic syndrome, ER stress markers are also elevated in tubular epithelial cells [22, 23]. In addition to glomerular cells, ER stress is also a key pathologic process leading to tubular cell injury in acute kidney injury (AKI) [24]. CHOP−/− mice or mice treated with 4-phenylbutyrate (4-PBA), an ER stress inhibitor, were protected against tunicamycin-induced AKI [25]. Heterozygous mutant-GRP78 mice also showed severe tubulointerstitial lesions with aging, indicating that this ER associated protein was important to maintaining the integrity of tubulointerstitial structure [26].
Cell Type-Specific ER Stress in Diabetic Nephropathy
A large body of evidence suggests that dysfunction of ER stress is associated with the onset and progression of DN. For example, microarray data showed upregulated expression of UPR genes, including ER chaperones GRP78, hypoxia up-regulated 1 (HYOU1), and XBP-1 from human biopsies of established DN compared with normal controls [7]. In this section, we have summarized some of the recent studies on the role of ER stress in different kidney cell types in DN.
Podocyte Injury and ER Stress
Podocytes are highly specialized and terminally differentiated glomerular epithelial cells, and their dysfunction causes defective glomerular filtration, resulting in the onset of proteinuria [27, 28]. Podocytes are thought to be highly susceptible to ER stress due to their high protein-folding capacity in the ER and their high levels of anabolic or catabolic activities [9]. High glucose concentrations can induce ER stress and apoptosis in podocytes [29, 30]. In addition, persistent proteinuria contributes to ER stress-mediated glomerular dysfunction in DN and is one of the important mechanisms in albumin-induced podocyte injury [7, 31]. It is well known that free fatty acids (FFAs) are critically involved in the pathogenesis of diabetes mellitus (DM). FFAs can induce podocyte ER stress, leading to the induction of ER chaperone BiP and proapoptotic transcription factor CHOP. FFAs can also increase apoptosis of podocytes in vitro, as well as in diabetic mice in vivo [8]. The mechanism of FFA-induced ER stress in podocytes may involve the activation of TRB3-mediated MAPK and NF-κB pathway [3]. Very recently, work by Madhusudhan et al. suggested that impaired insulin signaling impedes XBP1 activity in podocytes and exacerbates ATF6-dependent maladaptive ER response in diabetic mice [32•]. However, the exact role of diabetes-induced ER stress in podocytes remains unclear.
Mesangial Cell Injury and ER Stress
Mesangial cells (MCs) are essential in maintaining the structural integrity of the glomerular tuft and modulating the glomerular filtration. MCs are known to play an important role in DN progression [33]. However, the role of ER stress in mediating MC injury in DN remains largely unknown, as studies in this research area are still very limited.
Reactive oxygen species (ROS) and hyperglycemia are commonly considered to be triggers of ER stress in cultured mesangial cells, which can be alleviated by oleanolic acid treatment [34]. Studies have demonstrated that hyperglycemia and ROS-induced ER stress can result in MC proliferation and extracellular matrix (ECM) overproduction, whereas overexpression of XBP-1s suppresses the high glucose-induced ECM synthesis and ROS generation in cultured MCs [35]. Induction of ER stress was also shown to be involved in the AGE-induced MC apoptosis, while inhibition of ER stress by 4-PBA reversed AGE-induced apoptotic response in mesangial cells [6]. Fatty acid-binding protein 4 (FABP4), a carrier protein for fatty acids, has been linked to diabetes and diabetic nephropathy (DN). Very recently, Yao et al. reported that FABP4 was expressed mainly in glomerular mesangial cells and that its expression was increased in DN kidney biopsies [36]. The up-regulation of FABP4 was accompanied by increased expression of GRP78 and Caspase-12 and decreased expression of B-cell CLL/lymphoma 2 (Bcl-2) in the glomeruli, indicating that FABP4-mediated ER stress might be involved in mesangial cell apoptosis in DN.
Glomerular Endothelial Cell Injury and ER Stress
Endothelial cell injury is a central event in the development of diabetic vascular diseases, and DN is one of the most common microvascular complications. Prolonged ER stress response contributes to atherosclerosis and endothelial dysfunction [37, 38]. A number of studies in cultured endothelial cells and animal models have provided insights into the molecular mechanisms linking induction of ER stress to endothelial cell dysfunction [39,40,41]. High glucose-induced ER stress has been closely linked to various aspects of endothelial cell dysfunction in patients with diabetes [42]. Chen et al. [43] reported that high glucose-induced ER stress in retinal endothelial cells results in the activation of PERK–eIF2α-ATF4 pathway. Genetic inhibition of ATF4 ameliorated STAT3-mediated retinal inflammation and vascular injury in STZ-induced diabetic mice suggest a pivotal role of ER stress and the ATF4/STAT3 pathway in diabetic microvascular diseases. In glomerular endothelial cells, recent studies showed that advanced oxidation protein products (AOPPs) triggered endothelial-to-mesenchymal transition (EndMT) through the induction of ER stress [44]. Treatment with asymmetric dimethylarginine (ADMA), an endogenous competitive inhibitor of nitric oxide synthase (NOS) enzymes, activated PERK and IRE1α pathways, which induced CHOP expression and c-Jun N-terminal kinase (JNK) phosphorylation in glomerular endothelial cells [45]. Angiopoietin1 (Angpt1), a vascular growth factor, has been recently found to ameliorate glomerular endothelial cell dysfunction by suppression of PERK/CHOP pathway [46]. Since at this time only few reports focus on the role of ER stress in glomerular endothelial cells in DN, future research is needed to better understand the mechanism by which ER stress induces glomerular endothelial dysfunction in DN.
Renal Tubular Injury and ER Stress
Renal tubular epithelial cells (RTECs) are key target cells that are highly vulnerable to damage in conditions of glucose-induced metabolic disorder and play an important role in the progression of DN [7, 22]. ER stress has been recgnized to contribute to the development and progression of CKD, especially in tubulointerstitial injury [47, 48].
ER stress is closely associated with tubular injury in human DN. Microarray analysis of the tubulointerstitial compartment of renal biopsies obtained from DN patients revealed a significant upregulation of UPR-associated genes, including the ER chaperones BiP and HYOU1 [7]. It is well known that proteinuria contributes to tubulointerstitial injury in DN, as ER stress is one of the important mechanisms in albumin-induced tubular injury [7, 49]. In order to ascertain the central ER chaperone function of BiP, Mimura et al. generated a BiP knock-in mouse expressing BiP with a mutated retrieval sequence to cause a defect in ER function without completely eliminating BiP [50]. Although the heterozygous BiP-mutant mice showed no gross phenotype, aged BiP-mutant mice showed more severe tubulointerstitial lesions than in age-matched wildtype mice [26]. In addition, compared with age-matched wild-type controls, young heterozygous BiP-mutant mice showed more severe renal injury to albumin overload-induced proteinuric kidney disease. This injury was accompanied by caspase-12 activation and tubular cell apoptosis, suggesting that prolonged ER stress response is involved in the pathogenesis of chronic renal tubulointerstitial injury in proteinuric kidney diseases.
Recent studies have focused on some specific inducers that trigger UPR in renal tubular cells and investigated the possible mechanisms of ER stress in the progression of DN. Liu et al. reported that ATF4/p16 signaling was activated by AGE-induced ER stress in RTECs and was associated with DN progression [51]. Similarly, receptor of AGE (RAGE) mediated ER stress in RTECs to induce premature senescence via p21 signaling in DN [52]. Palmitic acid (PA) is an important promoter of tubulointerstitial damage in DN and activates ER stress and apoptosis in proximal tubular cells [53]. Activation of cannabinoid receptor 1 (CB1R) by PA results in ER stress and apoptosis in proximal tubular cells in DN [54]. A recent study found that urotensin II induces ER stress and increases extracellular matrix production in RTECs of early diabetic mice [55]. Taken together, these findings suggest that ER stress is an important mechanism of tubulointerstitial injury leading to the progression of DN.
Therapeutics by Targeting ER Stress
There has been considerable interest in developing compounds that modulate ER stress response in the recent years. FDA-approved chemical chaperones such as 4-PBA and taurine-conjugated ursodeoxycholic acid (TUDCA) are classic ER stress inhibitors that enhance ER folding capacity, alleviate ER stress, restore glucose tolerance, and improve insulin sensitivity in metabolic disorders such as diabetes, obesity, and insulin resistance [56, 57•]. Recent work showed that both 4-PBA and TUDCA can attenuate kidney injury not only in type I or type II diabetic murine model [58,59,60] but also in human subjects with obesity and insulin-resistance via ameliorating ER stress [61,62,63].
Of note, several recent studies demonstrated the beneficial effects of herbal or plant medicines in the treatment of DN via inhibition of ER stress. Astragaloside IV (AS-IV) has been shown to ameliorate albuminuria and kidney injury in diabetic rats, and this was associated with the inhibition of ER stress signaling [64•, 65]. Huangkui, a Chinese herbal extracted from Abelmoschus manihot (L.) medic (AM), was recently found to reduce ER stress and alleviate renal inflammation and glomerular injury in DN rats. Oleanolic acid (OA), a natural compound in fruits and vegetables, was shown to reduce ER stress and had therapeutic effects for DN [34]. Quercetin, a flavonoid found in a wide variety of plants and human diet, suppressed ER stress pathways and exhibited a promising effect in preventing glomerular endothelial cell apoptosis [45]. However, as the above-mentioned inhibitors and herbal medicines may have multiple biological functions and are not specific for inhibition of ER stress response, it is important to develop drugs that target specific molecules in the ER stress pathway that are activated in diabetic kidneys. In addition, the ER stress response may have both protective and deleterious features in different cellular contexts at various disease stages. Therefore, an improved understanding of the molecules regulating this process in a cell and disease stage-specific manner would expedite the identification of better therapeutic strategies targeting ER stress as a therapy for DN.
Role of Reticulon-1A in Diabetic Nephropathy
In a recent study, we identified a novel gene, reticulon-1 (RTN1), which was associated with the progression of kidney diseases [10••]. Reticulons were first described in neuroendocrine cells, localize primarily to the ER membrane, and are thought to function as ER-shaping proteins [66, 67]. Of the 3 known RTN1 isoforms (RTN1-A, RTN1-B, RTN-1C), upregulation of RTN1A was confirmed in the kidneys of both human and murine models of HIV-associated nephropathy (HIVAN) and DN. Interestingly, both mRNA and protein levels of RTN1A in the kidneys are associated with serum creatinine and eGFR in patients with DN [10••]. Genetic association studies showed that common variants in RTN1A gene were associated with ESRD in people with diabetes [68]. In vitro, RTN1A mediates hyperglycemia and albumin-induced ER stress in both podocytes and renal tubular cells. Overexpression of RTN1A leads to prolonged ER stress and apoptosis of kidney cells that is likely mediated through the sustained PERK activation and CHOP expression (Fig. 1b) [10••]. Global knockout of RTN1A attenuates albuminuria and kidney injury in an animal model of DN. In addition, knockdown of RTN1A specifically in kidney tubular cells ameliorates ER stress signaling and prevents apoptosis in mice with protein-overload nephropathy [69•]. Together, these findings suggest a pivotal role for RTN1A in ER-stress-induced kidney injury. RTN1A could be a new target for treatment of DN by specifically inhibiting the ER stress pathway.
Conclusions
A large body of evidence suggests that ER stress plays a major role in the development and progression of kidney disease, including DN, and that reducing ER stress may thwart the kidney disease progression. Evidence further suggests that the attenuation of maladaptive ER stress response decreases apoptosis of renal cells and attenuates kidney injury and may be a novel therapeutic approach in kidney disease, including diabetic nephropathy.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Jha V, Garcia-Garcia G, Iseki K, Li Z, Naicker S, Plattner B, et al. Chronic kidney disease: global dimension and perspectives. Lancet. 2013;382(9888):260–72.
Collins AJ, Foley RN, Herzog C, Chavers B, Gilbertson D, Ishani A, et al. US Renal Data System 2010 annual data report. Am J Kidney Dis. 2011;57(1 Suppl 1):A8, e1–526.
Morse E, Schroth J, You YH, Pizzo DP, Okada S, Ramachandrarao S, et al. TRB3 is stimulated in diabetic kidneys, regulated by the ER stress marker CHOP, and is a suppressor of podocyte MCP-1. Am J Physiol Renal Physiol. 2010;299(5):F965–72.
Cunard R, Sharma K. The endoplasmic reticulum stress response and diabetic kidney disease. Am J Physiol Renal Physiol. 2011;300(5):F1054–61.
Borsting E, Patel SV, Decleves AE, Lee SJ, Rahman QM, Akira S, et al. Tribbles homolog 3 attenuates mammalian target of rapamycin complex-2 signaling and inflammation in the diabetic kidney. J Am Soc Nephrol. 2014;25(9):2067–78.
Chiang CK, Wang CC, Lu TF, Huang KH, Sheu ML, Liu SH, et al. Involvement of endoplasmic reticulum stress, autophagy, and apoptosis in advanced glycation end products-induced glomerular mesangial cell injury. Sci Rep. 2016;6:34167.
Lindenmeyer MT, Rastaldi MP, Ikehata M, Neusser MA, Kretzler M, Cohen CD, et al. Proteinuria and hyperglycemia induce endoplasmic reticulum stress. J Am Soc Nephrol. 2008;19(11):2225–36.
Sieber J, Lindenmeyer MT, Kampe K, Campbell KN, Cohen CD, Hopfer H, et al. Regulation of podocyte survival and endoplasmic reticulum stress by fatty acids. Am J Physiol Renal Physiol. 2010;299(4):F821–9.
Inagi R, Nangaku M, Onogi H, Ueyama H, Kitao Y, Nakazato K, et al. Involvement of endoplasmic reticulum (ER) stress in podocyte injury induced by excessive protein accumulation. Kidney Int. 2005;68(6):2639–50.
•• Fan Y, Xiao W, Li Z, Li X, Chuang PY, Jim B, et al. RTN1 mediates progression of kidney disease by inducing ER stress. Nat Commun. 2015;6:7841. This study made an in-depth exploration of a previously undescribed role of RTN1 in ER stress mediated kidney injury and CKD progression.
Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clin Invest. 2002;110(10):1389–98.
Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13(2):89–102.
Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000;5(5):897–904.
Yoshida H, Nadanaka S, Sato R, Mori K. XBP1 is critical to protect cells from endoplasmic reticulum stress: evidence from Site-2 protease-deficient Chinese hamster ovary cells. Cell Struct Funct. 2006;31(2):117–25.
Chiang WC, Hiramatsu N, Messah C, Kroeger H, Lin JH. Selective activation of ATF6 and PERK endoplasmic reticulum stress signaling pathways prevent mutant rhodopsin accumulation. Invest Ophthalmol Vis Sci. 2012;53(11):7159–66.
• Cybulsky AV. The intersecting roles of endoplasmic reticulum stress, ubiquitin- proteasome system, and autophagy in the pathogenesis of proteinuric kidney disease. Kidney Int. 2013;84(1):25–33. This review provides a systemic and comprehensive interpretion on ER stress, ubiquitin–proteasome system, and autophagy in proteinuric glomerular diseases.
Sanchez-Nino MD, Benito-Martin A, Ortiz A. New paradigms in cell death in human diabetic nephropathy. Kidney Int. 2010;78(8):737–44.
Inagi R. Endoplasmic reticulum stress in the kidney as a novel mediator of kidney injury. Nephron Exp Nephrol. 2009;112(1):e1–9.
Cybulsky AV, Takano T, Papillon J, Bijian K. Role of the endoplasmic reticulum unfolded protein response in glomerular epithelial cell injury. J Biol Chem. 2005;280(26):24396–403.
Cybulsky AV, Takano T, Papillon J, Kitzler TM, Bijian K. Endoplasmic reticulum stress in glomerular epithelial cell injury. Am J Physiol Renal Physiol. 2011;301(3):F496–508.
Nakajo A, Khoshnoodi J, Takenaka H, Hagiwara E, Watanabe T, Kawakami H, et al. Mizoribine corrects defective nephrin biogenesis by restoring intracellular energy balance. J Am Soc Nephrol. 2007;18(9):2554–64.
Lhotak S, Sood S, Brimble E, Carlisle RE, Colgan SM, Mazzetti A, et al. ER stress contributes to renal proximal tubule injury by increasing SREBP-2-mediated lipid accumulation and apoptotic cell death. Am J Physiol Renal Physiol. 2012;303(2):F266–78.
Wu J, Zhang R, Torreggiani M, Ting A, Xiong H, Striker GE, et al. Induction of diabetes in aged C57B6 mice results in severe nephropathy: an association with oxidative stress, endoplasmic reticulum stress, and inflammation. Am J Pathol. 2010;176(5):2163–76.
Linkermann A, Chen G, Dong G, Kunzendorf U, Krautwald S, Dong Z. Regulated cell death in AKI. J Am Soc Nephrol. 2014;25(12):2689–701.
Carlisle RE, Brimble E, Werner KE, Cruz GL, Ask K, Ingram AJ, et al. 4-phenylbutyrate inhibits tunicamycin-induced acute kidney injury via CHOP/GADD153 repression. PLoS ONE. 2014;9(1):e84663.
Kimura K, Jin H, Ogawa M, Aoe T. Dysfunction of the ER chaperone BiP accelerates the renal tubular injury. Biochem Biophys Res Commun. 2008;366(4):1048–53.
Mundel P, Shankland SJ. Podocyte biology and response to injury. J Am Soc Nephrol. 2002;13(12):3005–15.
Nagata M. Podocyte injury and its consequences. Kidney Int. 2016;89(6):1221–30.
Fang L, Zhou Y, Cao H, Wen P, Jiang L, He W, et al. Autophagy attenuates diabetic glomerular damage through protection of hyperglycemia-induced podocyte injury. PLoS ONE. 2013;8(4):e60546.
Cao Y, Hao Y, Li H, Liu Q, Gao F, Liu W, et al. Role of endoplasmic reticulum stress in apoptosis of differentiated mouse podocytes induced by high glucose. Int J Mol Med. 2014;33(4):809–16.
Chen YM, Zhou Y, Go G, Marmerstein JT, Kikkawa Y, Miner JH. Laminin beta2 gene missense mutation produces endoplasmic reticulum stress in podocytes. J Am Soc Nephrol. 2013;24(8):1223–33.
• Madhusudhan T, Wang H, Dong W, Ghosh S, Bock F, Thangapandi VR, et al. Defective podocyte insulin signalling through p85-XBP1 promotes ATF6-dependent maladaptive ER-stress response in diabetic nephropathy. Nat Commun. 2015;6:6496. This study demonstrated that impaired insulin signaling directly impedes sXBP1 activity in podocytes, which was associated with maladaptive ER stress response in DN.
Abboud HE. Mesangial cell biology. Exp Cell Res. 2012;318(9):979–85.
Lee ES, Kim HM, Kang JS, Lee EY, Yadav D, Kwon MH, et al. Oleanolic acid and N-acetylcysteine ameliorate diabetic nephropathy through reduction of oxidative stress and endoplasmic reticulum stress in a type 2 diabetic rat model. Nephrol Dial Transplant. 2016;31(3):391–400.
Shao D, Liu J, Ni J, Wang Z, Shen Y, Zhou L, et al. Suppression of XBP1S mediates high glucose-induced oxidative stress and extracellular matrix synthesis in renal mesangial cell and kidney of diabetic rats. PLoS ONE. 2013;8(2):e56124.
Yao F, Li Z, Ehara T, Yang L, Wang D, Feng L, et al. Fatty acid-binding protein 4 mediates apoptosis via endoplasmic reticulum stress in mesangial cells of diabetic nephropathy. Mol Cell Endocrinol. 2015;411:232–42.
Kassan M, Galan M, Partyka M, Saifudeen Z, Henrion D, Trebak M, et al. Endoplasmic reticulum stress is involved in cardiac damage and vascular endothelial dysfunction in hypertensive mice. Arterioscler Thromb Vasc Biol. 2012;32(7):1652–61.
Tabas I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ Res. 2010;107(7):839–50.
Cimellaro A, Perticone M, Fiorentino TV, Sciacqua A, Hribal ML. Role of endoplasmic reticulum stress in endothelial dysfunction. Nutr Metab Cardiovasc Dis. 2016;26(10):863–71.
Lenna S, Han R, Trojanowska M. Endoplasmic reticulum stress and endothelial dysfunction. IUBMB Life. 2014;66(8):530–7.
Li WH, Li YZ, Song DD, Wang XR, Liu M, Wu XD, et al. Calreticulin protects rat microvascular endothelial cells against microwave radiation-induced injury by attenuating endoplasmic reticulum stress. Microcirculation. 2014;21(6):506–15.
Basha B, Samuel SM, Triggle CR, Ding H. Endothelial dysfunction in diabetes mellitus: possible involvement of endoplasmic reticulum stress? Exp Diabetes Res. 2012;2012:481840.
Chen Y, Wang JJ, Li J, Hosoya KI, Ratan R, Townes T, et al. Activating transcription factor 4 mediates hyperglycaemia-induced endothelial inflammation and retinal vascular leakage through activation of STAT3 in a mouse model of type 1 diabetes. Diabetologia. 2012;55(9):2533–45.
Liang X, Duan N, Wang Y, Shu S, Xiang X, Guo T, et al. Advanced oxidation protein products induce endothelial-to-mesenchymal transition in human renal glomerular endothelial cells through induction of endoplasmic reticulum stress. J Diabetes Complicat. 2016;30(4):573–9.
Guo W, Ding J, Zhang A, Dai W, Liu S, Diao Z, et al. The inhibitory effect of quercetin on asymmetric dimethylarginine-induced apoptosis is mediated by the endoplasmic reticulum stress pathway in glomerular endothelial cells. Int J Mol Sci. 2014;15(1):484–503.
Bi X, Niu J, Ding W, Zhang M, Yang M, Gu Y. Angiopoietin-1 attenuates angiotensin II-induced ER stress in glomerular endothelial cells via a Tie2 receptor/ERK1/2-p38 MAPK-dependent mechanism. Mol Cell Endocrinol. 2016;428:118–32.
Lee EK, Jeong JU, Chang JW, Yang WS, Kim SB, Park SK, et al. Activation of AMP-activated protein kinase inhibits albumin-induced endoplasmic reticulum stress and apoptosis through inhibition of reactive oxygen species. Nephron Exp Nephrol. 2012;121(1–2):e38–48.
Kim H, Moon SY, Kim JS, Baek CH, Kim M, Min JY, et al. Activation of AMP-activated protein kinase inhibits ER stress and renal fibrosis. Am J Physiol Renal Physiol. 2015;308(3):F226–36.
Lee JY, Chang JW, Yang WS, Kim SB, Park SK, Park JS, et al. Albumin-induced epithelial-mesenchymal transition and ER stress are regulated through a common ROS-c-Src kinase-mTOR pathway: effect of imatinib mesylate. Am J Physiol Renal Physiol. 2011;300(5):F1214–22.
Mimura N, Hamada H, Kashio M, Jin H, Toyama Y, Kimura K, et al. Aberrant quality control in the endoplasmic reticulum impairs the biosynthesis of pulmonary surfactant in mice expressing mutant BiP. Cell Death Differ. 2007;14(8):1475–85.
Liu J, Yang JR, Chen XM, Cai GY, Lin LR, He YN. Impact of ER stress-regulated ATF4/p16 signaling on the premature senescence of renal tubular epithelial cells in diabetic nephropathy. Am J Physiol Cell Physiol. 2015;308(8):C621–30.
Liu J, Huang K, Cai GY, Chen XM, Yang JR, Lin LR, et al. Receptor for advanced glycation end-products promotes premature senescence of proximal tubular epithelial cells via activation of endoplasmic reticulum stress-dependent p21 signaling. Cell Signal. 2014;26(1):110–21.
Sasaki H, Kamijo-Ikemori A, Sugaya T, Yamashita K, Yokoyama T, Koike J, et al. Urinary fatty acids and liver-type fatty acid binding protein in diabetic nephropathy. Nephron Clin Pract. 2009;112(3):c148–56.
Lim JC, Lim SK, Han HJ, Park SH. Cannabinoid receptor 1 mediates palmitic acid-induced apoptosis via endoplasmic reticulum stress in human renal proximal tubular cells. J Cell Physiol. 2010;225(3):654–63.
Pang XX, Bai Q, Wu F, Chen GJ, Zhang AH, Tang CS. Urotensin II induces ER stress and EMT and increase extracellular matrix production in renal tubular epithelial cell in early diabetic mice. Kidney Blood Press Res. 2016;41(4):434–49.
Nakatani Y, Kaneto H, Kawamori D, Yoshiuchi K, Hatazaki M, Matsuoka TA, et al. Involvement of endoplasmic reticulum stress in insulin resistance and diabetes. J Biol Chem. 2005;280(1):847–51.
• Lee J, Ozcan U. Unfolded protein response signaling and metabolic diseases. J Biol Chem. 2014;289(3):1203–11. A good review highlighting the role of ER stress in various metabolic disorders.
Luo ZF, Feng B, Mu J, Qi W, Zeng W, Guo YH, et al. Effects of 4-phenylbutyric acid on the process and development of diabetic nephropathy induced in rats by streptozotocin: regulation of endoplasmic reticulum stress-oxidative activation. Toxicol Appl Pharmacol. 2010;246(1–2):49–57.
Qi W, Mu J, Luo ZF, Zeng W, Guo YH, Pang Q, et al. Attenuation of diabetic nephropathy in diabetes rats induced by streptozotocin by regulating the endoplasmic reticulum stress inflammatory response. Metabolism. 2011;60(5):594–603.
Cao AL, Wang L, Chen X, Wang YM, Guo HJ, Chu S, et al. Ursodeoxycholic acid and 4-phenylbutyrate prevent endoplasmic reticulum stress-induced podocyte apoptosis in diabetic nephropathy. Lab Investig. 2016;96(6):610–22.
Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313(5790):1137–40.
Kars M, Yang L, Gregor MF, Mohammed BS, Pietka TA, Finck BN, et al. Tauroursodeoxycholic Acid may improve liver and muscle but not adipose tissue insulin sensitivity in obese men and women. Diabetes. 2010;59(8):1899–905.
Xiao C, Giacca A, Lewis GF. Sodium phenylbutyrate, a drug with known capacity to reduce endoplasmic reticulum stress, partially alleviates lipid-induced insulin resistance and beta-cell dysfunction in humans. Diabetes. 2011;60(3):918–24.
• Chen Y, Gui D, Chen J, He D, Luo Y, Wang N. Down-regulation of PERK-ATF4-CHOP pathway by Astragaloside IV is associated with the inhibition of endoplasmic reticulum stress-induced podocyte apoptosis in diabetic rats. Cell Physiol Biochem. 2014;33(6):1975–87. This study showed that the efficacy of Chinese traditional therapy by Astragaloside IV in DN may be through the inhibition of ER stress in kidney cells.
Wang ZS, Xiong F, Xie XH, Chen D, Pan JH, Cheng L. Astragaloside IV attenuates proteinuria in streptozotocin-induced diabetic nephropathy via the inhibition of endoplasmic reticulum stress. BMC Nephrol. 2015;16:44.
GrandPre T, Nakamura F, Vartanian T, Strittmatter SM. Identification of the Nogo inhibitor of axon regeneration as a Reticulon protein. Nature. 2000;403(6768):439–44.
Voeltz GK, Prinz WA, Shibata Y, Rist JM, Rapoport TA. A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell. 2006;124(3):573–86.
Bonomo JA, Palmer ND, He JC, Fan Y, Hicks PJ, Lea JP, et al. Association analysis of the Reticulon 1 gene in end-stage kidney disease. Am J Nephrol. 2015;42(4):259–64.
• Xiao W, Fan Y, Wang N, Chuang PY, Lee K, He JC. Knockdown of RTN1A attenuates ER stress and kidney injury in albumin overload-induced nephropathy. Am J Physiol Renal Physiol. 2016;310(5):F409–15. This study identified a role of RTN1A in mediating albumin overload-induced tubular cell injury through increased ER stress and apoptosis.
Acknowledgements
Y.F. is supported by National Natural Science Foundation of China (81400735) and Chinese Medical Association Funding (15020140602). K.L. is supported by NIH P30 DK079307 and 1R01DK098126. N.W. is supported by National Natural Science Foundation of China (81270824, 81670657). J.C.H. is supported by NIH 1R01DK109683, 1R01DK078897, 1R01DK088541, and P01-DK-56492.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Ying Fan, Kyung Lee, Niansong Wang, and John Cijiang He declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Microvascular Complications—Nephropathy
Rights and permissions
About this article

Cite this article
Fan, Y., Lee, K., Wang, N. et al. The Role of Endoplasmic Reticulum Stress in Diabetic Nephropathy. Curr Diab Rep 17, 17 (2017). https://doi.org/10.1007/s11892-017-0842-y
Published:
DOI: https://doi.org/10.1007/s11892-017-0842-y